


SLU7: A New Hub of Gene Expression Regulation—From Epigenetics to Protein Stability in Health and Disease

 ,
,  ,
,  ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
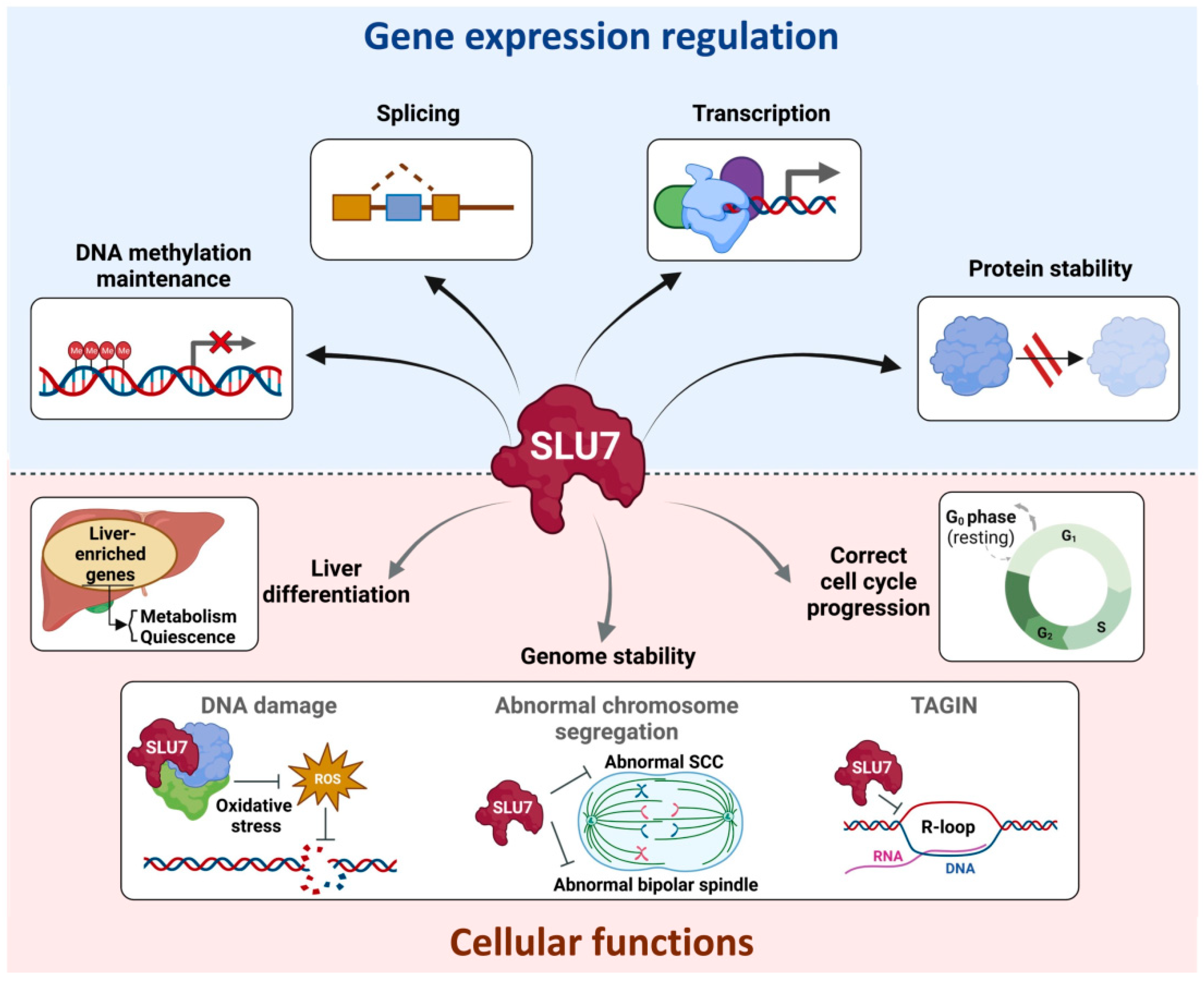{kind=link}
Abstract
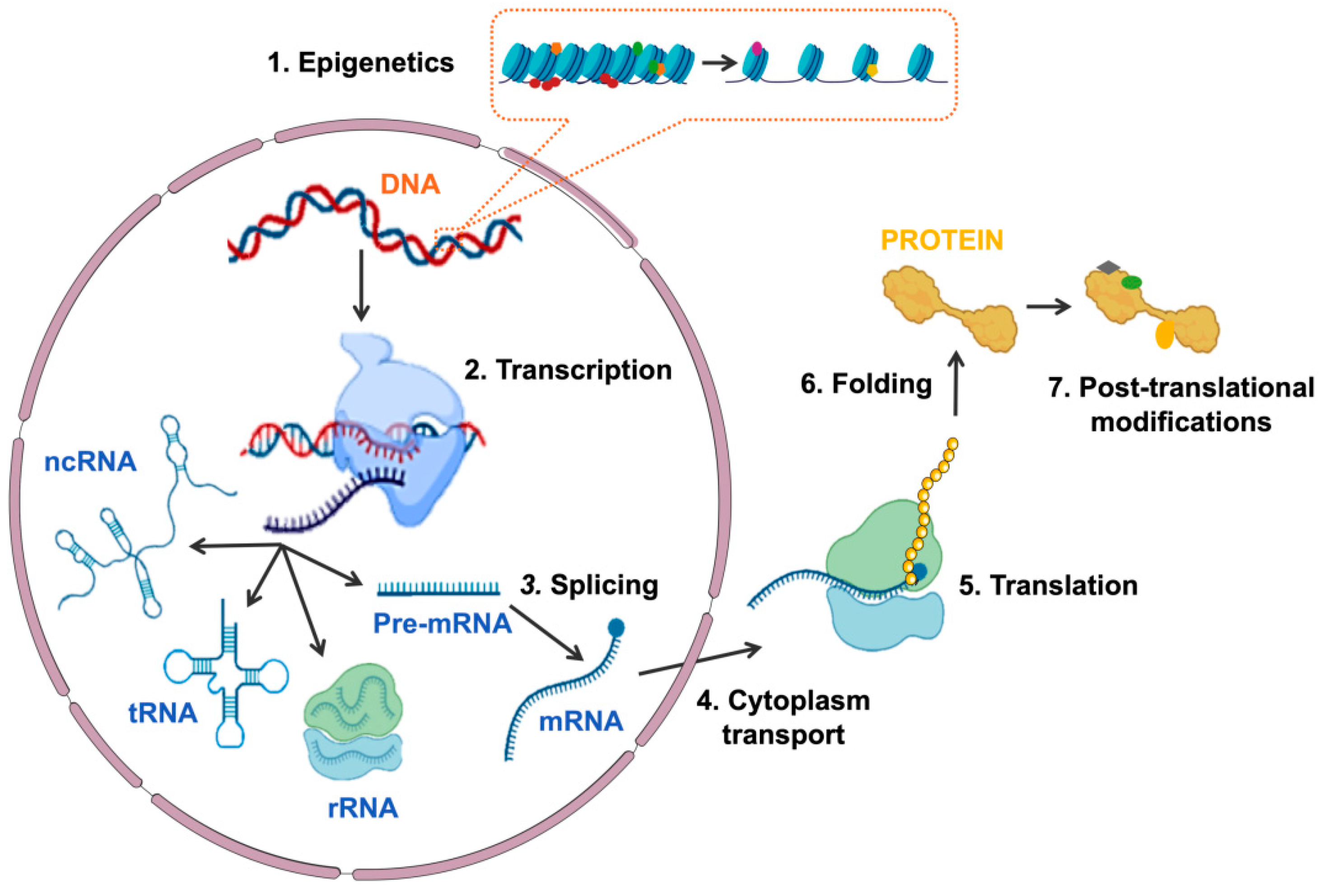
:1. Introduction
2. Characterization of SLU7
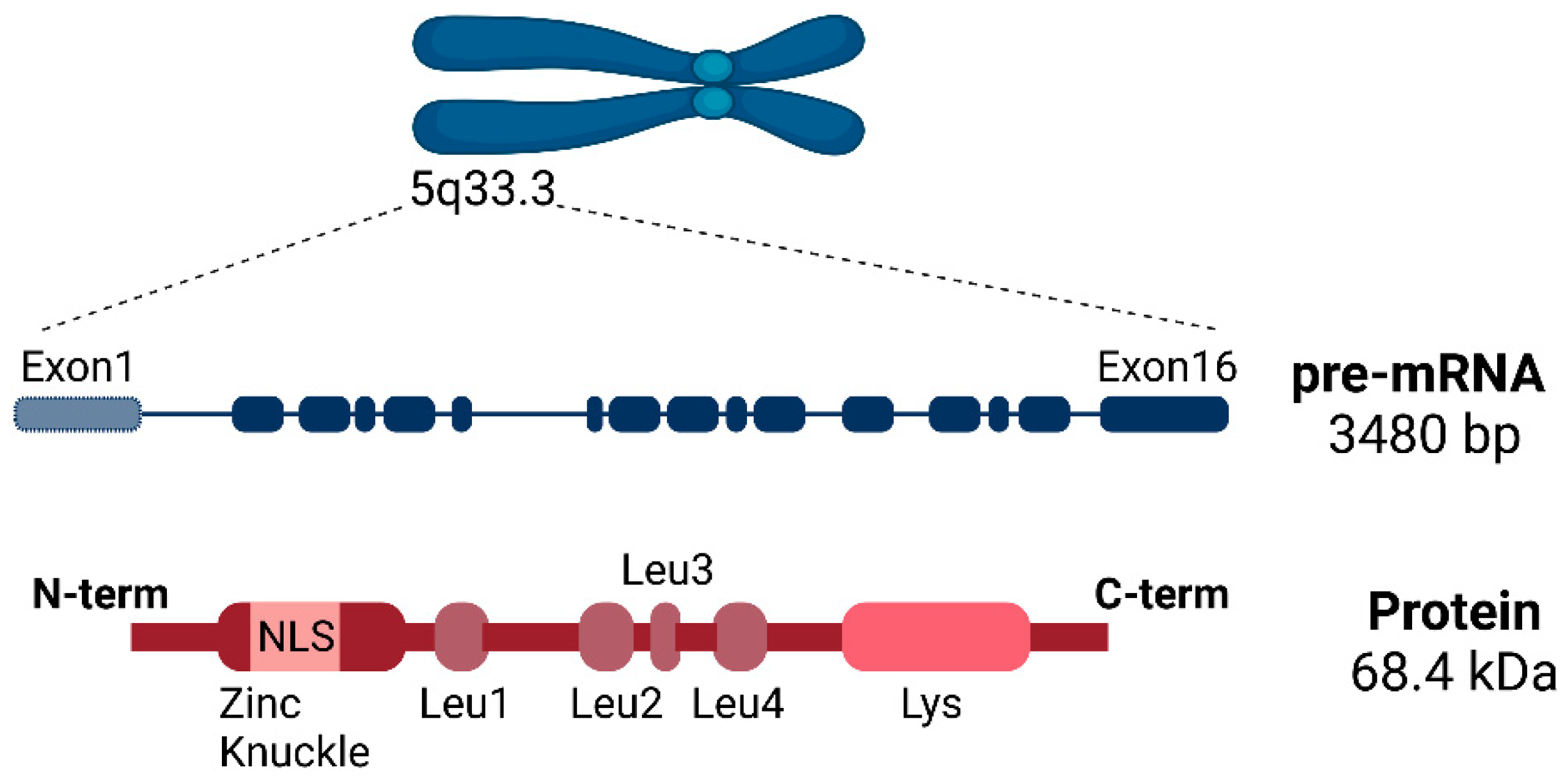
2.1. SLU7 Structure
2.2. SLU7 Expression and Subcellular Localization
3. SLU7 Regulates Gene Expression at Different Levels
3.1. SLU7 as a Splicing Factor
3.1.1. SLU7 and the Splicing Process
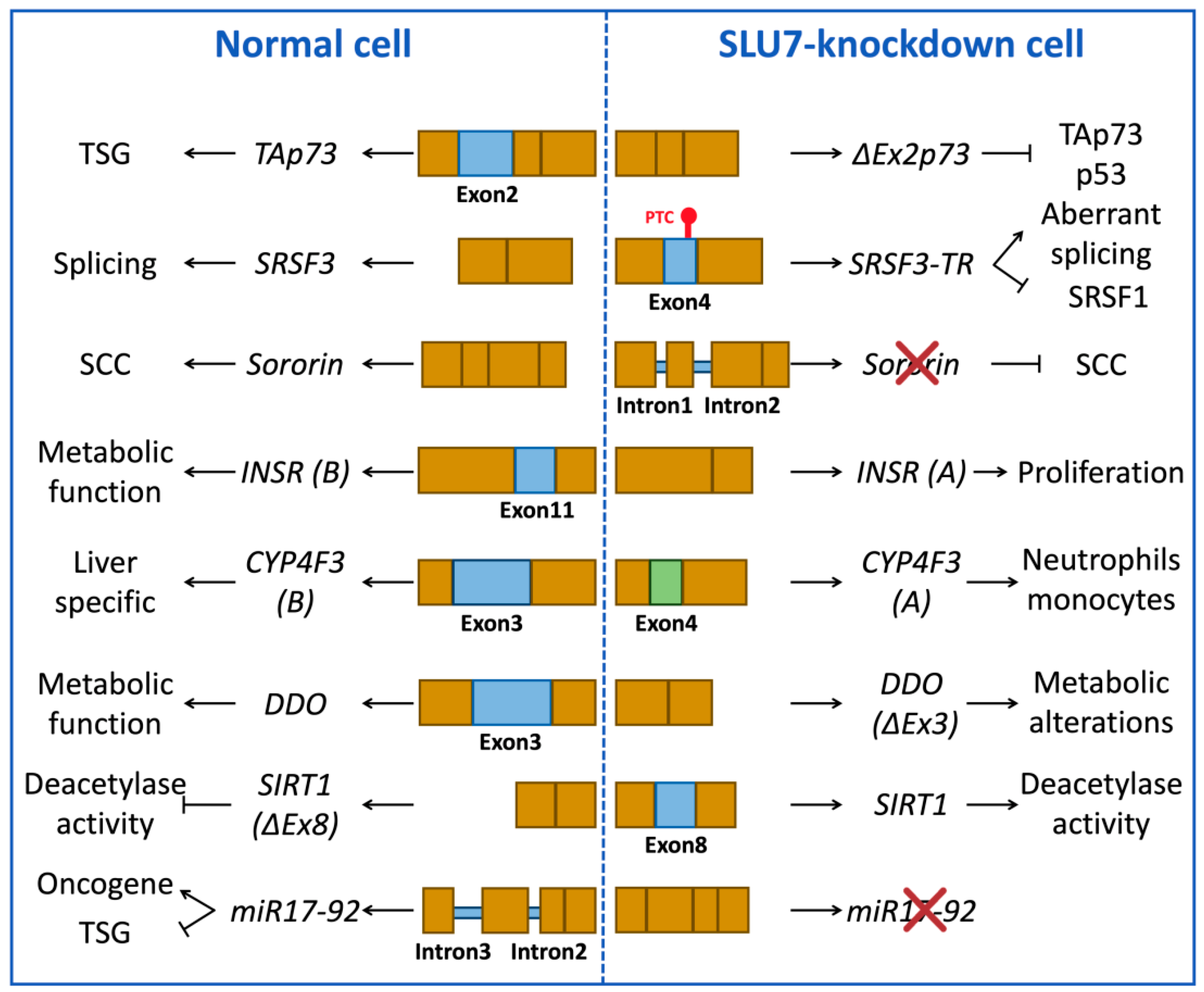
3.1.2. Examples of Splicing Events Associated with SLU7 Activity
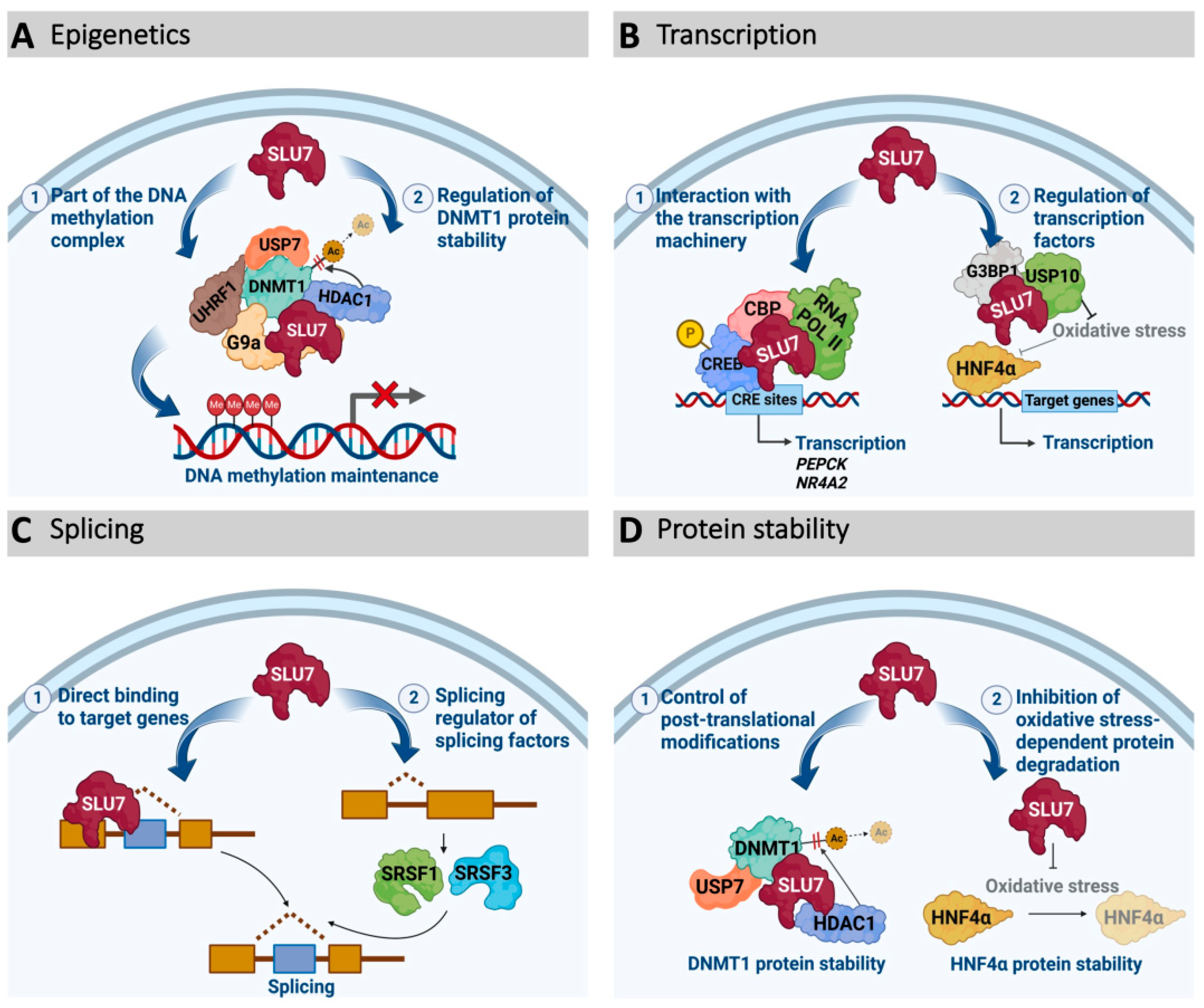
3.2. SLU7 as a Transcription Regulator
3.3. SLU7 as an Epigenetic Modulator
3.4. SLU7 as a Protein Stability Regulator
4. SLU7 Functions
4.1. SLU7 and Liver Differentiation
4.2. SLU7 and Cellular Response to Stress
4.3. SLU7 and Genome Stability
4.4. SLU7 and the Cell Cycle
5. SLU7 in Human Disease
5.1. SLU7 Expression in Pathological Conditions
5.2. SLU7 in Liver Pathology
5.3. SLU7 in Cancer
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Uzman, A. Molecular Biology of the Cell, 4th ed.; Alberts, B., Johnson, A., Lewis, J., Raff, M., Roberts, K., Walter, P., Eds.; Garland Science: New York, NY, USA, 2003. [Google Scholar] [CrossRef]
- Buchberger, E.; Reis, M.; Lu, T.-H.; Posnien, N. Cloudy with a Chance of Insights: Context Dependent Gene Regulation and Impli-cations for Evolutionary Studies. Genes 2019, 10, 492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- André, K.M.; Sipos, E.H.; Soutourina, J. Mediator Roles Going Beyond Transcription. Trends Genet. 2020, 37, 224–234. [Google Scholar] [CrossRef] [PubMed]
- Baylin, S.B.; Esteller, M.; Rountree, M.R.; Bachman, K.E.; Schuebel, K.; Herman, J.G. Aberrant patterns of DNA methylation, chromatin formation and gene expression in cancer. Hum. Mol. Genet. 2001, 10, 687–692. [Google Scholar] [CrossRef]
- Deutschman, C.S. Transcription. Crit. Care Med. 2005, 33, S400–S403. [Google Scholar] [CrossRef] [PubMed]
- Black, D.L. Protein Diversity from Alternative Splicing A Challenge for Bioinformatics and Post-Genome Biology. Cell 2000, 103, 367–370. [Google Scholar] [CrossRef] [Green Version]
- Kornblihtt, A.R.; Schor, I.E.; Alló, M.; Dujardin, G.; Petrillo, E.; Muñoz, M.J. Alternative splicing: A pivotal step between eukaryotic transcription and translation. Nat. Rev. Mol. Cell. Biol. 2013, 14, 153–165. [Google Scholar] [CrossRef]
- Frank, D.; Patterson, B.; Guthrie, C. Synthetic lethal mutations suggest interactions between U5 small nuclear RNA and four proteins required for the second step of splicing. Mol. Cell. Biol. 1992, 12, 5197–5205. [Google Scholar] [CrossRef]
- Chua, K.; Reed, R. Human step II splicing factor hSlu7 functions in restructuring the spliceosome between the catalytic steps of splicing. Genes Dev. 1999, 13, 841–850. [Google Scholar] [CrossRef] [Green Version]
- Chua, K.; Reed, R. The RNA splicing factor hSlu7 is required for correct 3′ splice-site choice. Nature 1999, 402, 207–210. [Google Scholar] [CrossRef]
- Zhang, X.; Yan, C.; Hang, J.; Finci, L.I.; Lei, J.; Shi, Y. An Atomic Structure of the Human Spliceosome. Cell 2017, 169, 918–929. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Zhan, X.; Yan, C.; Zhang, W.; Liu, D.; Lei, J.; Shi, Y. Structures of the human spliceosomes before and after release of the ligated exon. Cell Res. 2019, 29, 274–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhan, X.; Lu, Y.; Zhang, X.; Yan, C.; Shi, Y. Mechanism of exon ligation by human spliceosome. Mol. Cell. 2022, 82, 2769–2778. [Google Scholar] [CrossRef] [PubMed]
