
Delayed Impairment of Hippocampal Synaptic Plasticity after Pentylenetetrazole-Induced Seizures in Young Rats
 , , and
, , and 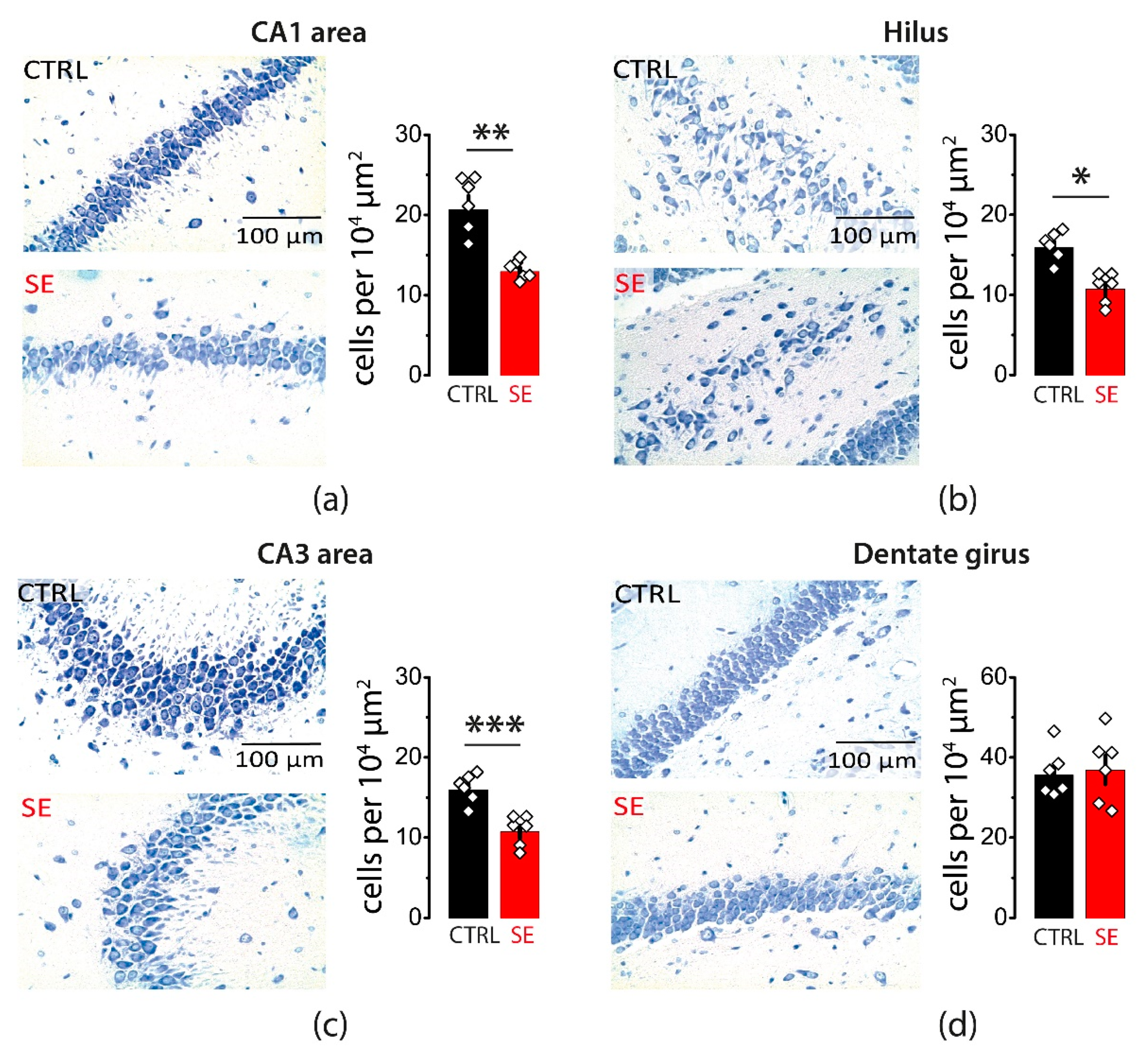{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
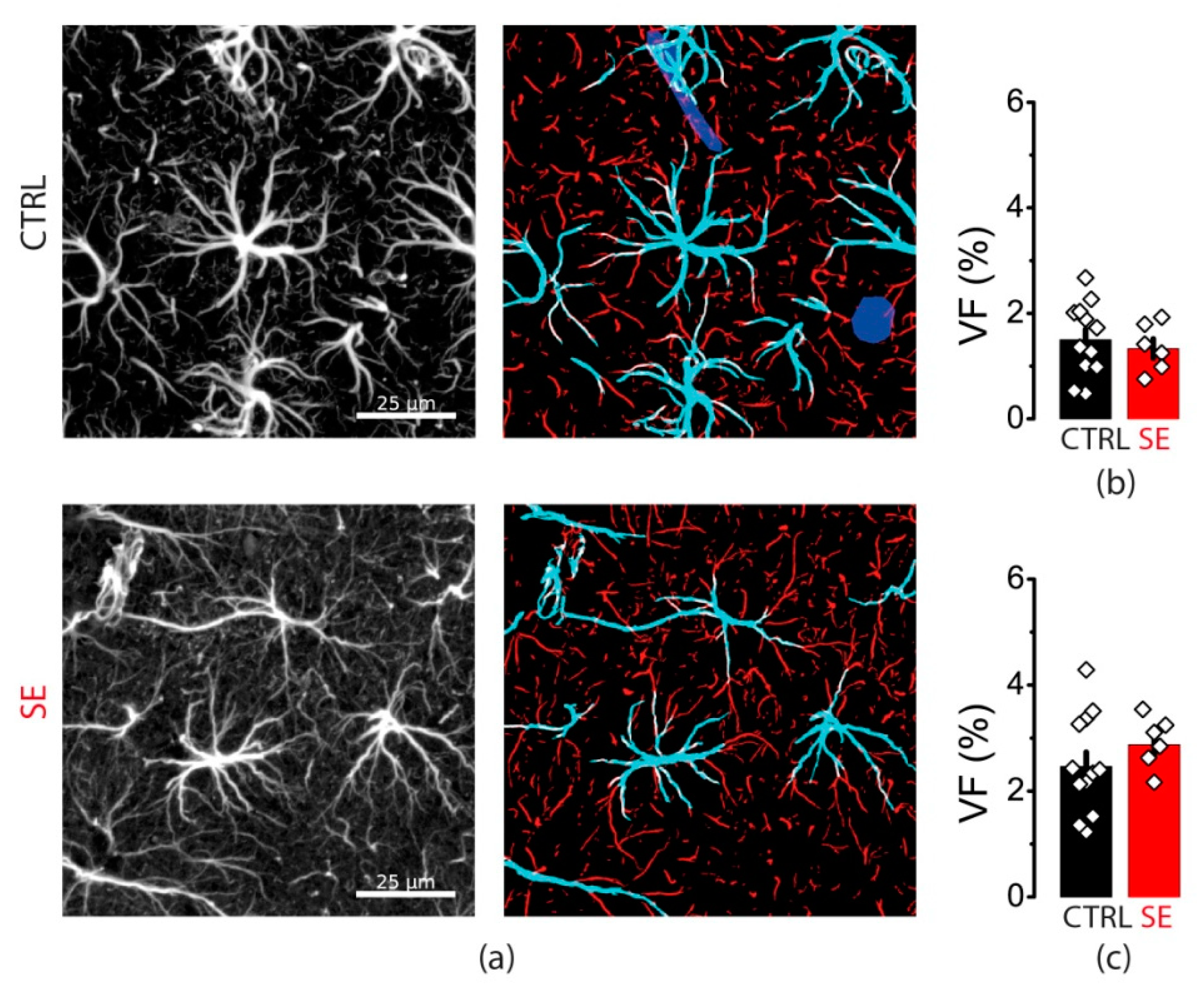
2.1. PTZ-Induced Seizures Resulted in a Decrease in the Number of Neurons in CA1 and CA3 Areas and Hilus of the Hippocampus
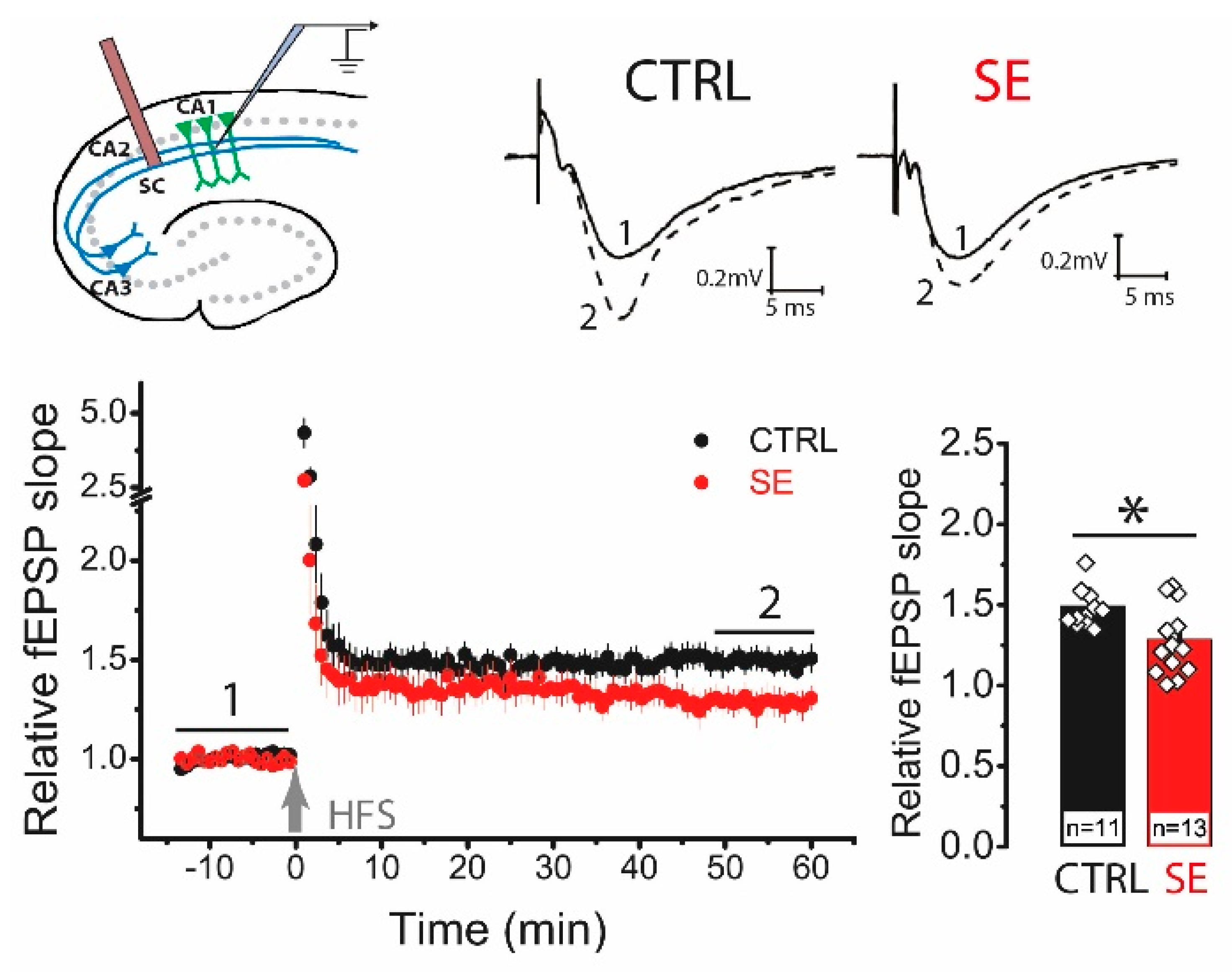
2.2. LTP Was Attenuated in the CA1 Hippocampal Area 30 Days after PTZ-Induced SE
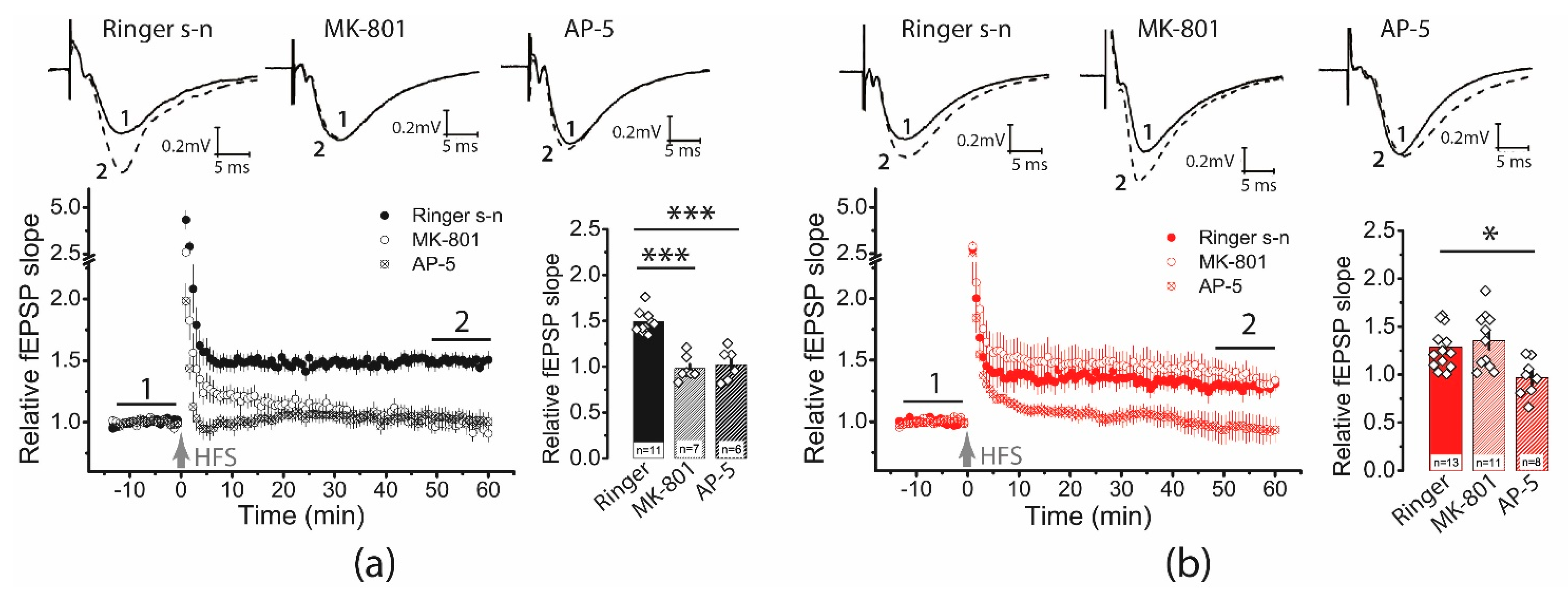
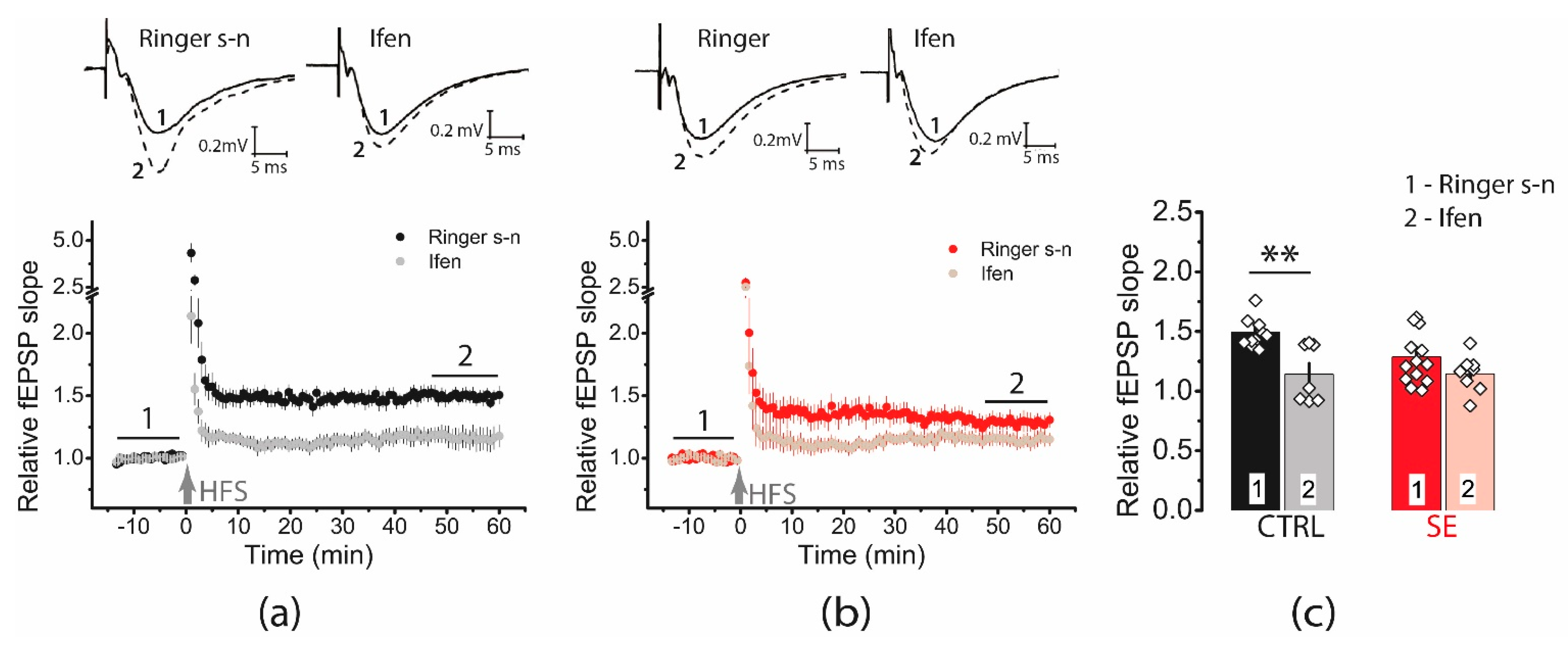
2.3. The Impact of Perisynaptic NMDA Receptors on Synaptic Plasticity Is Enhanced in Post-SE Rats
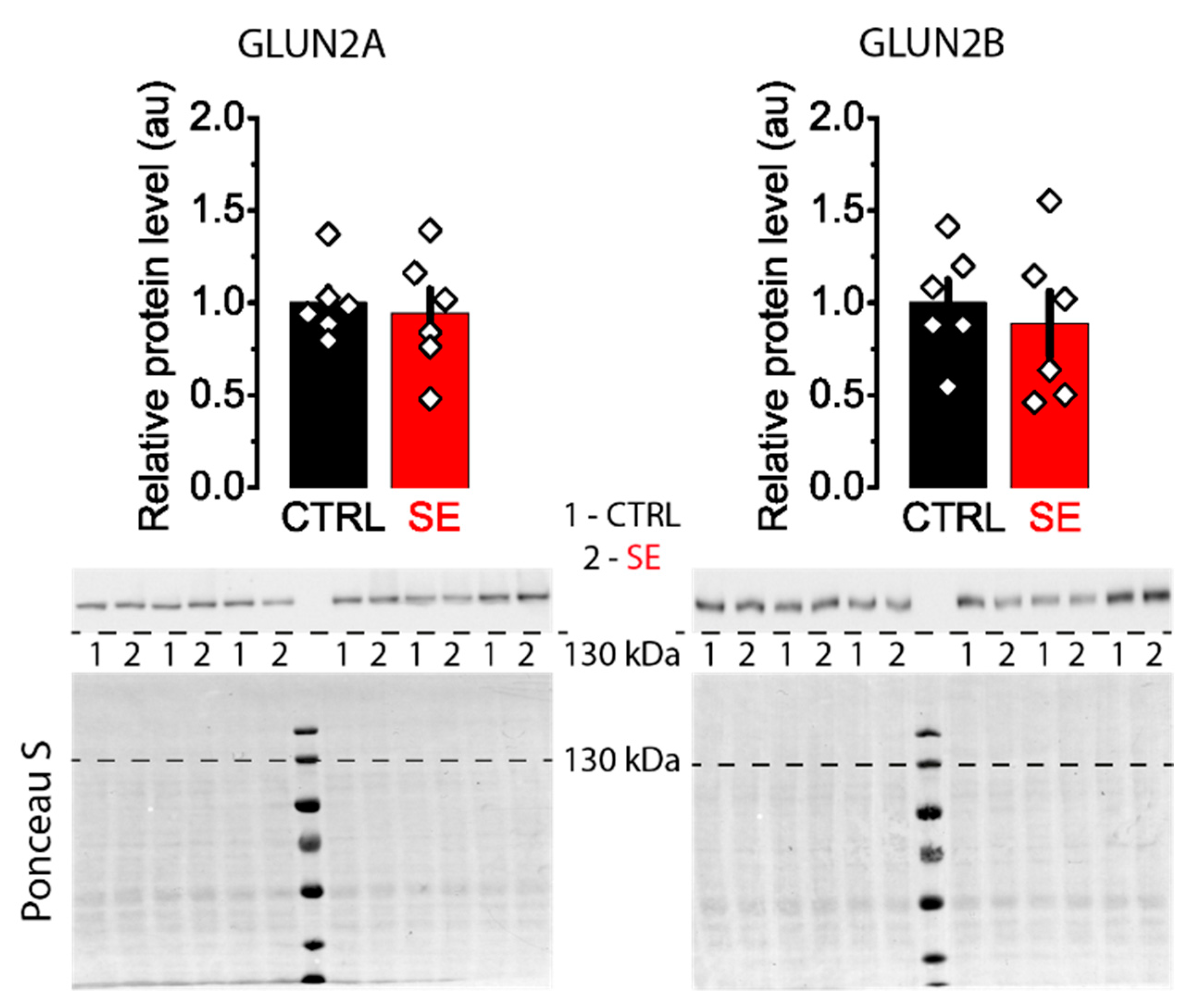
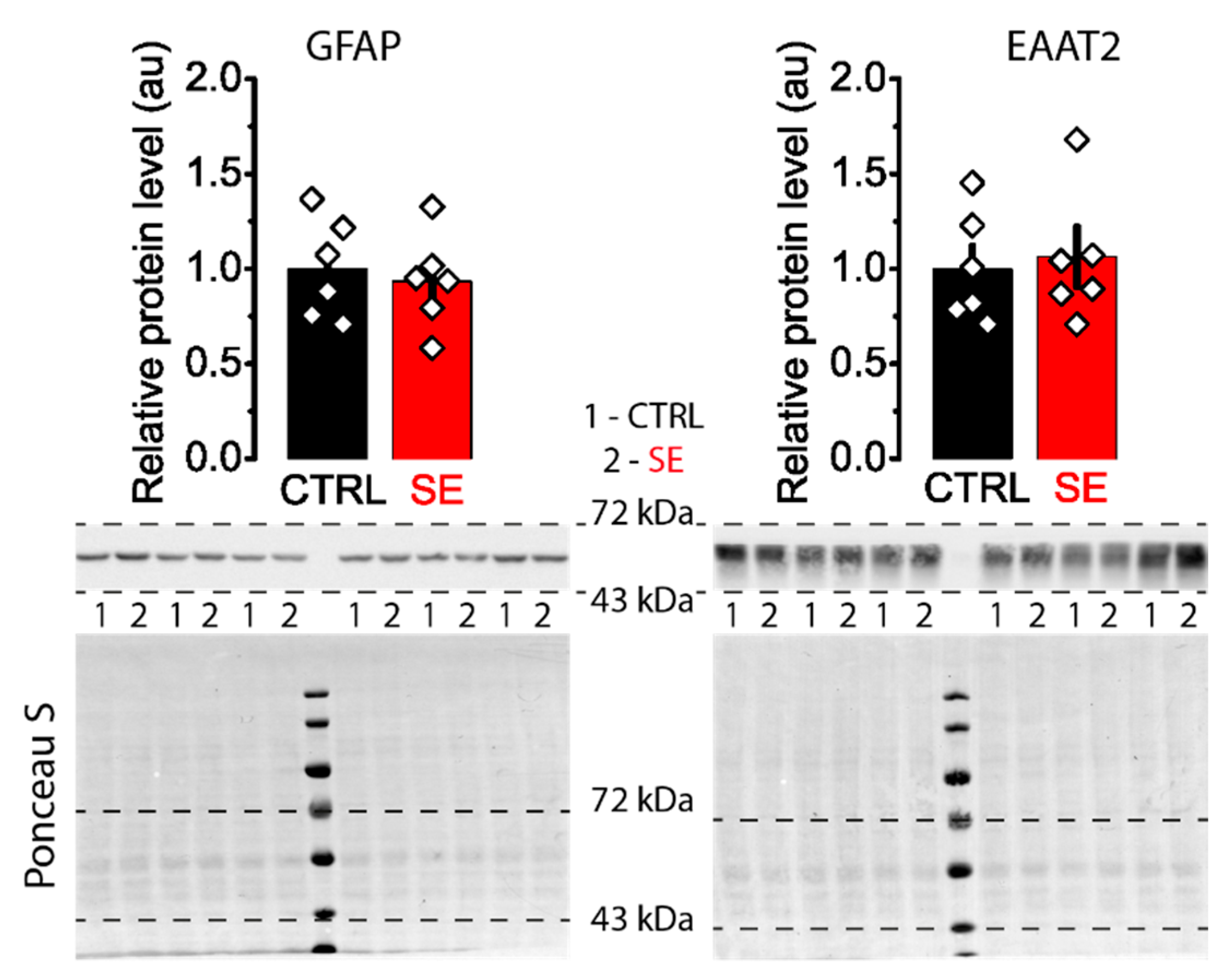
2.4. Subunit Composition of NMDA Receptors Is Unchanged in the Post-SE Group
2.5. Activation of mGluR1 Is Necessary for Induction of LTP in the Post-SE Group
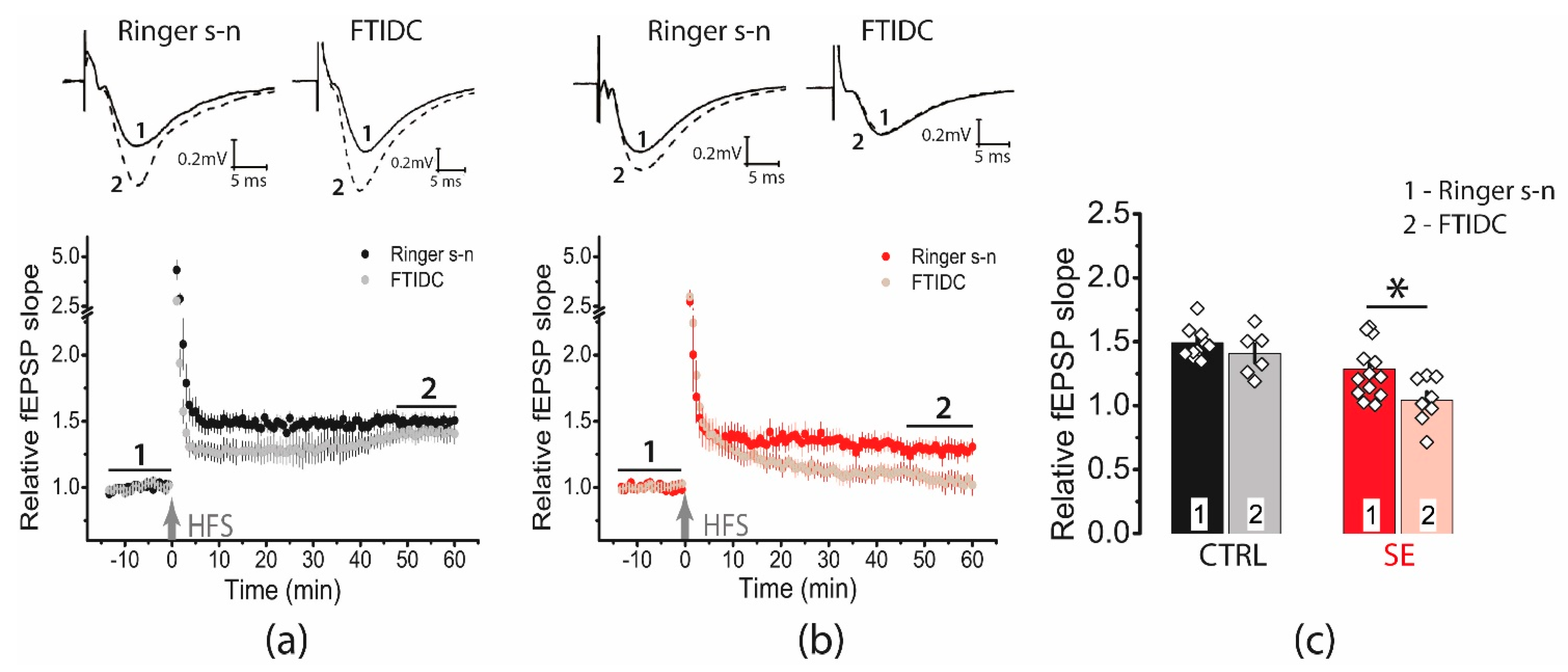
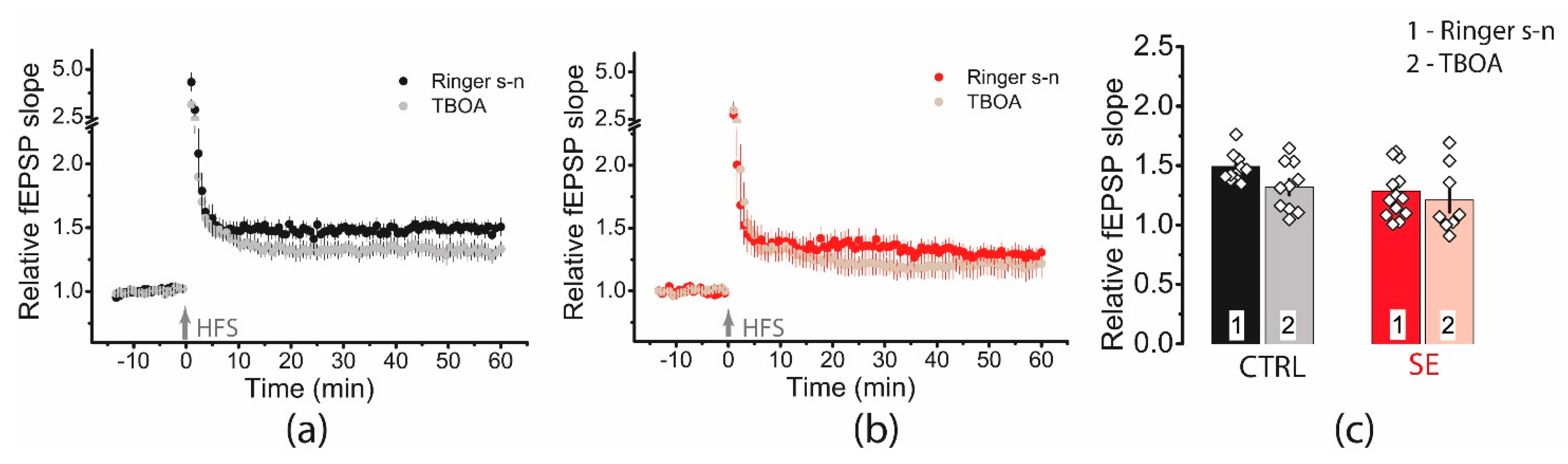
2.6. Blockade of Glutamate Transporters Has No Significant Effect on Synaptic Plasticity
3. Discussion
4. Materials and Methods
4.1. PTZ Model of Status Epilepticus
4.2. Brain Slices Preparation
4.3. Field Potential Recordings
4.4. Western Blot Assay
4.5. Nissl Staining
4.6. Immunocytochemical Method
4.7. Analysis of Immunostaining Data
4.8. Drugs
4.9. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pitkänen, A.; Lukasiuk, K. Molecular and Cellular Basis of Epileptogenesis in Symptomatic Epilepsy. Epilepsy Behav. 2009, 14, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Vasilev, D.S.; Tumanova, N.L.; Kim, K.K.; Lavrentyeva, V.V.; Lukomskaya, N.Y.; Zhuravin, I.A.; Magazanik, L.G.; Zaitsev, A.V. Transient Morphological Alterations in the Hippocampus after Pentylenetetrazole-Induced Seizures in Rats. Neurochem. Res. 2018, 43, 1671–1682. [Google Scholar] [CrossRef] [PubMed]
- Dudai, Y.; Karni, A.; Born, J. Perspective the Consolidation and Transformation of Memory. Neuron 2015, 88, 20–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pujar, S.S.; Martinos, M.M.; Cortina-Borja, M.; Chong, W.K.K.; De Haan, M.; Gillberg, C.; Neville, B.G.; Scott, R.C.; Chin, R.F. Long-Term Prognosis after Childhood Convulsive Status Epilepticus: A Prospective Cohort Study. Lancet Child Adolesc. Health 2018, 2, 103. [Google Scholar] [CrossRef] [Green Version]
- Cui, W.; Kobau, R.; Zack, M.M.; Helmers, S.; Yeargin-Allsopp, M. Seizures in Children and Adolescents Aged 6–17 Years—United States, 2010–2014. MMWR. Morb. Mortal. Wkly. Rep. 2015, 64, 1209–1214. [Google Scholar] [CrossRef]
- Massey, S.; Banwell, B. Clinical Implications of Status Epilepticus in Children. Lancet Child Adolesc. Health 2018, 2, 81–83. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.-L.; Huang, L.-T.; Liou, C.-W.; Wang, T.-J.; Tung, Y.-R.; Hsu, H.-Y.; Lai, M.-C. Lithium-Pilocarpine-Induced Status Epilepticus in Immature Rats Result in Long-Term Deficits in Spatial Learning and Hippocampal Cell Loss. Neurosci. Lett. 2001, 312, 113–117. [Google Scholar] [CrossRef]
