
Gut Microbiota Mediates Skin Ulceration Syndrome Outbreak by Readjusting Lipid Metabolism in Apostichopus japonicus



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. The Intestine Histology and Characteristics of A. japonicus under V. splendidus Infection
2.2. Overview of the Microbiota Composition of the Intestinal Contents of A. japonicus with V. splendidus Infection
2.3. V. splendidus Infection Altered the Major Microbiota Abundance of A. japonicus
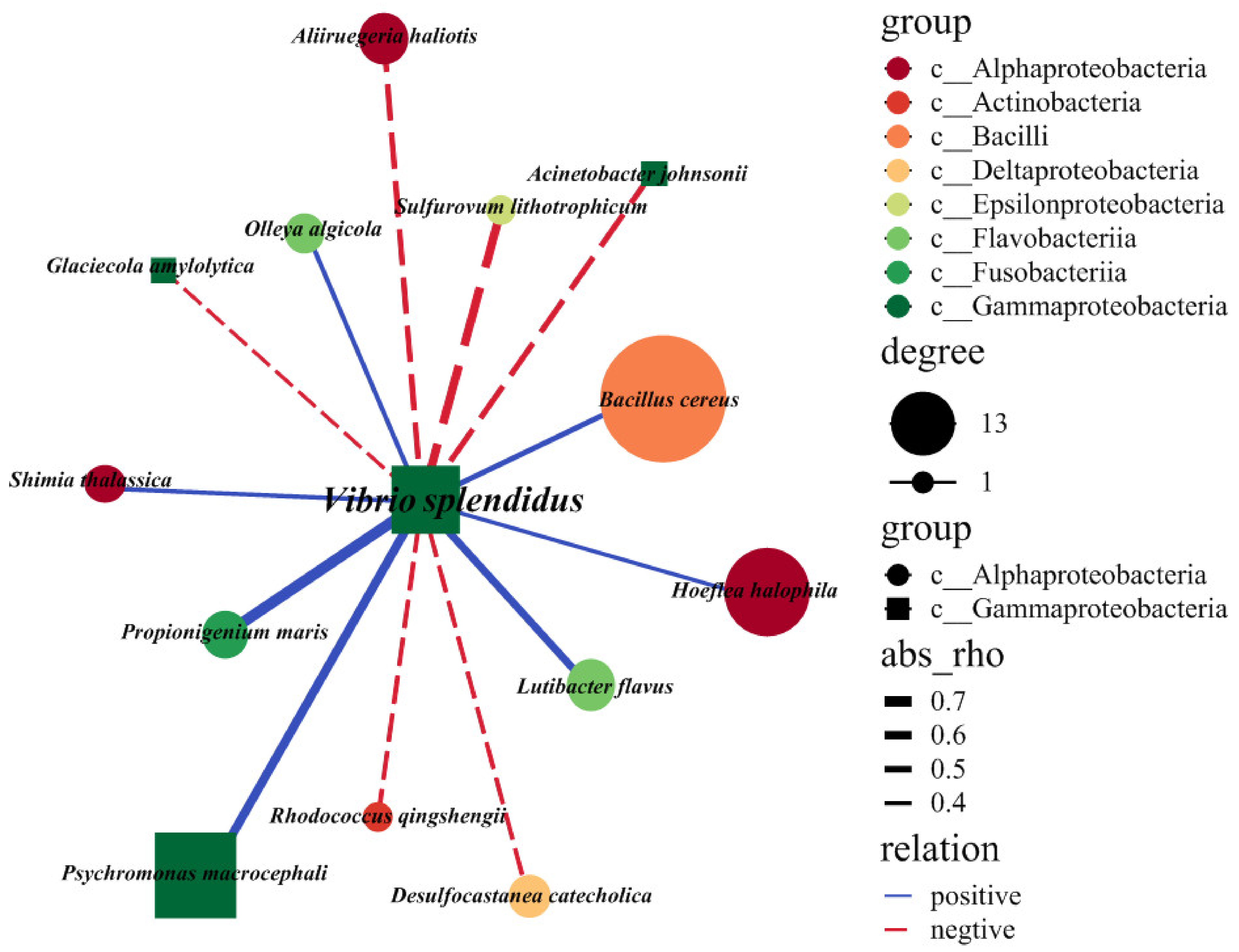
2.4. Exploring the Interspecies Interactions of the V. splendidus Pathogen
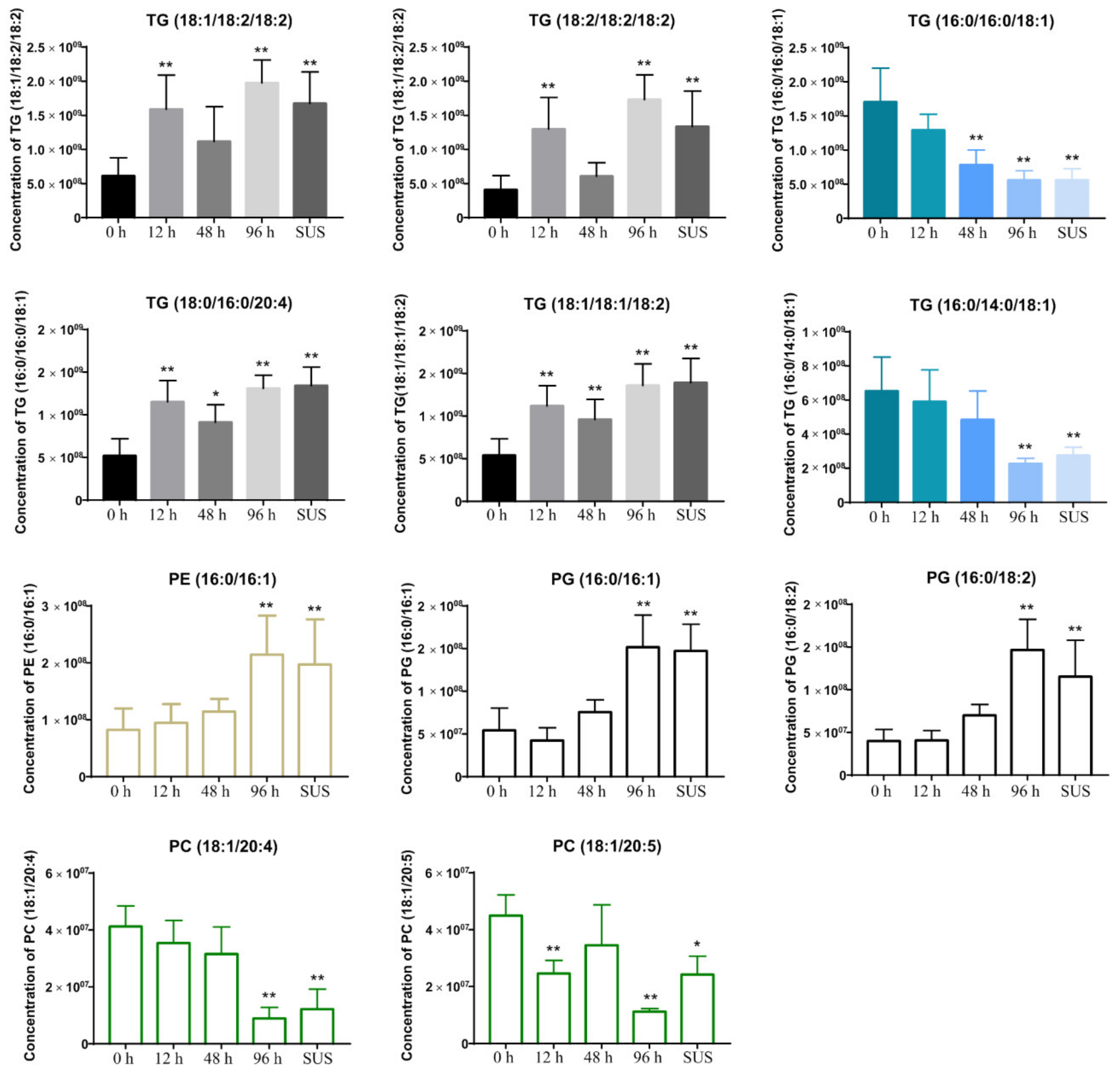
2.5. V. splendidus Infection Changed the Lipid Metabolism in the Sea Cucumber Gut Contents
2.6. The Relationship between the Gut Microbiota and Lipid Metabolite Profiles
3. Discussion
4. Materials and Methods
4.1. Ethics Statement
4.2. Animal Collection and Experimental Design
4.3. The Effects of V. splendidus Infection on the Sea Cucumber Intestine Histology
4.4. The Effects of V. splendidus on the Immune Genes
4.5. The Gut Microbiome Analysis
4.6. High-Performance Liquid Chromatography–Tandem Mass Spectrometry (HPLC–MS/MS) Assay
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Conflicts of Interest
References
- Wu, J.; Xiong, J.; Hu, C.; Shi, Y.; Wang, K.; Zhang, D. Temperature sensitivity of soil bacterial community along contrasting warming gradient. Appl. Soil Ecol. 2015, 94, 40–48. [Google Scholar] [CrossRef]
- Jin, C.; Luo, T.; Zhu, Z.; Pan, Z.; Yang, J.; Wang, W.; Fu, Z.; Jin, Y. Imazalil exposure induces gut microbiota dysbiosis and hepatic metabolism disorder in zebrafish. Comp. Biochem. Physiol. C Toxicol. Pharmacol. 2017, 202, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Mills, R.H.; Dulai, P.S.; Vázquez-Baeza, Y.; Sauceda, C.; Daniel, N.; Gerner, R.R.; Batachari, L.E.; Malfavon, M.; Zhu, Q.; Weldon, K.; et al. Multi-omics analyses of the ulcerative colitis gut microbiome link Bacteroides vulgatus proteases with disease severity. Nat. Microbiol. 2022, 7, 262–276. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Li, B.; Lou, P.; Dai, T.; Chen, Y.; Zhuge, A.; Yuan, Y.; Li, L. The relationship between the gut microbiome and neurodegenerative diseases. Neurosci. Bull. 2021, 37, 1510–1522. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Sun, L.; Gonzalez, F.J. Gut microbiota-derived bile acids in intestinal immunity, inflammation, and tumorigenesis. Cell Host Microbe. 2022, 30, 289–300. [Google Scholar] [CrossRef] [PubMed]