- Hegele, A.; Kamburov, A.; Grossmann, A.; Sourlis, C.; Wowro, S.; Weimann, M.; Will, C.L.; Pena, V.; Lührmann, R.; Stelzl, U. Dynamic Protein-Protein Interaction Wiring of the Human Spliceosome. Mol. Cell. 2012, 45, 567–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elizalde, M.; Urtasun, R.; Azkona, M.; Latasa, M.U.; Goñi, S.; García-Irigoyen, O.; Uriarte, I.; Segura, V.; Collantes, M.; Di Scala, M.; et al. Splicing regulator SLU7 is essential for main-taining liver homeostasis. J. Clin. Investig. 2014, 124, 2909–2920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urtasun, R.; Elizalde, M.; Azkona, M.; Latasa, M.U.; García-Irigoyen, O.; Uriarte, I.; Fernández-Barrena, M.G.; Vicent, S.; Alonso, M.M.; Muntané, J.; et al. Splicing regulator SLU7 preserves survival of hepatocellular carcinoma cells and other solid tumors via oncogenic miR-17-92 cluster expression. Oncogene 2016, 35, 4719–4729. [Google Scholar] [CrossRef]
- Jiménez, M.; Urtasun, R.; Elizalde, M.; Azkona, M.; Latasa, U.M.; Uriarte, I.; Arechederra, M.; Alignani, D.; Barcena-Varela, M.; Álvarez-Sola, G.; et al. Splicing events in the control of genome integrity: Role of SLU7 and truncated SRSF3 proteins. Nucleic Acids Res. 2019, 47, gkz014. [Google Scholar] [CrossRef] [Green Version]
- Recalde, M.; Gárate-Rascón, M.; Elizalde, M.; Azkona, M.; Latasa, M.U.; Bárcena-Varela, M.; Sangro, B.; Fernández-Barrena, M.G.; Ávila, M.A.; Arechederra, M.; et al. The splicing regulator SLU7 is re-quired to preserve DNMT1 protein stability and DNA methylation. Nucleic Acids Res. 2021, 49, gkab649. [Google Scholar] [CrossRef]
- Gárate-Rascón, M.; Recalde, M.; Jimenez, M.; Elizalde, M.; Azkona, M.; Uriarte, I.; Latasa, M.U.; Urtasun, R.; Bilbao, I.; Sangro, B.; et al. SLU7 prevents oxidative stress-mediated HNF4α degradation preserving hepatic differentiation and protecting from liver damage. Hepatology 2021, 74, 2791–2807. [Google Scholar] [CrossRef]
- Wang, J.; Kainrad, N.; Shen, H.; Zhou, Z.; Rote, P.; Zhang, Y.; Nagy, L.E.; Wu, J.; You, M. Hepatic Knockdown of Splicing Regulator Slu7 Ameliorates Inflammation and Attenuates Liver Injury in Ethanol-Fed Mice. Am. J. Pathol. 2018, 188, 1807–1819. [Google Scholar] [CrossRef]
- Alberstein, M.; Amit, M.; Vaknin, K.; O’Donnell, A.; Farhy, C.; Lerenthal, Y.; Shomron, N.; Shaham, O.; Sharrocks, A.D.; Ashery-Padan, R.; et al. Regulation of transcription of the RNA splicing factor hSlu7 by Elk-1 and Sp1 affects alternative splicing. RNA 2007, 13, 1988–1999. [Google Scholar] [CrossRef] [Green Version]
- Shomron, N.; Reznik, M.; Ast, G. Splicing Factor hSlu7 Contains a Unique Functional Domain Required to Retain the Protein within the Nucleus. Mol. Biol. Cell. 2004, 15, 3782–3795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hornbeck, P.V.; Zhang, B.; Murray, B.; Kornhauser, J.M.; Latham, V.; Skrzypek, E. PhosphoSitePlus, 2014: Mutations, PTMs and re-calibrations. Nucleic Acids Res. 2015, 43, D512–D520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frank, D.; Guthrie, C. An essential splicing factor, SLU7, mediates 3′ splice site choice in yeast. Genes Dev. 1992, 6, 2112–2124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Umen, J.G.; Guthrie, C. The second catalytic step of pre-mRNA splicing. RNA 1995, 9, 869–885. [Google Scholar]
- Angarola, B.L.; Anczuków, O. Splicing alterations in healthy aging and disease. WIREs RNA 2021, 12, e1643. [Google Scholar] [CrossRef]
- Tollervey, J.R.; Wang, Z.; Hortobágyi, T.; Witten, J.T.; Zarnack, K.; Kayikci, M.; Clark, T.A.; Schweitzer, A.C.; Rot, G.; Curk, T.; et al. Analysis of alternative splicing associated with aging and neurodegeneration in the human brain. Genome Res. 2011, 21, 1572–1582. [Google Scholar] [CrossRef] [Green Version]
- Fagerberg, L.; Hallström, B.M.; Oksvold, P.; Kampf, C.; Djureinovic, D.; Odeberg, J.; Habuka, M.; Tahmasebpoor, S.; Danielsson, A.; Edlund, K.; et al. Analysis of the Human Tissue-specific Expression by Genome-wide Integration of Transcriptomics and Antibody-based Proteomics*. Mol. Cell. Proteom. 2014, 13, 397–406. [Google Scholar] [CrossRef] [Green Version]
- Berasain, C.; Arechederra, M.; Argemí, J.; Fernández-Barrena, M.G.; Avila, M.A. Loss of liver function in chronic liver disease: An identity crisis. J. Hepatol. 2022. [Google Scholar] [CrossRef]