- Holmes, G.L.; Khazipov, R.; Ben-Ari, Y. New Concepts in Neonatal Seizures. Neuroreport 2002, 13, A3–A8. [Google Scholar] [CrossRef]
- Lynch, M.; Sayin, Ü.; Bownds, J.; Janumpalli, S.; Sutula, T. Long-Term Consequences of Early Postnatal Seizures on Hippocampal Learning and Plasticity. Eur. J. Neurosci. 2000, 12, 2252–2264. [Google Scholar] [CrossRef]
- Squires, R.F.; Saederup, E.; Crawley, J.N.; Skolnick, P.; Paul, S.M. Convulsant Potencies of Tetrazoles Are Highly Correlated with Actions on GABA/Benzodiazepine/Picrotoxin Receptor Complexes in Brain. Life Sci. 1984, 35, 1439–1444. [Google Scholar] [CrossRef]
- Huang, R.Q.; Bell-Horner, C.L.; Dibas, M.I.; Covey, D.F.; Drewe, J.A.; Dillon, G.H. Pentylenetetrazole-Induced Inhibition of Recombinant Gamma-Aminobutyric Acid Type A (GABA(A)) Receptors: Mechanism and Site of Action. J. Pharmacol. Exp. Ther. 2001, 298, 986–995. [Google Scholar] [PubMed]
- Han, T.; Qin, Y.; Mou, C.; Wang, M.; Jiang, M.; Liu, B. Seizure Induced Synaptic Plasticity Alteration in Hippocampus Is Mediated by IL-1β Receptor through PI3K/Akt Pathway. Am. J. Transl. Res. 2016, 8, 4499–4509. [Google Scholar] [PubMed]
- Mao, R.-R.; Tian, M.; Yang, Y.-X.; Zhou, Q.-X.; Xu, L.; Cao, J. Effects of Pentylenetetrazol-Induced Brief Convulsive Seizures on Spatial Memory and Fear Memory. Epilepsy Behav. 2009, 15, 441–444. [Google Scholar] [CrossRef] [PubMed]
- Socała, K.; Wlaź, P. Acute Seizure Tests Used in Epilepsy Research: Step-by-Step Protocol of the Maximal Electroshock Seizure (MES) Test, the Maximal Electroshock Seizure Threshold (MEST) Test, and the Pentylenetetrazole (PTZ)-Induced Seizure Test in Rodents. Neuromethods 2021, 167, 79–102. [Google Scholar]
- Velisek, L.; Kubova, H.; Pohl, M.; Stankova, L.; Mareš, P.; Schickerova, R. Pentylenetetrazol-Induced Seizures in Rats: An Ontogenetic Study. Naunyn. Schmiedebergs. Arch. Pharmacol. 1992, 346, 588–591. [Google Scholar] [CrossRef]
- Aniol, V.A.; Stepanichev, M.Y.; Lazareva, N.A.; Gulyaeva, N.V. An Early Decrease in Cell Proliferation after Pentylenetetrazole-Induced Seizures. Epilepsy Behav. 2011, 22, 433–441. [Google Scholar] [CrossRef]
- Zaitsev, A.V.; Kim, K.K.; Vasilev, D.S.; Lukomskaya, N.Y.; Lavrentyeva, V.V.; Tumanova, N.L.; Zhuravin, I.A.; Magazanik, L.G. N-Methyl-D-Aspartate Receptor Channel Blockers Prevent Pentylenetetrazole-Induced Convulsions and Morphological Changes in Rat Brain Neurons. J. Neurosci. Res. 2015, 93, 454–465. [Google Scholar] [CrossRef]
- Mohamed, H.K.; Eltony, S.A. Effect of Acute Pentylenetetrazol Injection Induced Epileptic Seizures on Rat Dentate Gyrus at Different Postnatal Ages. Anat. Cell Biol. 2020, 53, 84. [Google Scholar] [CrossRef] [Green Version]
- Zhvania, M.G.; Ksovreli, M.; Japaridze, N.J.; Lordkipanidze, T.G. Ultrastructural Changes to Rat Hippocampus in Pentylenetetrazol- and Kainic Acid-Induced Status Epilepticus: A Study Using Electron Microscopy. Micron 2015, 74, 22–29. [Google Scholar] [CrossRef]