- Ziarati, M.; Zorriehzahra, M.J.; Hassantabar, F.; Mehrabi, Z.; Dhawan, M.; Sharun, K.; Emran, T.B.; Dhama, K.; Chaicumpa, W.; Shamsi, S. Zoonotic diseases of fish and their prevention and control. Vet. Q. 2022, 42, 95–118. [Google Scholar] [CrossRef]
- Pickard, J.M.; Zeng, M.Y.; Caruso, R.; Núñez, G. Gut microbiota: Role in pathogen colonization, immune responses, and inflammatory disease. Immunol. Rev. 2017, 279, 70–89. [Google Scholar] [CrossRef]
- Brogden, K.A.; Guthmiller, J.M.; Taylor, C.E. Human polymicrobial infections. Lancet 2005, 365, 253–255. [Google Scholar] [CrossRef]
- Round, J.L.; Mazmanian, S.K. The gut microbiota shapes intestinal immune responses during health and disease. Nat. Rev. Immunol. 2009, 9, 313–323. [Google Scholar] [CrossRef]
- Sato, Y.; Civiello, M.; Bell, S.C.; Willis, B.L.; Bourne, D.G. Integrated approach to understanding the onset and pathogenesis of black band disease in corals. Environ. Microbiol. 2016, 18, 752–765. [Google Scholar] [CrossRef]
- Kogut, M.H. Role of diet-microbiota interactions in precision nutrition of the chicken: Facts; gaps; and new concepts. Poult. Sci. 2022, 101, 101673. [Google Scholar] [CrossRef]
- Collins, S.L.; Patterson, A.D. The gut microbiome: An orchestrator of xenobiotic metabolism. Acta Pharm. Sin. B. 2020, 10, 19–32. [Google Scholar] [CrossRef]
- Houtman, T.A.; Eckermann, H.A.; Smidt, H.; de Weerth, C. Gut microbiota and BMI throughout childhood: The role of firmicutes; bacteroidetes; and short-chain fatty acid producers. Sci. Rep. 2022, 12, 3140. [Google Scholar] [CrossRef]
- Liu, W.; Li, C.; Li, B.; Shang, Q.; Han, Z.; Zhang, Y.; Liu, X.; Fan, H.; Zhang, J.; Chen, Y.; et al. Lactiplantibacillus plantarum P9 improved gut microbial metabolites and alleviated inflammatory response in pesticide exposure cohorts. iScience 2022, 25, 104472. [Google Scholar] [CrossRef]
- Wilson, K.R.; Gressier, E.; McConville, M.J.; Bedoui, S. Microbial metabolites in the maturation and activation of dendritic cells and their relevance for respiratory immunity. Front. Immunol. 2022, 13, 897462. [Google Scholar] [CrossRef]
- Ghazalpour, A.; Cespedes, I.; Bennett, B.J.; Allayee, H. Expanding role of gut microbiota in lipid metabolism. Curr. Opin. Lipidol. 2016, 27, 141–147. [Google Scholar] [CrossRef]
