
Detergent-Assisted Protein Digestion—On the Way to Avoid the Key Bottleneck of Shotgun Bottom-Up Proteomics



Abstract
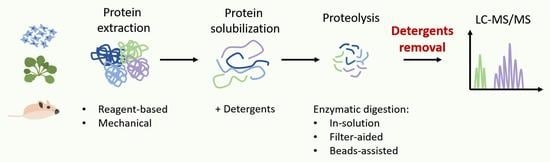
:
1. Introduction
2. Proteases Used for Gel-Free Proteomics

{kind=link}
{kind=link}
{kind=link}
| Protease | Family | Cleavage Site | Application | Reference |
|---|---|---|---|---|
| LysC | Serine protease | C-terminal of K | Used in combination with trypsin to improve digestion efficiency | [57] |
| GluC | Serine protease | C-terminal of E (at pH 4) C-terminal of D (at pH 8) | Analysis of glycated proteins | [42] |
| Chymotrypsin | Serine protease | C-terminal of F, Y, L, W and M | Analysis of proteins transmembrane regions | [51,52] |
| LysN | Metalloprotease | N-terminal of K | Analysis of N-terminal modifications | [44] |
| AspN | Metalloprotease | N-terminal of D | Used in combination with other enzymes to improve digestion efficiency | [49,58] |
| ArgC | Cysteine protease | C-terminal of R | Used in combination with other enzymes to improve digestion efficiency and analyze C-termini of proteins | [47,50] |
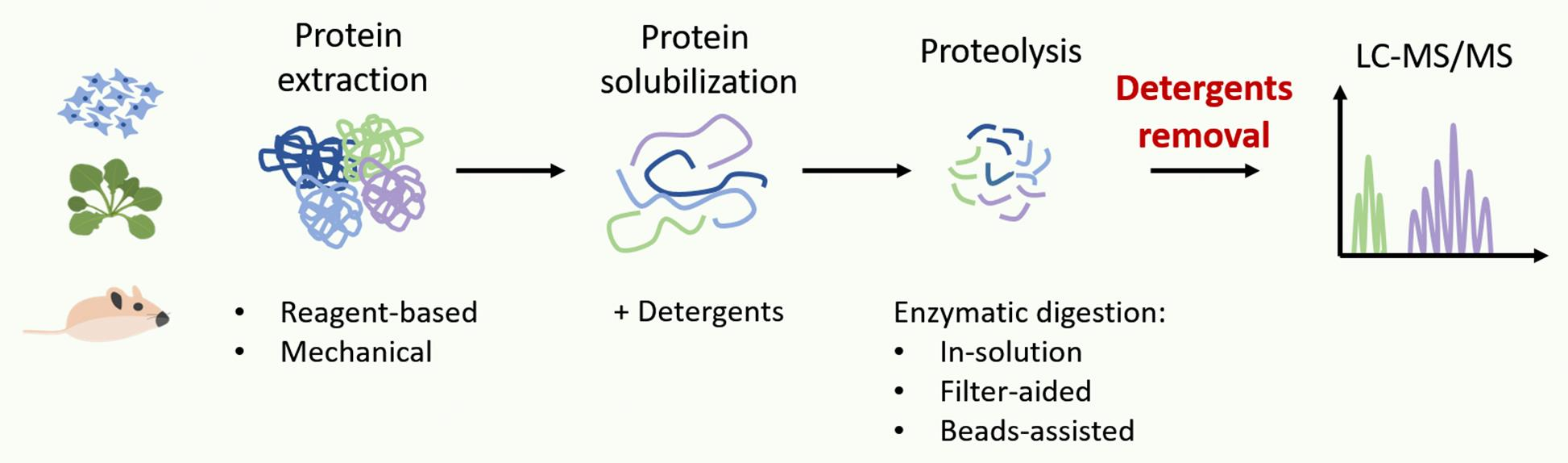
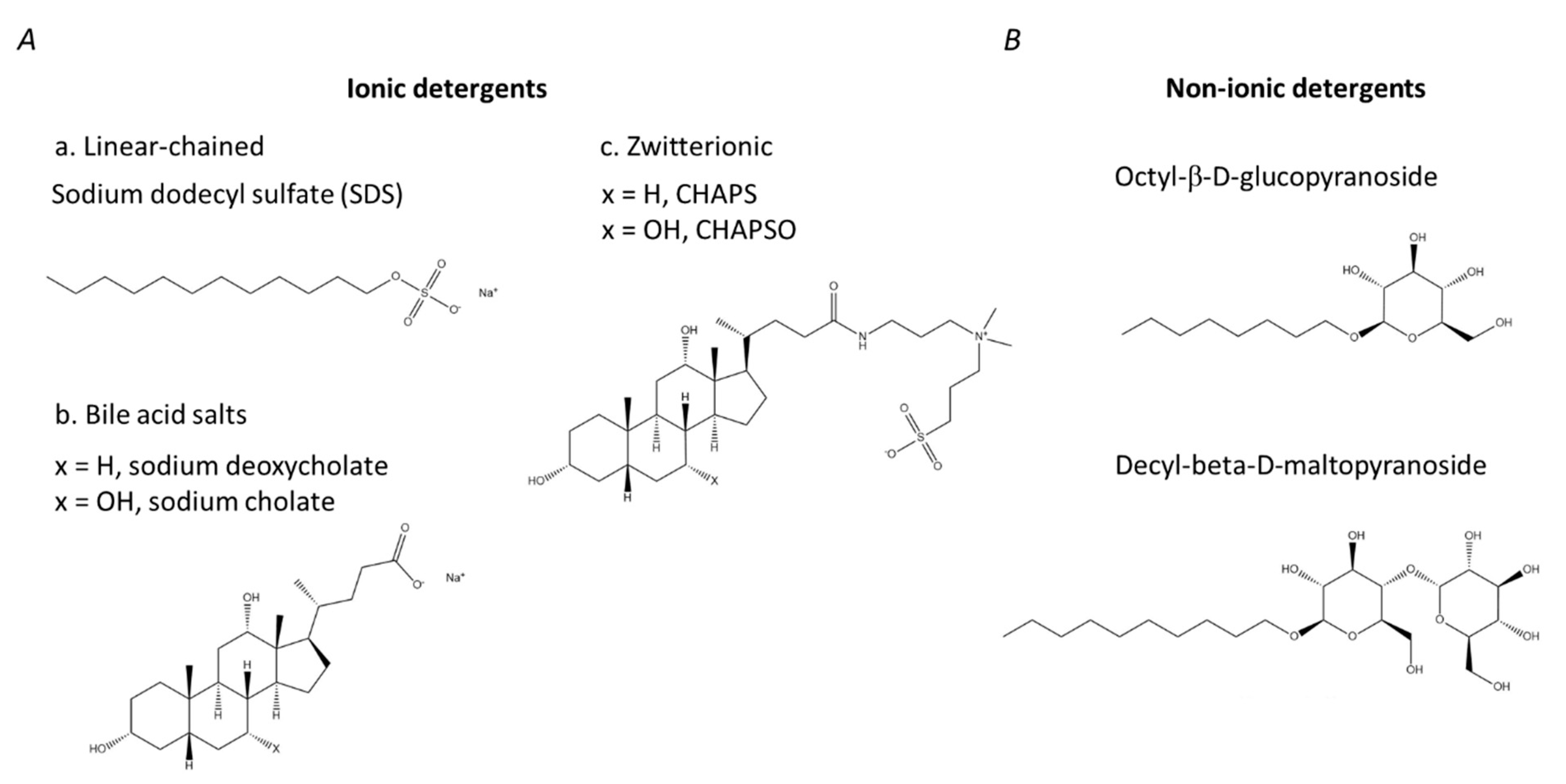
3. Detergents
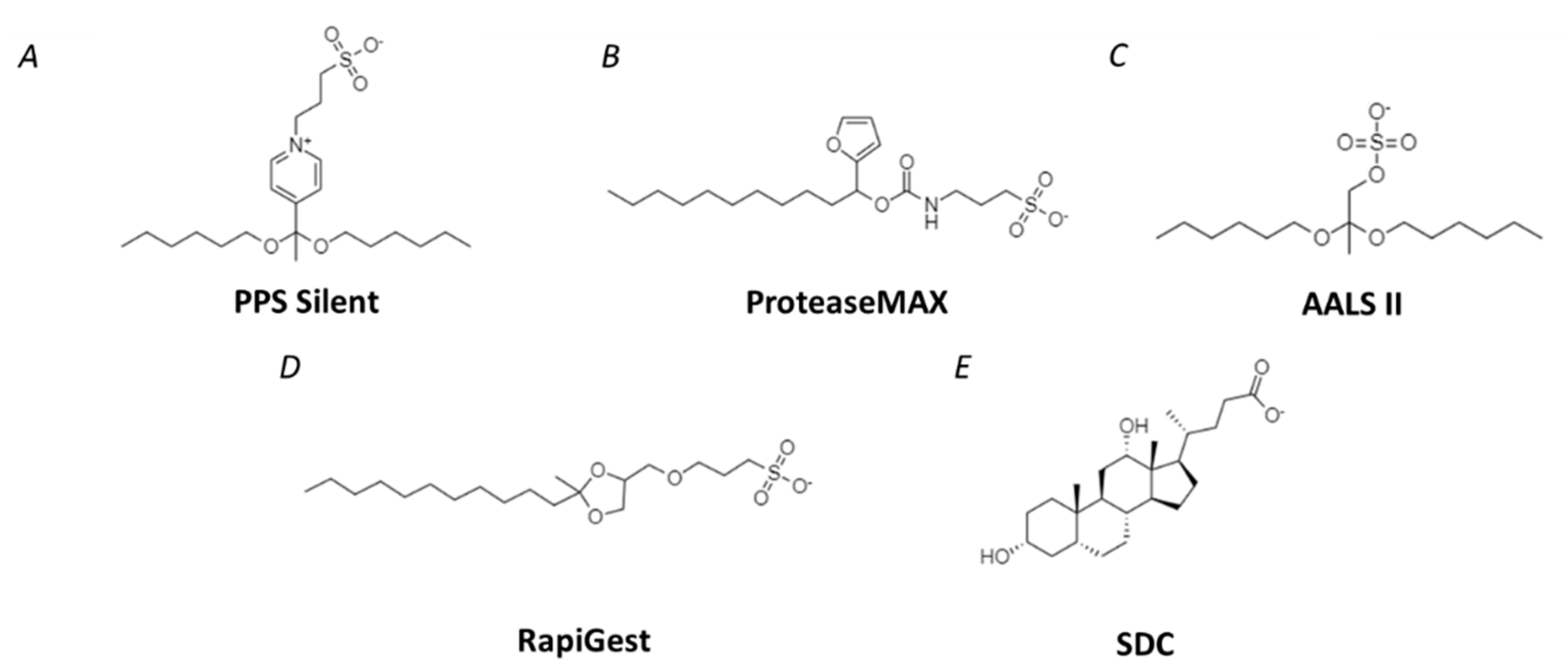
4. Strategies for the Removal of Detergents during Sample Preparation
4.1. Methods for Removal of SDS
4.2. Detergents Compatible with LC-MS
5. Comparison of Different Protein Digestion Strategies in Terms of Their Efficiency
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A
| # | Type of Sample | Methodology | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Lysis/Protein Isolation | Detergent or Chaotrope | Reduction/ Alkylation | Protease | Detergent Removal Strategy | Chromatographic System | MS | Reference | ||
| 1 | S. cerevisiae lysate (insoluble fraction) | Ultrasound | SDS | +/+ | trypsin | Pierce detergent removal spin columns | RP, C18 Phenomenex, water-ACN grad., 0.1% (v/v) FA | ESI-LIT-MS | [77] |