- Aniol, V.A.; Ivanova-Dyatlova, A.Y.; Keren, O.; Guekht, A.B.; Sarne, Y.; Gulyaeva, N.V. A Single Pentylenetetrazole-Induced Clonic-Tonic Seizure Episode Is Accompanied by a Slowly Developing Cognitive Decline in Rats. Epilepsy Behav. 2013, 26, 196–202. [Google Scholar] [CrossRef]
- Jandová, K.; Riljak, V.; Pokorný, J.; Langmeier, M. Pentylentetrazol Associated Changes of Hippocampal Neurons in Immature Rats. Prague Med. Rep. 2007, 108, 67–74. [Google Scholar] [PubMed]
- Postnikova, T.Y.; Amakhin, D.V.; Trofimova, A.M.; Smolensky, I.V.; Zaitsev, A.V. Changes in Functional Properties of Rat Hippocampal Neurons Following Pentylenetetrazole-Induced Status Epilepticus. Neuroscience 2019, 399, 103–116. [Google Scholar] [CrossRef] [PubMed]
- Postnikova, T.Y.; Trofimova, A.M.; Ergina, J.L.; Zubareva, O.E.; Kalemenev, S.V.; Zaitsev, A.V. Transient Switching of NMDA-Dependent Long-Term Synaptic Potentiation in CA3-CA1 Hippocampal Synapses to MGluR1-Dependent Potentiation After Pentylenetetrazole-Induced Acute Seizures in Young Rats. Cell. Mol. Neurobiol. 2019, 39, 287–300. [Google Scholar] [CrossRef] [PubMed]
- Zucker, R.S.; Regehr, W.G. Short-Term Synaptic Plasticity. Annu. Rev. Physiol. 2002, 64, 355–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaitsev, A.V.; Anwyl, R. Inhibition of the Slow Afterhyperpolarization Restores the Classical Spike Timing-Dependent Plasticity Rule Obeyed in Layer 2/3 Pyramidal Cells of the Prefrontal Cortex. J. Neurophysiol. 2012, 107, 205–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luscher, C.; Malenka, R.C. NMDA Receptor-Dependent Long-Term Potentiation and Long-Term Depression (LTP/LTD). Cold Spring Harb. Perspect. Biol. 2012, 4, a005710. [Google Scholar] [CrossRef] [Green Version]
- Li, R.; Huang, F.S.; Abbas, A.K.; Wigström, H. Role of NMDA Receptor Subtypes in Different Forms of NMDA-Dependent Synaptic Plasticity. BMC Neurosci. 2007, 8, 55. [Google Scholar] [CrossRef] [Green Version]
- Harris, A.Z.; Pettit, D.L. Extrasynaptic and Synaptic NMDA Receptors Form Stable and Uniform Pools in Rat Hippocampal Slices. J. Physiol. 2007, 584, 509–519. [Google Scholar] [CrossRef]
- Huettner, J.E.; Bean, B.P. Block of N-Methyl-D-Aspartate-Activated Current by the Anticonvulsant MK-801: Selective Binding to Open Channels. Proc. Natl. Acad. Sci. USA 1988, 85, 1307–1311. [Google Scholar] [CrossRef] [Green Version]
- Scheefhals, N.; MacGillavry, H.D. Functional Organization of Postsynaptic Glutamate Receptors. Mol. Cell. Neurosci. 2018, 91, 82–94. [Google Scholar] [CrossRef]
- Liu, L.; Wong, T.P.; Pozza, M.F.; Lingenhoehl, K.; Wang, Y.; Sheng, M.; Auberson, Y.P.; Wang, Y.T. Role of NMDA Receptor Subtypes in Governing the Direction of Hippocampal Synaptic Plasticity. Science 2004, 304, 1021–1024. [Google Scholar] [CrossRef]
- Zhang, L.; Meng, K.; Li, Y.-H.; Han, T.-Z. NR2A-Containing NMDA Receptors Are Required for L-LTP Induction and Depotentiation in CA1 Region of Hippocampal Slices. Eur. J. Neurosci. 2009, 29, 2137–2144. [Google Scholar] [CrossRef] [PubMed]