- Nicholson, J.K.; Holmes, E.; Kinross, J.; Burcelin, R.; Gibson, G.; Jia, W.; Pettersson, S. Host-gut microbiota metabolic interactions. Science 2012, 336, 1262–1267. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.J.; Zeng, B.H.; Li, W.W.; Zhou, C.J.; Fan, S.H.; Cheng, K.; Zeng, L.; Zheng, P.; Fang, L.; Wei, H.; et al. Effects of gut microbiota on the microRNA and mRNA expression in the hippocampus of mice. Behav. Brain Res. 2017, 322, 34–41. [Google Scholar] [CrossRef]
- Diaz Heijtz, R.; Wang, S.; Anuar, F.; Qian, Y.; Björkholm, B.; Samuelsson, A.; Hibberd, M.L.; Forssberg, H.; Pettersson, S. Normal gut microbiota modulates brain development and behavior. Proc. Natl. Acad. Sci. USA 2011, 108, 3047–3052. [Google Scholar] [CrossRef] [Green Version]
- Yilmaz, B.; Juillerat, P.; Øyås, O.; Ramon, C.; Bravo, F.D.; Franc, Y.; Fournier, N.; Michetti, P.; Mueller, C.; Geuking, M.; et al. Microbial network disturbances in relapsing refractory Crohn’s disease. Nat. Med. 2019, 25, 323–336. [Google Scholar] [CrossRef]
- Schirmer, M.; Garner, A.; Vlamakis, H.; Xavier, R.J. Microbial genes and pathways in inflammatory bowel disease. Nat. Rev. Microbiol. 2019, 17, 497–511. [Google Scholar] [CrossRef] [PubMed]
- Surana, N.K.; Kasper, D.L. Moving beyond microbiome-wide associations to causal microbe identification. Nature 2017, 552, 244–247. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.H.; Yu, J. Gut microbiota in colorectal cancer: Mechanisms of action and clinical applications. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 690–704. [Google Scholar] [CrossRef] [PubMed]
- Behary, J.; Amorim, N.; Jiang, X.T.; Raposo, A.; Gong, L.; McGovern, E.; Ibrahim, R.; Chu, F.; Stephens, C.; Jebeili, H.; et al. Gut microbiota impact on the peripheral immune response in non-alcoholic fatty liver disease related hepatocellular carcinoma. Nat. Commun. 2021, 12, 187. [Google Scholar] [CrossRef] [PubMed]
- Jones, K.E.; Patel, N.G.; Levy, M.A.; Storeygard, A.; Balk, D.; Gittleman, J.L.; Daszak, P. Global trends in emerging infectious diseases. Nature 2008, 451, 990–993. [Google Scholar] [CrossRef]
- Han, Q.; Keesing, J.K.; Liu, D.Y. A review of sea cucumber aquaculture; ranching; and stock enhancement in China. Rev. Fish. Sci. Aquac. 2016, 24, 326–341. [Google Scholar] [CrossRef]