| C. elegans lysate (soluble fraction) | Mechanically | ||||||||
| Human embryonic kidney cell line (HEK293T) | Freeze/thaw | ||||||||
| 2 | HeLa cell line, BSA | Freeze/thaw, sonication | SDS, urea | DTT/IAA | (1) trypsin (2) LysC (3) ArgC (4) GluC (5) AspN (6) Chymotrypsin | FASP | RP, C18, 5–40% MeCN in 0.5% (v/v) acetic acid | ESI-LIT (LTQ)-Orbitrap-MS | [78] |
| Mouse brain tissue | Mechanically, freeze/thaw, ultrasound | ||||||||
| Mouse liver tissue | Mechanical, freeze/thaw, ultrasound | ||||||||
| 3 | Yeast | (1) French press 20,000 psi, GELFrEE Frac (2) Acetone precipitation (3) Chloroform/methanol-water precipitation | SDS | none | trypsin | FASP | RP, C18 Phenomenex | ESI-LIT(LTQ)- MS | [79] |
| 4 | Shewanella oneidensis MR-1 | Barocycler and pressure 35,000 psi | SDS, Urea | DTT/IAA | trypsin | (1) FASP (2) KDS precipitation | RP, C18 Phenomenex | ESI-LIT-MS | [80] |
| Mouse brain tissue | Mechanically | ||||||||
| Mouse liver tissue | Mechanically | ||||||||
| 5 | Rat liver membrane enriched fraction | Mechanically, centrifugation, sucrose density fractionation | (1) SDS (2) Urea (3) RapiGest | DTT/IAA | trypsin | (1) FASP (2) KDS precipitation | RP, C18, PepMap, water-ACN grad., 0.1% (v/v) FA | ESI-HCIT-MS | [81] |
| 6 | Escherichia coli membrane fraction | Cell disruptor at 25,000 psi | (1) SDS (2) RapiGest (3) PPS Silent | DTT/IAA | trypsin | SCXX | RP, C18, Atlantis, water-ACN grad., 0.1% (v/v) FA | ESI-QqTOF-MS | [82] |
| Breast cancer cell line (MCF-7) membrane fraction | Triton X-114-based lysis buffer, centrifugation | ||||||||
| 7 | Escherichia coli | Sonication | (1) DCA/ N-lauroyl sarcosine (2) SDS | (1) DTT/IAA (2) DTT/4-VP (3) TCEP/IAA (4) TCEP/4-VP | trypsin | FASP | RP, C18, water-ACN grad., 0.1% (v/v) FA | ESI-QqTOF HDMS or ESI-QqTOF-MS | [83] |
| 8 | Normal human cholangiocyte (H69) cell line | SDS, CHAPS Urea | DTT/IAA | trypsin | FASP | RP, C18, water-ACN grad., 0.1% (v/v) FA | ESI-TripleTOF (QqTOF-MS) | [84] | |