- Franchini, L.; Carrano, N.; Di Luca, M.; Gardoni, F. Synaptic GluN2A-Containing NMDA Receptors: From Physiology to Pathological Synaptic Plasticity. Int. J. Mol. Sci. 2020, 21, 1538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Köhr, G.; Jensen, V.; Koester, H.J.; Mihaljevic, A.L.A.; Utvik, J.K.; Kvello, A.; Ottersen, O.P.; Seeburg, P.H.; Sprengel, R.; Hvalby, Ø. Intracellular Domains of NMDA Receptor Subtypes Are Determinants for Long-Term Potentiation Induction. J. Neurosci. 2003, 23, 10791–10799. [Google Scholar] [CrossRef] [Green Version]
- Gardoni, F.; Mauceri, D.; Malinverno, M.; Polli, F.; Costa, C.; Tozzi, A.; Siliquini, S.; Picconi, B.; Cattabeni, F.; Calabresi, P.; et al. Decreased NR2B Subunit Synaptic Levels Cause Impaired Long-Term Potentiation but Not Long-Term Depression. J. Neurosci. 2009, 29, 669–677. [Google Scholar] [CrossRef] [Green Version]
- Kovalenko, A.A.; Zakharova, M.V.; Zubareva, O.E.; Schwarz, A.P.; Postnikova, T.Y.; Zaitsev, A.V. Alterations in MRNA and Protein Expression of Glutamate Receptor Subunits Following Pentylenetetrazole-Induced Acute Seizures in Young Rats. Neuroscience 2021, 468, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Anwyl, R. Metabotropic Glutamate Receptor-Dependent Long-Term Potentiation. Neuropharmacology 2009, 56, 735–740. [Google Scholar] [CrossRef] [PubMed]
- Zaitsev, A.V.; Smolensky, I.V.; Jorratt, P.; Ovsepian, S.V. Neurobiology, Functions, and Relevance of Excitatory Amino Acid Transporters (EAATs) to Treatment of Refractory Epilepsy. CNS Drugs 2020, 34, 1089–1103. [Google Scholar] [CrossRef]
- Verhoog, Q.P.; Holtman, L.; Aronica, E.; van Vliet, E.A. Astrocytes as Guardians of Neuronal Excitability: Mechanisms Underlying Epileptogenesis. Front. Neurol. 2020, 11, 1541. [Google Scholar] [CrossRef] [PubMed]
- Steward, O.; Torre, E.R.; Tomasulo, R.; Lothman, E. Seizures and the Regulation of Astroglial Gene Expression. Epilepsy Res. Suppl. 1992, 7, 197–209. [Google Scholar]
- Alese, O.O.; Mabandla, M.V. Upregulation of Hippocampal Synaptophysin, GFAP and MGluR3 in a Pilocarpine Rat Model of Epilepsy with History of Prolonged Febrile Seizure. J. Chem. Neuroanat. 2019, 100, 101659. [Google Scholar] [CrossRef] [PubMed]
- Simani, L.; Elmi, M.; Asadollahi, M. Serum GFAP Level: A Novel Adjunctive Diagnostic Test in Differentiate Epileptic Seizures from Psychogenic Attacks. Seizure 2018, 61, 41–44. [Google Scholar] [CrossRef] [PubMed]
- Melone, M.; Bellesi, M.; Ducati, A.; Iacoangeli, M.; Conti, F. Cellular and Synaptic Localization of EAAT2a in Human Cerebral Cortex. Front. Neuroanat. 2011, 4, 151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valtcheva, S.; Venance, L. Control of Long-Term Plasticity by Glutamate Transporters. Front. Synaptic Neurosci. 2019, 11, 10. [Google Scholar] [CrossRef] [Green Version]