- Castillo, J.; Goñi, S.; Latasa, M.U.; Perugorría, M.J.; Calvo, A.; Muntané, J.; Bioulac-Sage, P.; Balabaud, C.; Prieto, J.; Ávila, M.A.; et al. Amphiregulin induces the alternative splicing of p73 into its oncogenic isoform DeltaEx2p73 in human hepatocellular tumors. Gastroenterology 2009, 137, 1805–1815. [Google Scholar] [CrossRef]
- Shomron, N.; Alberstein, M.; Reznik, M.; Ast, G. Stress alters the subcellular distribution of hSlu7 and thus modulates alternative splicing. J. Cell. Sci. 2005, 118, 1151–1159. [Google Scholar] [CrossRef]
- Spector, D.L.; Lamond, A.I. Nuclear Speckles. Cold Spring Harb. Perspect. Biol. 2011, 3, a000646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janowicz, A.; Michalak, M.; Krebs, J. Stress induced subcellular distribution of ALG-2, RBM22 and hSlu7. Biochim. Biophys. Acta 2011, 1813, 1045–1049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Licatalosi, D.D.; Darnell, R.B. RNA processing and its regulation: Global insights into biological networks. Nat. Rev. Genet. 2010, 11, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Manley, J.L. Mechanisms of alternative splicing regulation: Insights from molecular and genomics approaches. Nat. Rev. Mol. Cell. Biol. 2009, 10, 741–754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conti, L.D.; Baralle, M.; Buratti, E. Exon and intron definition in pre-mRNA splicing. WIREs RNA 2013, 4, 49–60. [Google Scholar] [CrossRef]
- Berasain, C.; Goñi, S.; Castillo, J.; Latasa, M.U.; Prieto, J.; Aacute, V.M.A. Impairment of pre-mRNA splicing in liver disease: Mechanisms and consequences. World. J. Gastroenterol. 2010, 16, 3091–3102. [Google Scholar] [CrossRef]
- Fabrizio, P.; Dannenberg, J.; Dube, P.; Kastner, B.; Stark, H.; Urlaub, H.; Lührmann, R. The Evolutionarily Conserved Core Design of the Catalytic Activation Step of the Yeast Spliceosome. Mol. Cell. 2009, 36, 593–608. [Google Scholar] [CrossRef] [Green Version]
- Brys, A.; Schwer, B. Requirement for SLU7 in yeast pre-mRNA splicing is dictated by the distance between the branchpoint and the 3′ splice site. RNA 1996, 7, 707–717. [Google Scholar]
- Fica, S.M.; Oubridge, C.; Galej, W.P.; Wilkinson, M.E.; Bai, X.-C.; Newman, A.J.; Nagai, K. Structure of a spliceosome remodelled for exon ligation. Nature 2017, 542, 377–380. [Google Scholar] [CrossRef] [Green Version]
- Ohrt, T.; Odenwälder, P.; Dannenberg, J.; Prior, M.; Warkocki, Z.; Schmitzová, J.; Karaduman, R.; Gregor, I.; Enderlein, J.; Fabrizio, P.; et al. Molecular dissection of step 2 catalysis of yeast pre-mRNA splicing investigated in a purified system. RNA 2013, 19, 902–915. [Google Scholar] [CrossRef] [Green Version]
- Dvinge, H. Regulation of alternative mRNA splicing: Old players and new perspectives. FEBS Lett. 2018, 592, 2987–3006. [Google Scholar] [CrossRef] [PubMed]
- Baralle, F.E.; Giudice, J. Alternative splicing as a regulator of development and tissue identity. Nat. Rev. Mol. Cell. Biol. 2017, 18, 437–451. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.; Aifantis, I. RNA Splicing and Cancer. Trends Cancer 2020, 6, 631–644. [Google Scholar] [CrossRef] [PubMed]
- Anczuków, O.; Krainer, A.R. Splicing-factor alterations in cancers. RNA 2016, 22, 1285–1301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berasain, C.; Elizalde, M.; Urtasun, R.; Castillo, J.; Garca-Irigoyen, O.; Uriarte, I.; Latasa, M.U.; Prieto, J.; Ávila, M.A. Alterations in the expression and activity of pre-mRNA splicing factors in hepatocarcinogenesis. Hepatic Oncol. 2014, 1, 241–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oswald, C.; Stiewe, T. In good times and bad: p73 in cancer. Cell Cycle 2008, 7, 1726–1731. [Google Scholar] [CrossRef]
- Buhlmann, S.; Pützer, B.M. DNp73 a matter of cancer: Mechanisms and clinical implications. Biochim. Biophys. Acta 2008, 1785, 207–216. [Google Scholar] [CrossRef]
- Stiewe, T. The p53 family in differentiation and tumorigenesis. Nat. Rev. Cancer 2007, 7, 165–167. [Google Scholar] [CrossRef]