| 9 | Mytilus galloprovincialis hepatopancreas | Sonication | SDS | (1) none (2) DTT/IAA | (1) trypsin (2) trypsin/Lys-C | (1) FASP (2) SP3 (3) S-Trap | RP, C18, water-ACN grad., 0.1% (v/v) FA | ESI-Q-Exactive HF (Q-Orbitrap-MS) | [85] |
| Scophthalmus maximus liver | |||||||||
| 10 | HEK293 cell line | CelLytic NuCLEAR Extraction kit | SDS | TCEP+DTT/IAA | trypsin | FASP | RP, C18, water-ACN grad., 0.1% (v/v) FA | ESI-LIT(LCQ)-MS | [86] |
| BSA | |||||||||
| 11 | HeLa cell line | Freeze/thaw, sonication | SDS | IAA | (1) GluC (2) ArgC (3) LysC (4) AspN (5) trypsin (6) chymotrypsin | FASP | RP, C18 ReproSil-Pur, water-ACN grad., 0.1% (v/v) acetic acid | ESI-LIT(LTQ)-Orbitrap-MS | [87] |
| Mouse brain tissue | Mechanically, sonication | ||||||||
| Mouse liver tissue | Mechanically, sonication | ||||||||
| 12 | HeLa cell line membrane enriched fraction | Mechanically | (1) RapiGest (2) SDC | DTT/IAA | LysC/trypsin | (1) SDC: acid precipitation (2) SDC: phase transfer surfactant | RP, C18 ReproSil-Pur, water-ACN grad., 0.1% (v/v) acetic acid | ESI-LIT(LTQ)-Orbitrap MS or QSTAR QqTOF-MS | [88] |
| Escherichia coli membrane enriched fraction | Sonication | (1) RapiGest (2) SDC | DTT/IAA | LysC/trypsin | (1) SDC: acidprecipitation (2) SDC: phase transfer surfactant | C18 ReproSil-Pur, water-ACN grad., 0.1% (v/v) acetic acid | ESI-LTQ-Orbitrap MS or QSTAR QqTOF-MS | [88] | |
| 13 | Escherichia coli | Freeze/thaw | (1) SDS (2) DCA | DTT/IAA | trypsin | FASP | RP, C18 Luna, water-ACN grad., 0.1% (v/v) FA | ESI-Q-Exactive (Q-Orbitrap-MS) | [89] |
| 14 | Hordeum vulgare L. cv. Golden promise (barley) leaves | Mechanically, sonication | (1) SDS (2) SDC (3) SDC/CAA | IAA | trypsin | FASP | RP, C18 ReproSil-Pur, water-ACN grad., 0.1% (v/v) FA | ESI-Q-Exactive (Q-Orbitrap-MS) | [90] |
| 15 | Arabidopsis thaliana shoots, roots, seeds | (1) Chloroform/methanol-water precipitation (2) Phenol extraction (3) Urea-based extraction (4) SDS-based extraction | (1) Urea (2) SDS | TCEP/IAA | LysC/trypsin | FASP | RP, C18, water-ACN grad., 0.1% (v/v) FA | ESI-Q-Exactive HF (Q-Orbitrap-MS) | [91] |