- Deng, H.; He, C.B.; Zhou, Z.C.; Liu, C.; Tan, K.F.; Wang, N.B.; Jiang, B.; Gao, X.G.; Liu, W.D. Isolation and pathogenicity of pathogens from skin ulceration disease and viscera ejection syndrome of the sea cucumber Apostichopus japonicas. Aquaculture 2009, 287, 18–27. [Google Scholar] [CrossRef]
- Stojanov, S.; Berlec, A.; Štrukelj, B. The influence of probiotics on the Firmicutes/Bacteroidetes ratio in the treatment of obesity and inflammatory bowel disease. Microorganisms 2020, 8, 1715. [Google Scholar] [CrossRef]
- Wiklund, S.; Johansson, E.; Sjöström, L.; Mellerowicz, E.J.; Edlund, U.; Shockcor, J.P.; Gottfries, J.; Moritz, T.; Trygg, J. Visualization of GC/TOF-MS-based metobolomics data for identification of biochemically interesting compounds using OPLS class models. Anal. Chem. 2008, 80, 115–122. [Google Scholar] [CrossRef]
- Lee, J.Y.; Tsolis, R.M.; Bäumler, A.J. The microbiome and gut homeostasis. Science 2022, 377, 115–122. [Google Scholar] [CrossRef]
- Cui, X.; Zhang, Q.; Zhang, Q.; Zhang, Y.; Chen, H.; Liu, G.; Zhu, L. Research progress of the Gut microbiome in hybrid fish. Microorganisms 2022, 10, 891. [Google Scholar] [CrossRef]
- Martel, J.; Chang, S.H.; Ko, Y.F.; Hwang, T.L.; Young, J.D.; Ojcius, D.M. Gut barrier disruption and chronic disease. Trends Endocrinol. Metab. 2022, 33, 247–265. [Google Scholar] [CrossRef]
- Jiao, L.; Dai, T.; Zhong, S.; Jin, M.; Sun, P.; Zhou, Q. Vibrio parahaemolyticus infection influenced trace element homeostasis; impaired antioxidant function; and induced inflammation response in Litopenaeus vannamei. Biol. Trace. Elem. Res. 2021, 199, 329–337. [Google Scholar] [CrossRef]
- Amoroso, C.; Perillo, F.; Strati, F.; Fantini, M.C.; Caprioli, F.; Facciotti, F. The role of gut microbiota biomodulators on mucosal immunity and intestinal inflammation. Cells 2020, 9, 1234. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, A.; Hugerth, L.W.; Hases, L.; Saxena, A.; Seifert, M.; Thomas, Q.; Gustafsson, J.Å.; Engstrand, L.; Williams, C. Colitis-induced colorectal cancer and intestinal epithelial estrogen receptor beta impact gut microbiota diversity. Int. J. Cancer 2019, 144, 3086–3098. [Google Scholar] [CrossRef] [Green Version]
- Tran, N.T.; Zhang, J.; Xiong, F.; Wang, G.T.; Li, W.X.; Wu, S.G. Altered gut microbiota associated with intestinal disease in grass carp (Ctenopharyngodon idellus). World J. Microbiol. Biotechnol. 2018, 34, 71. [Google Scholar] [CrossRef]