- Armbruster, M.; Hanson, E.; Dulla, C.G. Glutamate Clearance Is Locally Modulated by Presynaptic Neuronal Activity in the Cerebral Cortex. J. Neurosci. 2016, 36, 10404–10415. [Google Scholar] [CrossRef] [Green Version]
- Rusakov, D.A.; Kullmann, D.M. Extrasynaptic Glutamate Diffusion in the Hippocampus: Ultrastructural Constraints, Uptake, and Receptor Activation. J. Neurosci. 1998, 18, 3158–3170. [Google Scholar] [CrossRef] [Green Version]
- Katagiri, H.; Tanaka, K.; Manabe, T. Requirement of Appropriate Glutamate Concentrations in the Synaptic Cleft for Hippocampal LTP Induction. Eur. J. Neurosci. 2001, 14, 547–553. [Google Scholar] [CrossRef]
- Scimemi, A.; Tian, H.; Diamond, J.S. Neuronal Transporters Regulate Glutamate Clearance, NMDA Receptor Activation, and Synaptic Plasticity in the Hippocampus. J. Neurosci. 2009, 29, 14581–14595. [Google Scholar] [CrossRef] [Green Version]
- Shimamoto, K.; Sakai, R.; Takaoka, K.; Yumoto, N.; Nakajima, T.; Amara, S.G.; Shigeri, Y. Characterization of Novel L- Threo -β-Benzyloxyaspartate Derivatives, Potent Blockers of the Glutamate Transporters. Mol. Pharmacol. 2004, 65, 1008–1015. [Google Scholar] [CrossRef] [Green Version]
- Zaitsev, A.V.; Amakhin, D.V.; Dyomina, A.V.; Zakharova, M.V.; Ergina, J.L.; Postnikova, T.Y.; Diespirov, G.P.; Magazanik, L.G. Synaptic Dysfunction in Epilepsy. J. Evol. Biochem. Physiol. 2021, 57, 542–563. [Google Scholar] [CrossRef]
- Zhang, Y.; Cai, G.E.; Yang, Q.; Lu, Q.C.; Li, S.T.; Ju, G. Time-Dependent Changes in Learning Ability and Induction of Long-Term Potentiation in the Lithium–Pilocarpine-Induced Epileptic Mouse Model. Epilepsy Behav. 2010, 17, 448–454. [Google Scholar] [CrossRef] [PubMed]
- Cruz Del Angel, Y.; Orfila, J.E.; Herson, P.S.; Brooks-Kayal, A.; González, M.I. Down-Regulation of AMPA Receptors and Long-Term Potentiation during Early Epileptogenesis. Epilepsy Behav. 2021, 124, 108320. [Google Scholar] [CrossRef] [PubMed]
- Kryukov, K.A.; Kim, K.K.; Magazanik, L.G.; Zaitsev, A.V. Status Epilepticus Alters Hippocampal Long-Term Synaptic Potentiation in a Rat Lithium-Pilocarpine Model. Neuroreport 2016, 27, 1191–1195. [Google Scholar] [CrossRef] [PubMed]
- Plata, A.; Lebedeva, A.; Denisov, P.; Nosova, O.; Postnikova, T.Y.; Pimashkin, A.; Brazhe, A.; Zaitsev, A.V.; Rusakov, D.A.; Semyanov, A. Astrocytic Atrophy Following Status Epilepticus Parallels Reduced Ca2+ Activity and Impaired Synaptic Plasticity in the Rat Hippocampus. Front. Mol. Neurosci. 2018, 11, 215. [Google Scholar] [CrossRef]
- Grosser, S.; Buck, N.; Braunewell, K.H.; Gilling, K.E.; Wozny, C.; Fidzinski, P.; Behr, J. Loss of Long-Term Potentiation at Hippocampal Output Synapses in Experimental Temporal Lobe Epilepsy. Front. Mol. Neurosci. 2020, 13, 143. [Google Scholar] [CrossRef]
- Debanne, D.; Thompson, S.M.; Gähwiler, B.H. A Brief Period of Epileptiform Activity Strengthens Excitatory Synapses in the Rat Hippocampus in Vitro. Epilepsia 2006, 47, 247–256. [Google Scholar] [CrossRef]