| 16 | Ramonda serbica (Phenix plant) | (1) TCA/acetone precipitation (2) Phenol-based extraction (3) Detergents-based extraction | (1) SDS (2) Triton X-100 (3) dodecyl-β-D-maltoside | DTT/IAA | trypsin | FASP | RP, C18, water-ACN grad., 0.1% (v/v) FA | ESI-LIT(LTQ)-Orbitrap-MS | [92] |
| 17 | Triticum aestivum L. (wheat) roots | TCA/acetone precipitation | SDS | IAA | trypsin | FASP | RP, C18, water-ACN grad., 0.1% (v/v) FA | ESI-Q-Exactive (Q-Orbitrap-MS) | [94] |
| 18 | S. lycopersicum (tomato) fruits | Sonication | SDS | DTT/IAA | trypsin | FASP | RP, C18, water-ACN grad., 0.1% (v/v) FA | ESI-Q-Exactive (Q-Orbitrap-MS) | [95] |
| 19 | Ziziphus jujuba Mill. flowers | TCA/acetone precipitation | SDS | DTT/IAA | trypsin | FASP | RP, C18, water-ACN grad., 0.1% (v/v) FA | ESI-Q-Exactive (Q-Orbitrap-MS) | [96] |
| 20 | Human prostate cancer (PC-3) cell line | (1) Phenol-based extraction (2) Detergent-based extraction | SDS, urea | DTT/IAA | trypsin | FASP | RP, C18, PepMap, water-ACN grad., 0.1% (v/v) FA | ESI-LIT(LTQ)-Orbitrap-MS | [97] |
| Arabidopsis thaliana shoots | |||||||||
| Pisum sativum (pea) seeds | (1) Phenol-based extraction (2) Detergent-based extraction | SDS, urea | DTT/IAA | trypsin | FASP | RP, C18, PepMap, water-ACN grad., 0.1% (v/v) FA | ESI-LIT(LTQ)-Orbitrap-MS | [97] | |
| 21 | HeLa cell line, membrane enriched fraction | Sonication | (1) SDS (2) Triton X-114 | DTT/IAA | (1) trypsin (2) trypsin/LysC | S-Trap | RP, C18, water-ACN grad., 0.1% (v/v) FA | ESI-LIT(LTQ)-Orbitrap-MS | [98] |
| 22 | Colorectal cancer (SW480) cell line | Sonication | Urea, SDS | DTT/IAA | trypsin | (1) FASP (2) S-Trap | RP, C18, water-ACN grad., 0.1% (v/v) FA | ESI-Q-Exactive (Q-Orbitrap-MS) | [99] |
| 23 | Human blood plasma | none | SDS | TCEP/IAA | trypsin | Chromatography | RP, C18, water-ACN grad., 0.1% (v/v) FA | ESI-LIT(LTQ)-Orbitrap-MS | [108] |