- Stiewe, T.; Theseling, C.C.; Pützer, B.M. Transactivation-deficient ΔTA-p73 Inhibits p53 by Direct Competition for DNA Binding IMPLICATIONS FOR TUMORIGENESIS*. J. Biol. Chem. 2002, 277, 14177–14185. [Google Scholar] [CrossRef] [Green Version]
- Tannapfel, A.; John, K.; Miše, N.; Schmidt, A.; Buhlmann, S.; Ibrahim, S.M.; Pützer, B.M. Autonomous growth and hepatocarcinogenesis in transgenic mice expressing the p53 family inhibitor DNp73. Carcinogenesis 2008, 29, 211–218. [Google Scholar] [CrossRef] [Green Version]
- Jeong, S. SR Proteins: Binders, Regulators, and Connectors of RNA. Mol. Cells 2017, 40, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Kano, S.; Nishida, K.; Kurebe, H.; Nishiyama, C.; Kita, K.; Akaike, Y.; Kajita, K.; Kurokawa, K.; Masuda, K.; Kuwano, Y.; et al. Oxidative stress-inducible truncated serine/arginine-rich splicing factor 3 regulates interleukin-8 production in human colon cancer cells. Am. J. Physiol. Cell. Physiol. 2014, 306, C250–C262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belfiore, A.; Malaguarnera, R.; Vella, V.; Lawrence, M.C.; Sciacca, L.; Frasca, F.; Morrione, A.; Vigneri, R. Insulin Receptor Isoforms in Physiology and Disease: An Updated View. Endocr. Rev. 2017, 38, 379–431. [Google Scholar] [CrossRef] [PubMed]
- Savkur, R.S.; Philips, A.V.; Cooper, T.A.; Dalton, J.C.; Moseley, M.L.; Ranum, L.P.W.; Day, J.W. Insulin Receptor Splicing Alteration in Myo-tonic Dystrophy Type 2. Am. J. Hum. Genet. 2004, 74, 1309–1313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frasca, F.; Pandini, G.; Scalia, P.; Sciacca, L.; Mineo, R.; Costantino, A.; Goldfine, I.D.; Belfiore, A.; Vigneri, R. Insulin Receptor Isoform A, a Newly Recognized, High-Affinity Insulin-Like Growth Factor II Receptor in Fetal and Cancer Cells. Mol. Cell Biol. 1999, 19, 3278–3288. [Google Scholar] [CrossRef] [Green Version]
- Khamzina, L.; Gruppuso, P.A.; Wands, J.R. Insulin signaling through insulin receptor substrate 1 and 2 in normal liver develop-ment. Gastroenterology 2003, 125, 572–585. [Google Scholar] [CrossRef]
- Christmas, P.; Jones, J.P.; Patten, C.J.; Rock, D.A.; Zheng, Y.; Cheng, S.-M.; Weber, B.M.; Carlesso, N.; Scadden, D.T.; Rettie, A.E.; et al. Alternative Splicing Determines the Function of CYP4F3 by Switching Substrate Specificity*. J. Biol. Chem. 2001, 276, 38166–38172. [Google Scholar] [CrossRef]
- Christmas, P.; Ursino, S.R.; Fox, J.W.; Soberman, R.J. Expression of the CYP4F3 Gene tissue-specific splicing and alter-native promoters generate high and low Km forms of leukotriene B4ω-hydroxylase*. J. Biol. Chem. 1999, 274, 21191–21199. [Google Scholar] [CrossRef] [Green Version]
- Corcos, L.; Lucas, D.; Jossic-Corcos, C.L.; Dréano, Y.; Simon, B.; Plée-Gautier, E.; Amet, Y.; Salaün, J.-P. Human cytochrome P450 4F3: Structure, func-tions, and prospects. Drug Metabol. Drug Interact. 2012, 27, 63–71. [Google Scholar] [CrossRef]
- Zhu, H.; Han, C.; Lu, D.; Wu, T. miR-17-92 Cluster Promotes Cholangiocarcinoma Growth Evidence for PTEN as Downstream Target and IL-6/Stat3 as Upstream Activator. Am. J. Pathol. 2014, 184, 2828–2839. [Google Scholar] [CrossRef] [Green Version]
- Tan, W.; Li, Y.; Lim, S.-G.; Tan, T.M. miR-106b-25/miR-17-92 clusters: Polycistrons with oncogenic roles in hepatocellular carcino-ma. World. J. Gastroenterol. 2014, 20, 5962–5972. [Google Scholar] [CrossRef] [PubMed]
- Olive, V.; Li, Q.; He, L. mir-17-92: A polycistronic oncomir with pleiotropic functions. Immunol. Rev. 2013, 253, 158–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendell, J.T. miRiad Roles for the miR-17-92 Cluster in Development and Disease. Cell 2008, 133, 217–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ota, A.; Tagawa, H.; Karnan, S.; Tsuzuki, S.; Karpas, A.; Kira, S.; Yoshida, Y.; Seto, M. Identification and Characterization of a Novel Gene, C13orf25, as a Target for 13q31-q32 Amplification in Malignant Lymphoma. Cancer Res. 2004, 64, 3087–3095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Onn, I.; Heidinger-Pauli, J.M.; Guacci, V.; Ünal, E.; Koshland, D.E. Sister Chromatid Cohesion: A Simple Concept with a Complex Reality. Annu. Rev. Cell. Dev. Biol. 2008, 24, 105–129. [Google Scholar] [CrossRef] [Green Version]