- Di Maio, R.; Mastroberardino, P.G.; Hu, X.; Montero, L.; Greenamyre, J.T. Pilocapine Alters NMDA Receptor Expression and Function in Hippocampal Neurons: NADPH Oxidase and ERK1/2 Mechanisms. Neurobiol. Dis. 2011, 42, 482–495. [Google Scholar] [CrossRef]
- Postnikova, T.Y.; Diespirov, G.P.; Amakhin, D.V.; Vylekzhanina, E.N.; Soboleva, E.B.; Zaitsev, A.V. Impairments of Long-Term Synaptic Plasticity in the Hippocampus of Young Rats during the Latent Phase of the Lithium-Pilocarpine Model of Temporal Lobe Epilepsy. Int. J. Mol. Sci. 2021, 22, 13355. [Google Scholar] [CrossRef]
- Miltiadous, P.; Stamatakis, A.; Koutsoudaki, P.N.; Tiniakos, D.G.; Stylianopoulou, F. IGF-I Ameliorates Hippocampal Neurodegeneration and Protects against Cognitive Deficits in an Animal Model of Temporal Lobe Epilepsy. Exp. Neurol. 2011, 231, 223–235. [Google Scholar] [CrossRef]
- Yang, H.; Shan, W.; Zhu, F.; Yu, T.; Fan, J.; Guo, A.; Li, F.; Yang, X.; Wang, Q. C-Fos Mapping and EEG Characteristics of Multiple Mice Brain Regions in Pentylenetetrazol-Induced Seizure Mice Model. Neurol. Res. 2019, 41, 749–761. [Google Scholar] [CrossRef]
- O’Neill, N.; McLaughlin, C.; Komiyama, N.; Sylantyev, S. Biphasic Modulation of NMDA Receptor Function by Metabotropic Glutamate Receptors. J. Neurosci. 2018, 38, 9840–9855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lan, J.Y.; Skeberdis, V.A.; Jover, T.; Zheng, X.; Bennett, M.V.L.; Zukin, R.S. Activation of Metabotropic Glutamate Receptor 1 Accelerates NMDA Receptor Trafficking. J. Neurosci. 2001, 21, 6058–6068. [Google Scholar] [CrossRef] [PubMed]
- Reiner, A.; Levitz, J. Glutamatergic Signaling in the Central Nervous System: Ionotropic and Metabotropic Receptors in Concert. Neuron 2018, 98, 1080–1098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skeberdis, V.A.; Lan, J.; Opitz, T.; Zheng, X.; Bennett, M.V.; Zukin, R.S. MGluR1-Mediated Potentiation of NMDA Receptors Involves a Rise in Intracellular Calcium and Activation of Protein Kinase C. Neuropharmacology 2001, 40, 856–865. [Google Scholar] [CrossRef]
- Bertaso, F.; Roussignol, G.; Worley, P.; Bockaert, J.; Fagni, L.; Ango, F. Homer1a-Dependent Crosstalk between NMDA and Metabotropic Glutamate Receptors in Mouse Neurons. PLoS ONE 2010, 5, e9755. [Google Scholar] [CrossRef] [Green Version]
- Paoletti, P.; Bellone, C.; Zhou, Q. NMDA Receptor Subunit Diversity: Impact on Receptor Properties, Synaptic Plasticity and Disease. Nat. Rev. Neurosci. 2013, 14, 383–400. [Google Scholar] [CrossRef]
- Proper, E.A.; Hoogland, G.; Kappen, S.M.; Jansen, G.H.; Rensen, M.G.A.; Schrama, L.H.; van Veelen, C.W.M.; van Rijen, P.C.; van Nieuwenhuizen, O.; Gispen, W.H.; et al. Distribution of Glutamate Transporters in the Hippocampus of Patients with Pharmaco-resistant Temporal Lobe Epilepsy. Brain 2002, 125, 32–43. [Google Scholar] [CrossRef]
- Mathern, G.W.; Mendoza, D.; Lozada, A.; Pretorius, J.K.; Dehnes, Y.; Danbolt, N.C.; Nelson, N.; Leite, J.P.; Chimelli, L.; Born, D.E.; et al. Hippocampal GABA and Glutamate Transporter Immunoreactivity in Patients with Temporal Lobe Epilepsy. Neurology 1999, 52, 453. [Google Scholar] [CrossRef]