- Xing, M.X.; Hou, Z.H.; Qu, Y.M.; Liu, B. Enhancing the culturability of bacteria from the gastrointestinal tract of farmed adult turbot Scophthalmus maximus. Chin. J. Oceanol. Limnol. 2014, 32, 316–325. [Google Scholar] [CrossRef]
- Guo, X.Y.; Liu, X.J.; Hao, J.Y. Gut microbiota in ulcerative colitis: Insights on pathogenesis and treatment. J. Dig. Dis. 2020, 21, 147–159. [Google Scholar] [CrossRef]
- Zhu, W.; Winter, M.G.; Byndloss, M.X.; Spiga, L.; Duerkop, B.A.; Hughes, E.R.; Büttner, L.; Romão, E.D.L.; Behrendt, C.L.; Lopez, C.A.; et al. Precision editing of the gut microbiota ameliorates colitis. Nature 2018, 553, 208–211. [Google Scholar] [CrossRef]
- Jensen, S.; Samuelsen, O.B.; Andersen, K.; Torkildsen, L.; Lambert, C.; Choquet, G.; Paillard, C.; Bergh, O. Characterization of strains of Vibrio splendidus and V. tapetis isolated from corkwing wrasse Symphodus melops suffering vibriosis. Dis. Aquat. Organ. 2003, 53, 25–31. [Google Scholar] [CrossRef]
- González, R.; Brokordt, K.; Cárcamo, C.B.; Coba de la Peña, T.; Oyanedel, D.; Mercado, L.; Schmitt, P. Molecular characterization and protein localization of the antimicrobial peptide big defensin from the scallop Argopecten purpuratus after Vibrio splendidus challenge. Fish. Shellfish Immunol. 2017, 68, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Cao, R.; Liu, Y.; Wang, Q.; Yang, D.; Liu, H.; Ran, W.; Qu, Y.; Zhao, J. Seawater acidification reduced the resistance of Crassostrea gigas to Vibrio splendidus Challenge: An Energy Metabolism Perspective. Front. Physiol. 2018, 9, 880. [Google Scholar] [CrossRef] [PubMed]
- Thomson, R.; Macpherson, H.L.; Riaza, A.; Birkbeck, T.H. Vibrio splendidus biotype 1 as a cause of mortalities in hatchery-reared larval turbot, Scophthalmus maximus (L.). J. Appl. Microbiol. 2005, 99, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Hosseini, E.; Grootaert, C.; Verstraete, W.; Van de Wiele, T. Propionate as a health-promoting microbial metabolite in the human gut. Nutr. Rev. 2011, 69, 245–258. [Google Scholar] [CrossRef] [PubMed]
- Bottone, E.J. Bacillus cereus, a volatile human pathogen. Clin. Microbiol. Rev. 2010, 23, 382–398. [Google Scholar] [CrossRef] [Green Version]