- Nasmyth, K.; Haering, C.H. Cohesin: Its Roles and Mechanisms. Annu. Rev. Genet. 2009, 43, 525–558. [Google Scholar] [CrossRef] [Green Version]
- Nishiyama, T.; Ladurner, R.; Schmitz, J.; Kreidl, E.; Schleiffer, A.; Bhaskara, V.; Bando, M.; Shirahige, K.; Hyman, A.A.; Mechtler, K.; et al. Sororin Mediates Sister Chromatid Cohesion by Antagonizing Wapl. Cell 2010, 143, 737–749. [Google Scholar] [CrossRef] [Green Version]
- Rankin, S.; Ayad, N.G.; Kirschner, M.W. Sororin, a Substrate of the Anaphase-Promoting Complex, Is Required for Sister Chro-matid Cohesion in Vertebrates. Mol. Cell 2005, 18, 185–200. [Google Scholar] [CrossRef]
- Beavo, J.A.; Brunton, L.L. Cyclic nucleotide research—till expanding after half a century. Nat. Rev. Mol. Cell. Biol. 2002, 3, 710–717. [Google Scholar] [CrossRef]
- Wahlang, B.; McClain, C.; Barve, S.; Gobejishvili, L. Role of cAMP and phosphodiesterase signaling in liver health and disease. Cell. Signal 2018, 49, 105–115. [Google Scholar] [CrossRef]
- Zhang, X.; Odom, D.T.; Koo, S.-H.; Conkright, M.D.; Canettieri, G.; Best, J.; Chen, H.; Jenner, R.; Herbolsheimer, E.; Jacobsen, E.; et al. Genome-wide analysis of cAMP-response element binding protein occupancy, phosphorylation, and target gene activation in human tissues. Proc. Natl. Acad. Sci. USA 2005, 102, 4459–4464. [Google Scholar] [CrossRef] [PubMed]
- Briançon, N.; Bailly, A.; Clotman, F.; Jacquemin, P.; Lemaigre, F.P.; Weiss, M.C. Expression of the α7 Isoform of Hepatocyte Nuclear Factor (HNF) 4 Is Activated by HNF6/OC-2 and HNF1 and Repressed by HNF4α1 in the Liver*. J. Biol. Chem. 2004, 279, 33398–33408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kyrmizi, I.; Hatzis, P.; Katrakili, N.; Tronche, F.; Gonzalez, F.J.; Talianidis, I. Plasticity and expanding complexity of the hepatic transcription factor network during liver development. Genes. Dev. 2006, 20, 2293–2305. [Google Scholar] [CrossRef] [Green Version]
- Strahl, B.D.; Allis, C.D. The language of covalent histone modifications. Nature 2000, 403, 41–45. [Google Scholar] [CrossRef]
- Hardy, T.; Mann, D.A. Epigenetics in liver disease: From biology to therapeutics. Gut 2016, 65, 1895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arechederra, M.; Daian, F.; Yim, A.; Bazai, S.K.; Richelme, S.; Dono, R.; Saurin, A.J.; Habermann, B.H.; Maina, F. Hypermethylation of gene body CpG islands predicts high dosage of functional oncogenes in liver cancer. Nat. Commun. 2018, 9, 3164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jjingo, D.; Conley, A.B.; Yi, S.V.; Lunyak, V.V.; Jordan, I.K. On the presence and role of human gene-body DNA methylation. Oncotarget 2012, 3, 462–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hermann, A.; Gowher, H.; Jeltsch, A. Biochemistry and biology of mammalian DNA methyltransferases. Cell. Mol. Life Sci. 2004, 61, 2571–2587. [Google Scholar] [CrossRef]
- Bird, A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002, 16, 6–21. [Google Scholar] [CrossRef] [Green Version]
- Hervouet, E.; Nadaradjane, A.; Gueguen, M.; Vallette, F.M.; Cartron, P.-F. Kinetics of DNA methylation inheritance by the Dnmt1-including complexes during the cell cycle. Cell. Div. 2012, 7, 5. [Google Scholar] [CrossRef] [Green Version]
- Song, J.; Teplova, M.; Ishibe-Murakami, S.; Patel, D.J. Structure-Based Mechanistic Insights into DNMT1-Mediated Maintenance DNA Methylation. Science 2012, 335, 709–712. [Google Scholar] [CrossRef] [PubMed]
- Sen, S.; Jumaa, H.; Webster, N.J.G. Splicing factor SRSF3 is crucial for hepatocyte differentiation and metabolic function. Nat. Commun. 2013, 4, 1336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubois, V.; Staels, B.; Lefebvre, P.; Verzi, M.P.; Eeckhoute, J. Control of Cell Identity by the Nuclear Receptor HNF4 in Organ Pathophysiology. Cells 2020, 9, 2185. [Google Scholar] [CrossRef] [PubMed]