- Zubareva, O.E.; Kovalenko, A.A.A.; Kalemenev, S.V.; Schwarz, A.P.; Karyakin, V.B.; Zaitsev, A.V. Alterations in MRNA Expression of Glutamate Receptor Subunits and Excitatory Amino Acid Transporters Following Pilocarpine-Induced Seizures in Rats. Neurosci. Lett. 2018, 686, 94–100. [Google Scholar] [CrossRef]
- Lopes, M.W.; Soares, F.M.S.; De Mello, N.; Nunes, J.C.; Cajado, A.G.; De Brito, D.; De Cordova, F.M.; Da Cunha, R.M.S.; Walz, R.; Leal, R.B. Time-Dependent Modulation of AMPA Receptor Phosphorylation and MRNA Expression of NMDA Receptors and Glial Glutamate Transporters in the Rat Hippocampus and Cerebral Cortex in a Pilocarpine Model of Epilepsy. Exp. Brain Res. 2013, 226, 153–163. [Google Scholar] [CrossRef]
- Clarkson, C.; Smeal, R.M.; Hasenoehrl, M.G.; White, J.A.; Rubio, M.E.; Wilcox, K.S. Ultrastructural and Functional Changes at the Tripartite Synapse during Epileptogenesis in a Model of Temporal Lobe Epilepsy. Exp. Neurol. 2020, 326, 113196. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, D.K.; Vargas, J.R.; Wilcox, K.S. Increased Coupling and Altered Glutamate Transport Currents in Astrocytes Following Kainic-Acid-Induced Status Epilepticus. Neurobiol. Dis. 2010, 40, 573–585. [Google Scholar] [CrossRef] [PubMed]
- Postnikova, T.Y.; Griflyuk, A.V.; Amakhin, D.V.; Kovalenko, A.A.; Soboleva, E.B.; Zubareva, O.E.; Zaitsev, A.V. Early Life Febrile Seizures Impair Hippocampal Synaptic Plasticity in Young Rats. Int. J. Mol. Sci. 2021, 22, 8218. [Google Scholar] [CrossRef] [PubMed]
- Sankar, R.; Shin, D.H.; Liu, H.; Mazarati, A.; Pereira de Vasconcelos, A.; Wasterlain, C.G. Patterns of Status Epilepticus-Induced Neuronal Injury during Development and Long-Term Consequences. J. Neurosci. 1998, 18, 8382–8393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riban, V.; Bouilleret, V.; Pham-Lê, B.T.; Fritschy, J.-M.; Marescaux, C.; Depaulis, A. Evolution of Hippocampal Epileptic Activity during the Development of Hippocampal Sclerosis in a Mouse Model of Temporal Lobe Epilepsy. Neuroscience 2002, 112, 101–111. [Google Scholar] [CrossRef]
- Fang, M.; Xi, Z.-Q.; Wu, Y.; Wang, X.-F. A New Hypothesis of Drug Refractory Epilepsy: Neural Network Hypothesis. Med. Hypotheses 2011, 76, 871–876. [Google Scholar] [CrossRef]
- Fattorusso, A.; Matricardi, S.; Mencaroni, E.; Dell’Isola, G.B.; Di Cara, G.; Striano, P.; Verrotti, A. The Pharmacoresistant Epilepsy: An Overview on Existant and New Emerging Therapies. Front. Neurol. 2021, 12, 1030. [Google Scholar] [CrossRef]
- Schmidt, D.; Sillanpää, M. Prevention of Epilepsy: Issues and Innovations. Curr. Neurol. Neurosci. Rep. 2016, 16, 95. [Google Scholar] [CrossRef]
- Paxinos, G.; Watson, C. The Rat Brain in Stereotaxic Coordinates; Elsevier: Amsterdam, The Netherlands, 2007. [Google Scholar]