| 24 | Human blood plasma | none | SDS | TCEP/IAA | trypsin | Boronic acid affinity chromatography | RP, C18, water-ACN grad., 0.1% (v/v) FA | ESI-LIT(LTQ)-Orbitrap-MS | [111] |
| 25 | Mouse kidney tissue | Microdissection | PPS Silent | DTT/CAA | trypsin | degradation under acidic conditions | RP, C18 | ESI-LIT(LTQ)-Orbitrap-MS | [113] |
| Mouse pancreas/pancreatic islets | Sonication | ||||||||
| 26 | Salmonella enterica outer membrane vesicles | LPI™ FlowCells | PPS Silent | none | trypsin | degradation under acidic conditions | RP, C18 ReproSil-Pur, water-ACN grad., 0.1% (v/v) FA | ESI-LIT(LTQ)-FT-MS | [114] |
| 27 | Arabidopsis thaliana shoots | (1) SDS/Phenol-based extraction (2) Methanol/Chloroform extraction | PPS Silent | none (in-gel digest) | trypsin | degradation under acidic conditions | RP, C18 ReproSil-Pur, water-ACN grad., 0.1% (v/v) FA | ESI-QqTOF-MS | [116] |
| 28 | Insect Sf9 cell line, His-tagged RGS4 | Sonication, CHAPS | ProteaseMAX | DTT/IAA | trypsin | degradation under acidic conditions | RP, C18 ReproSil-Pur, water-ACN grad., 0.1% (v/v) FA | MALDI-TOF/TOF or ESI-LIT(LTQ)-Orbitrap-MS | [117] |
| 29 | Mouse liver tissue | Mechanically, Extraction kit | ProteaseMAX | DTT/IAA | LysC/trypsin | degradation under acidic conditions | RP, C18 Easy-Spray, water-ACN grad., 0.1% (v/v) FA | ESI-LIT(LTQ)-Orbitrap-MS | [120] |
| 30 | Mouse brain tissue | Liquid microjunction (LMJ) technique | ProteaseMAX | DTT | trypsin | heating | PLRP-S, water-ACN grad | MALDI-MSI and ESI-Q-Exactive (Q-Orbitrap-MS) | [121] |
| 31 | Human glioblastoma tissue (whole tissue lysate, cytosol, microsomes, and plasma membrane fractions) | Mechanically | Urea, ProteaseMAX | DTT/IAA | (1) trypsin (2) LysC/trypsin | degradation under acidic conditions | RP, C18 ReproSil-Pur, water-ACN grad., 0.1% (v/v) FA | ESI-TripleTOF QqTOF-MS | [122] |
| Human blood plasma | |||||||||