- Bolotin, E.; Liao, H.; Ta, T.C.; Yang, C.; Hwang-Verslues, W.; Evans, J.R.; Jiang, T.; Sladek, F.M. Integrated approach for the identification of human hepatocyte nuclear factor 4α target genes using protein binding microarrays. Hepatology 2010, 51, 642–653. [Google Scholar] [CrossRef] [Green Version]
- Ebi, H.; Sato, T.; Sugito, N.; Hosono, Y.; Yatabe, Y.; Matsuyama, Y.; Yamaguchi, T.; Osada, H.; Suzuki, M.; Takahashi, T. Counterbalance between RB inactivation and miR-17–92 overexpression in reactive oxygen species and DNA damage induction in lung cancers. Oncogene 2009, 28, 3371–3379. [Google Scholar] [CrossRef] [Green Version]
- Kedersha, N.; Ivanov, P.; Anderson, P. Stress granules and cell signaling: More than just a passing phase? Trends Biochem. Sci. 2013, 38, 494–506. [Google Scholar] [CrossRef] [Green Version]
- Protter, D.S.W.; Parker, R. Principles and Properties of Stress Granules. Trends Cell Biol. 2016, 26, 668–679. [Google Scholar] [CrossRef] [Green Version]
- Markmiller, S.; Soltanieh, S.; Server, K.L.; Mak, R.; Jin, W.; Fang, M.Y.; Luo, E.C.; Krach, F.; Yang, D.; Sen, A.; et al. Context-Dependent and Disease-Specific Diversity in Protein Interactions within Stress Granules. Cell 2018, 172, 590–604. [Google Scholar] [CrossRef] [Green Version]
- Youn, J.-Y.; Dyakov, B.J.A.; Zhang, J.; Knight, J.D.R.; Vernon, R.M.; Forman-Kay, J.D.; Gingras, A.C. Properties of Stress Granule and P-Body Proteomes. Mol. Cell. 2019, 76, 286–294. [Google Scholar] [CrossRef]
- Takahashi, M.; Higuchi, M.; Matsuki, H.; Yoshita, M.; Ohsawa, T.; Oie, M.; Fujii, M. Stress Granules Inhibit Apoptosis by Reducing Re-active Oxygen Species Production. Mol. Cell. Biol. 2013, 33, 815–829. [Google Scholar] [CrossRef] [Green Version]
- Cho, E.; Than, T.T.; Kim, S.-H.; Park, E.-R.; Kim, M.-Y.; Lee, K.H.; Shin, H.J. G3BP1 Depletion Increases Radiosensitisation by Inducing Oxidative Stress in Response to DNA Damage. Anticancer Res. 2019, 39, 6087–6095. [Google Scholar] [CrossRef] [PubMed]
- Aguilera, A.; Gómez-González, B. Genome instability: A mechanistic view of its causes and consequences. Nat. Rev. Genet. 2008, 9, 204–217. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Manley, J.L. Inactivation of the SR Protein Splicing Factor ASF/SF2 Results in Genomic Instability. Cell 2005, 122, 365–378. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A., Jr.; Kinzler, K.W. Cancer Genome Landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef]
- McCord, J.M. The evolution of free radicals and oxidative stress. Am. J. Med. 2000, 108, 652–659. [Google Scholar] [CrossRef]
- Ajiro, M.; Jia, R.; Yang, Y.; Zhu, J.; Zheng, Z.-M. A genome landscape of SRSF3-regulated splicing events and gene expression in human osteosarcoma U2OS cells. Nucleic Acids Res. 2016, 44, 1854–1870. [Google Scholar] [CrossRef]
- Muñoz, Ú.; Puche, J.E.; Hannivoort, R.; Lang, U.E.; Cohen-Naftaly, M.; Friedman, S.L. Hepatocyte Growth Factor Enhances Alterna-tive Splicing of the Krüppel-like Factor 6 (KLF6) Tumor Suppressor to Promote Growth through SRSF1. Mol. Cancer Res. 2012, 10, 1216–1227. [Google Scholar] [CrossRef] [Green Version]
- Maslon, M.M.; Heras, S.R.; Bellora, N.; Eyras, E.; Cáceres, J.F. The translational landscape of the splicing factor SRSF1 and its role in mitosis. eLife 2014, 3, e02028. [Google Scholar] [CrossRef]
- Zhang, J.; Dai, Q.; Park, D.; Deng, X. Targeting DNA Replication Stress for Cancer Therapy. Genes 2016, 7, 51. [Google Scholar] [CrossRef] [Green Version]
- Ubhi, T.; Brown, G.W. Exploiting DNA Replication Stress for Cancer Treatment. Cancer Res. 2019, 79, 1730–1739. [Google Scholar] [CrossRef] [PubMed]