- Inagaki, F.; Takai, K.; Nealson, K.H.; Horikoshi, K. Sulfurovum lithotrophicum gen. nov.; sp. nov.; a novel sulfur-oxidizing chemolithoautotroph within the ε-Proteobacteria isolated from Okinawa Trough hydrothermal sediments. Int. J. Syst. Evol. Microbiol. 2004, 54, 1477–1482. [Google Scholar] [CrossRef] [Green Version]
- Galushko, A.; Kuever, J. Desulfocastanea gen. nov. In Bergey’s Manual of Systematics of Archaea and Bacteria; Whitman, W.B., Ed.; John Wiley & Sons: New York, NY, USA, 2019; pp. 1–4. [Google Scholar]
- Jia, J.; Xue, X.; Guan, Y.; Fan, X.; Wang, Z. Biofilm characteristics and transcriptomic profiling of Acinetobacter johnsonii defines signatures for planktonic and biofilm cells. Environ. Res. 2022, 213, 113714. [Google Scholar] [CrossRef]
- Kimura, T.; Matsumoto, T.; Akiyoshi, M.; Owa, Y.; Miyasaka, N.; Aso, T.; Moritani, T. Body fat and blood lipids in postmenopausal women are related to resting autonomic nervous system activity. Eur. J. Appl. Physiol. 2006, 97, 542–547. [Google Scholar] [CrossRef]
- Greer, R.L.; Morgun, A.; Shulzhenko, N. Bridging immunity and lipid metabolism by gut microbiota. J. Allergy Clin. Immunol. 2013, 132, 253–262. [Google Scholar] [CrossRef]
- Lee, J.G.; Lee, J.; Lee, A.R.; Jo, S.V.; Park, C.H.; Han, D.S.; Eun, C.S. Impact of short-chain fatty acid supplementation on gut inflammation and microbiota composition in a murine colitis model. J. Nutr. Biochem. 2022, 101, 108926. [Google Scholar] [CrossRef]
- Kovács, T.; Varga, G.; Erces, D.; Tőkés, T.; Tiszlavicz, L.; Ghyczy, M.; Boros, M.; Kaszaki, J. Dietary phosphatidylcholine supplementation attenuates inflammatory mucosal damage in a rat model of experimental colitis. Shock 2012, 38, 177–185. [Google Scholar] [CrossRef]
- Parlesak, A.; Schaeckeler, S.; Moser, L.; Bode, C. Conjugated primary bile salts reduce permeability of endotoxin through intestinal epithelial cells and synergize with phosphatidylcholine in suppression of inflammatory cytokine production. Crit. Care Med. 2007, 35, 2367–2374. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Li, L.; Liu, R.; Zhu, L.; Zhou, B.; Xiao, Y.; Hou, G.; Lin, L.; Chen, X.; Peng, C. Integrative lipidomic features identify plasma lipid signatures in chronic urticaria. Front. Immunol. 2022, 13, 933312. [Google Scholar] [CrossRef]
- Treede, I.; Braun, A.; Sparla, R.; Kühnel, M.; Giese, T.; Turner, J.R.; Anes, E.; Kulaksiz, H.; Füllekrug, J.; Stremmel, W.; et al. Anti-inflammatory effects of phosphatidylcholine. J. Biol. Chem. 2007, 282, 27155–27164. [Google Scholar] [CrossRef] [Green Version]