| 32 | PLRV-infected Solanum tuberosum (potato) leaves | Beads-assisted extraction | ProteaseMAX | TCEP/methanethiosulfonate | trypsin | none | RP, C18 ReproSil-Pur, water-ACN grad., 0.1% (v/v) FA | ESI-LIT(LTQ)-Orbitrap-MS | [123] |
| 33 | Phaseolus vulgaris L. (running bean) leaves | Methanol/acetone | Urea, ProteaseMAX | DTT/IAA | trypsin | none | RP, C18 Easy-Spray, water-ACN grad., 0.1% (v/v) FA | ESI-LIT(LTQ)-Orbitrap-MS | [124] |
| 34 | Mixture of myoglobin, BSA, B-casein, and ovalbumin | none | (1) SDS (2) ALSI | none (in-gel digest) | trypsin | degradation under acidic conditions | none | MALDI-TOF-MS | [127] |
| 35 | Arabidopsis thaliana | Phenol-based extraction | Urea, thiourea, AALS II | TCEP/IAA | trypsin | degradation under acidic conditions | RP, C18 PepMap, water-ACN grad., 0.1% (v/v) FA | ESI-LIT-Q-Orbitrap-MS | [128] |
| 36 | Pisum sativum (pea) seeds | Phenol-based extraction | Urea, thiourea, AALS II | TCEP/IAA | trypsin | degradation under acidic conditions | RP, C18 PepMap, water-ACN grad., 0.1% (v/v) FA | ESI-Q-Orbitrap-MS | [129] |
| 37 | Pisum sativum (pea) seeds | Phenol-based extraction | Urea, thiourea, AALS II | TCEP/IAA | trypsin | degradation under acidic conditions | RP, C18 PepMap, water-ACN grad., 0.1% (v/v) FA | ESI-LIT-Q-Orbitrap-MS | [130] |
| 38 | Phaseolus vulgaris L. (running bean) nodules | Phenol-based extraction | AALS II | TCEP/IAA | trypsin | degradation under acidic conditions | RP, C18, water-ACN grad., 0.1% (v/v) FA | ESI-LTQ-Orbitrap-ETD-MS | [131] |
| 39 | Mouse eye retina | Sonication | AALS II | TCEP/IAA | trypsin | degradation under acidic conditions | nanoUPLC | ESI-QqTOF-MS | [132] |
| 40 | MDCK II Tet-off cell line | Chloroform/methanol extraction | AALS II | DTT/IAA (in-gel digest) | trypsin | degradation under acidic conditions | RP, PicoFrit, water-ACN grad., 0.1% (v/v) FA | ESI-LIT(LTQ)-Orbitrap-MS | [133] |