- Panasyuk, G.; Espeillac, C.; Chauvin, C.; Pradelli, L.A.; Horie, Y.; Suzuki, A.; Annicotte, J.S.; Fajas, L.; Foretz, M.; Verdeguer, F.; et al. PPARγ contributes to PKM2 and HK2 expression in fatty liver. Nat. Commun. 2012, 3, 672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofmann, J.C.; Husedzinovic, A.; Gruss, O.J. The function of spliceosome components in open mitosis. Nucleus 2010, 1, 447–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karamysheva, Z.; Díaz-Martínez, L.A.; Warrington, R.; Yu, H. Graded requirement for the spliceosome in cell cycle progression. Cell Cycle 2015, 14, 1873–1883. [Google Scholar] [CrossRef] [Green Version]
- Petasny, M.; Bentata, M.; Pawellek, A.; Baker, M.; Kay, G.; Salton, M. Splicing to Keep Cycling: The Importance of Pre-mRNA Splicing during the Cell Cycle. Trends Genet. 2021, 37, 266–278. [Google Scholar] [CrossRef]
- Gartel, A.L.; Serfas, M.S.; Tyner, A.L. p21—Negative Regulator of the Cell Cycle. Proc. Soc. Exp. Biol. Med. 1996, 213, 138–149. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Hevi, S.; Gay, F.; Tsujimoto, N.; He, T.; Zhang, B.; Ueda, Y.; Li, E. Complete inactivation of DNMT1 leads to mitotic catastrophe in human cancer cells. Nat. Genet 2007, 39, 391–396. [Google Scholar] [CrossRef]
- Häsler, R.; Kerick, M.; Mah, N.; Hultschig, C.; Richter, G.; Bretz, F.; Sina, C.; Lehrach, H.; Nietfeld, W.; Schreiber, S.; et al. Alterations of pre-mRNA splicing in human inflammato-ry bowel disease. Eur. J. Cell. Biol. 2011, 90, 603–611. [Google Scholar] [CrossRef]
- Yadav, P.; Ellinghaus, D.; Rémy, G.; Freitag-Wolf, S.; Cesaro, A.; Degenhardt, F.; Boucher, G.; Delacre, M.; Peyrin-Biroulet, L.; Pichavant, M.; et al. Genetic Factors Interact with Tobacco Smoke to Modify Risk for Inflammatory Bowel Disease in Humans and Mice. Gastroenterology 2017, 153, 550–565. [Google Scholar] [CrossRef] [Green Version]
- Berasain, C.; García-Trevijano, E.R.; Castillo, J.; Erroba, E.; Santamaría, M.; Lee, D.C.; Prieto, J.; Avila, M.A. Novel Role for Amphiregulin in Protec-tion from Liver Injury. J. Biol. Chem. 2005, 280, 19012–19020. [Google Scholar] [CrossRef] [Green Version]
- Berasain, C.; Avila, M.A. Amphiregulin. Semin. Cell Dev. Biol. 2014, 28, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Berasain, C.; Castillo, J.; Perugorría, M.J.; Prieto, J.; Avila, M.A. Amphiregulin: A new growth factor in hepatocarcinogenesis. Cancer Lett. 2007, 254, 30–41. [Google Scholar] [CrossRef] [PubMed]
- Douet-Guilbert, N.; Soubise, B.; Bernard, D.G.; Troadec, M.-B. Cytogenetic and Genetic Abnormalities with Diagnostic Value in Myelodysplastic Syndromes (MDS): Focus on the Pre-Messenger RNA Splicing Process. Diagnostics 2022, 12, 1658. [Google Scholar] [CrossRef]
- Lessard, C.J.; Adrianto, I.; Ice, J.A.; Wiley, G.B.; Kelly, J.A.; Glenn, S.B.; Adler, A.J.; Li, H.; Rasmussen, A.; Williams, A.H.; et al. Identification of IRF8, TMEM39A, and IKZF3-ZPBP2 as Susceptibility Loci for Systemic Lupus Erythematosus in a Large-Scale Multiracial Replication Study. Am. J. Hum. Genet. 2012, 90, 648–660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Cheung, S.T.; So, S.; Fan, S.T.; Barry, C.; Higgins, J.; Lai, K.M.; Ji, J.; Dudoit, S.; Ng, I.; et al. Gene Expression Patterns in Human Liver Cancers. Mol. Biol. Cell. 2002, 13, 1929–1939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niu, Z.-S.; Niu, X.-J.; Wang, W.-H. Genetic alterations in hepatocellular carcinoma: An update. World. J. Gastroenterol. 2016, 22, 9069–9095. [Google Scholar] [CrossRef] [PubMed]
- Feinberg, A.P.; Ohlsson, R.; Henikoff, S. The epigenetic progenitor origin of human cancer. Nat. Rev. Genet. 2006, 7, 21–33. [Google Scholar] [CrossRef]
- Tafaleng, E.N.; Mukherjee, A.; Bell, A.; Morita, K.; Guzman-Lepe, J.; Haep, N.; Florentino, N.; Diaz-Aragon, R.; Frau, C.; Ostrowska, A.; et al. Hepatocyte Nuclear Factor 4 alpha 2 Messenger RNA Reprograms Liver-Enriched Transcription Factors and Functional Proteins in End-Stage Cirrhotic Human Hepatocytes. Hepatol. Commun. 2021, 5, 1911–1926. [Google Scholar] [CrossRef]