- Kopec, A.M.; Rivera, P.D.; Lacagnina, M.J.; Hanamsagar, R.; Bilbo, S.D. Optimized Solubilization of TRIzol-Precipitated Protein Permits Western Blotting Analysis to Maximize Data Available from Brain Tissue. J. Neurosci. Methods 2017, 280, 64–76. [Google Scholar] [CrossRef] [Green Version]
- Harrington, C.R. Lowry Protein Assay Containing Sodium Dodecyl Sulfate in Microtiter Plates for Protein Determinations on Fractions from Brain Tissue. Anal. Biochem. 1990, 186, 285–287. [Google Scholar] [CrossRef]
- Laemmli, U.K. Cleavage of Structural Proteins during the Assembly of the Head of Bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef] [PubMed]
- Thacker, J.S.; Yeung, D.H.; Staines, W.R.; Mielke, J.G. Total Protein or High-Abundance Protein: Which Offers the Best Loading Control for Western Blotting? Anal. Biochem. 2016, 496, 76–78. [Google Scholar] [CrossRef] [PubMed]
- Korzhevskii, D.E.; Sukhorukova, E.G.; Kirik, O.V.; Grigorev, I.P. Immunohistochemical Demonstration of Specific Antigens in the Human Brain Fixed in Zinc-Ethanol-Formaldehyde. Eur. J. Histochem. 2015, 59, 5–9. [Google Scholar] [CrossRef] [PubMed]
- Frangi, A.F.; Niessen, W.J.; Vincken, K.L.; Viergever, M.A. Multiscale Vessel Enhancement Filtering. In Lecture Notes in Computer Science (Including Subseries Lecture Notes in Artificial Intelligence and Lecture Notes in Bioinformatics); Springer: Berlin/Heidelberg, Germany, 1998; Volume 1496, pp. 130–137. ISBN 3540651365. [Google Scholar]
- Koenderink, J.J.; van Doorn, A.J. Surface Shape and Curvature Scales. Image Vis. Comput. 1992, 10, 557–564. [Google Scholar] [CrossRef]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Postnikova, T.Y.; Trofimova, A.M.; Zakharova, M.V.; Nosova, O.I.; Brazhe, A.R.; Korzhevskii, D.E.; Semyanov, A.V.; Zaitsev, A.V. Delayed Impairment of Hippocampal Synaptic Plasticity after Pentylenetetrazole-Induced Seizures in Young Rats. Int. J. Mol. Sci. 2022, 23, 13461. https://doi.org/10.3390/ijms232113461
Postnikova TY, Trofimova AM, Zakharova MV, Nosova OI, Brazhe AR, Korzhevskii DE, Semyanov AV, Zaitsev AV. Delayed Impairment of Hippocampal Synaptic Plasticity after Pentylenetetrazole-Induced Seizures in Young Rats. International Journal of Molecular Sciences. 2022; 23(21):13461. https://doi.org/10.3390/ijms232113461
Chicago/Turabian StylePostnikova, Tatyana Y., Alina M. Trofimova, Maria V. Zakharova, Olga I. Nosova, Alexey R. Brazhe, Dmitry E. Korzhevskii, Alexey V. Semyanov, and Aleksey V. Zaitsev. 2022. "Delayed Impairment of Hippocampal Synaptic Plasticity after Pentylenetetrazole-Induced Seizures in Young Rats" International Journal of Molecular Sciences 23, no. 21: 13461. https://doi.org/10.3390/ijms232113461
APA StylePostnikova, T. Y., Trofimova, A. M., Zakharova, M. V., Nosova, O. I., Brazhe, A. R., Korzhevskii, D. E., Semyanov, A. V., & Zaitsev, A. V. (2022). Delayed Impairment of Hippocampal Synaptic Plasticity after Pentylenetetrazole-Induced Seizures in Young Rats. International Journal of Molecular Sciences, 23(21), 13461. https://doi.org/10.3390/ijms232113461