- Bo, T.B.; Wen, J.; Zhao, Y.C.; Tian, S.J.; Zhang, X.Y.; Wang, D.H. Bifidobacterium pseudolongum reduces triglycerides by modulating gut microbiota in mice fed high-fat food. J. Steroid. Biochem. Mol. Biol. 2020, 198, 105602. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Kozich, J.J.; Westcott, S.L.; Baxter, N.T.; Highlander, S.K.; Schloss, P.D. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl. Environ. Microbiol. 2013, 79, 5112–5120. [Google Scholar] [CrossRef] [Green Version]
- Magoč, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Quince, C.; Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 2011, 27, 2194–2200. [Google Scholar] [CrossRef] [Green Version]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kenkel, N.C.; Orloci, L. Applying metric and nonmetric multidimensional scaling to ecological studies: Some new results. Ecology 1986, 67, 919–928. [Google Scholar] [CrossRef]
- Core Team. R: A Language and Environment for Statistical Computing. In The R Foundation for Statistical Computing; R Core Team: Vienna, Austria, 2015; ISBN 3-900051-07-0. Available online: http://www.R-project.org/ (accessed on 30 March 2012).







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Z.; Song, M.; Lv, Z.; Guo, M.; Li, C. Gut Microbiota Mediates Skin Ulceration Syndrome Outbreak by Readjusting Lipid Metabolism in Apostichopus japonicus. Int. J. Mol. Sci. 2022, 23, 13583. https://doi.org/10.3390/ijms232113583
Zhang Z, Song M, Lv Z, Guo M, Li C. Gut Microbiota Mediates Skin Ulceration Syndrome Outbreak by Readjusting Lipid Metabolism in Apostichopus japonicus. International Journal of Molecular Sciences. 2022; 23(21):13583. https://doi.org/10.3390/ijms232113583
Chicago/Turabian StyleZhang, Zhen, Mingshan Song, Zhimeng Lv, Ming Guo, and Chenghua Li. 2022. "Gut Microbiota Mediates Skin Ulceration Syndrome Outbreak by Readjusting Lipid Metabolism in Apostichopus japonicus" International Journal of Molecular Sciences 23, no. 21: 13583. https://doi.org/10.3390/ijms232113583
APA StyleZhang, Z., Song, M., Lv, Z., Guo, M., & Li, C. (2022). Gut Microbiota Mediates Skin Ulceration Syndrome Outbreak by Readjusting Lipid Metabolism in Apostichopus japonicus. International Journal of Molecular Sciences, 23(21), 13583. https://doi.org/10.3390/ijms232113583