- Yang, T.; Poenisch, M.; Khanal, R.; Hu, Q.; Dai, Z.; Li, R.; Song, G.; Yuan, Q.; Yao, Q.; Shen, X.; et al. Therapeutic HNF4A mRNA attenuates liver fibrosis in a preclinical model. J. Hepatol. 2021, 75, 1420–1433. [Google Scholar] [CrossRef]
- Zhao, X.; Voutila, J.; Ghobrial, S.; Habib, N.A.; Reebye, V. RNA Activation. Adv. Exp. Med. Biol. 2017, 983, 189–194. [Google Scholar] [CrossRef]
- Reebye, V.; Huang, K.-W.; Lin, V.; Jarvis, S.; Cutilas, P.; Dorman, S.; Ciriello, S.; Andrikakou, P.; Voutila, J.; Saetrom, P.; et al. Gene activation of CEBPA using saRNA: Preclinical studies of the first in human saRNA drug candidate for liver cancer. Oncogene 2018, 37, 3216–3228. [Google Scholar] [CrossRef] [PubMed]
- Stone, M.L.; Chiappinelli, K.B.; Li, H.; Murphy, L.M.; Travers, M.E.; Topper, M.J.; Mathios, D.; Lim, M.; Shih, I.M.; Wang, T.; et al. Reply to Haffner et al.: DNA hypomethylation renders tumors more immunogenic. Proc. Natl. Acad. Sci. USA 2018, 115, E8583-4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Topper, M.J.; Vaz, M.; Marrone, K.A.; Brahmer, J.R.; Baylin, S.B. The emerging role of epigenetic therapeutics in immuno-oncology. Nat. Rev. Clin. Oncol. 2020, 17, 75–90. [Google Scholar] [CrossRef] [PubMed]
- Forment, J.V.; O’Connor, M.J. Targeting the replication stress response in cancer. Pharmacol. Ther. 2018, 188, 155–167. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.D.; Zou, L.; Graubert, T.A. Targeting R-loop-associated ATR response in myelodysplastic syndrome. Oncotarget 2019, 10, 2581–2582. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, G.K.; Shah, M.A. Targeting the Cell Cycle: A New Approach to Cancer Therapy. J. Clin. Oncol. 2005, 23, 9408–9421. [Google Scholar] [CrossRef] [PubMed]
- Kumar, G.; Mittal, S.; Parashar, D.; Jadhav, K.; Geethadevi, A.; Cheema, P.S.; Tuli, H.S. Drug Targets in Cellular Processes of Cancer: From Nonclinical to Preclinical Models; Springer: Singapore, 2020; pp. 45–63. [Google Scholar] [CrossRef]
- Janku, F.; McConkey, D.J.; Hong, D.S.; Kurzrock, R. Autophagy as a target for anticancer therapy. Nat. Rev. Clin. Oncol. 2011, 8, 528–539. [Google Scholar] [CrossRef]
- Tschan, M.P.; Simon, H.-U. The role of autophagy in anticancer therapy: Promises and uncertainties. J. Intern. Med. 2010, 268, 410–418. [Google Scholar] [CrossRef]
- Pfeffer, C.M.; Singh, A.T.K. Apoptosis: A Target for Anticancer Therapy. Int. J. Mol. Sci. 2018, 19, 448. [Google Scholar] [CrossRef] [Green Version]
- Pistritto, G.; Trisciuoglio, D.; Ceci, C.; Garufi, A.; D’Orazi, G. Apoptosis as anticancer mechanism: Function and dysfunction of its modulators and targeted therapeutic strategies. Aging 2016, 8, 603–619. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gárate-Rascón, M.; Recalde, M.; Rojo, C.; Fernández-Barrena, M.G.; Ávila, M.A.; Arechederra, M.; Berasain, C. SLU7: A New Hub of Gene Expression Regulation—From Epigenetics to Protein Stability in Health and Disease. Int. J. Mol. Sci. 2022, 23, 13411. https://doi.org/10.3390/ijms232113411
Gárate-Rascón M, Recalde M, Rojo C, Fernández-Barrena MG, Ávila MA, Arechederra M, Berasain C. SLU7: A New Hub of Gene Expression Regulation—From Epigenetics to Protein Stability in Health and Disease. International Journal of Molecular Sciences. 2022; 23(21):13411. https://doi.org/10.3390/ijms232113411
Chicago/Turabian StyleGárate-Rascón, María, Miriam Recalde, Carla Rojo, Maite G. Fernández-Barrena, Matías A. Ávila, María Arechederra, and Carmen Berasain. 2022. "SLU7: A New Hub of Gene Expression Regulation—From Epigenetics to Protein Stability in Health and Disease" International Journal of Molecular Sciences 23, no. 21: 13411. https://doi.org/10.3390/ijms232113411
APA StyleGárate-Rascón, M., Recalde, M., Rojo, C., Fernández-Barrena, M. G., Ávila, M. A., Arechederra, M., & Berasain, C. (2022). SLU7: A New Hub of Gene Expression Regulation—From Epigenetics to Protein Stability in Health and Disease. International Journal of Molecular Sciences, 23(21), 13411. https://doi.org/10.3390/ijms232113411