| 41 | Escherichia coli membrane fraction | French press 20,000 psi | (1) SDS (2) RapiGest (3) PPS | DTT/IAA | trypsin | (1) SDS: SCX (2) RapiGest and PPS Silent: degradation under acidic conditions | RP, C18, water-ACN grad., 0.1% (v/v) FA | ESI-QqTOF-MS | [135] |
| Human breast cancer (MCF7) cell line membrane fraction | French press 20,000 psi, aceton | ||||||||
| 42 | Human blood plasma | none | RapiGest | DTT/IAA | trypsin | degradation under acidic conditions | RP, C18, water-ACN grad., 0.1% (v/v) FA | ESI-QqTOF-MS | [136] |
| 43 | U-2 OS osteosarcoma cell line | Sonication | RapiGest | TCEP/IAA | trypsin | degradation under acidic conditions | RP, C18, water-ACN grad., 0.1% (v/v) FA | ESI-LIT(LTQ)-Orbitrap MS | [137] |
| U251 glioblastoma cell line | |||||||||
| HeLa CCL2 cell line | |||||||||
| Human (BT-474) breast cancer cell line | |||||||||
| Jurkat cell line | |||||||||
| Murine pre-adipocytes | |||||||||
| 44 | Rat ligodendro- cytes, oligodendrocyte precursor cells | Sonication, chloroform/methanol extraction | (1) SDS (2) RapiGest | DTT/acrylamide | trypsin | FASP | RP, C18 ReploSil-Pur, water-ACN grad., 0.1% (v/v) FA | ESI-LIT(LTQ)-Orbitrap MS | [138] |
| 45 | Hordeum vulgare L (barley) caryopse | TCA/acetone precipitation | RapiGest | DTT/IAA | trypsin | degradation under acidic conditions | RP, C18, water-ACN grad., 0.1% (v/v) FA | ESI-QqTOF-MS | [139] |
| 46 | Rat liver membrane enriched fraction | Mechanically, centrifugation, sucrose density fractionation | SDC | DTT/IAA | trypsin | (1) acid precipitation (2) phase transfer surfactant | RP, C18 PepMap, water-ACN grad., 0.1% (v/v) FA | ESI-3D-HCIT-MS | [142] |
| 47 | Human blood plasma | none | SDC | DTT/IAA | trypsin | acid precipitation | RP, C18 PepMap, water-ACN grad., 0.1% (v/v) FA | ESI-LIT(LTQ)-FT-MS | [143] |
| 48 | Human blood plasma | none | SDS | TCEP/IAA | trypsin | trap column | RP, C18 Aquity, water-ACN grad., 0.1% (v/v) FA | ESI-LIT-Q-Orbitrap-MS | [144] |
| 49 | Human blood plasma | none | SDS | TCEP/IAA | trypsin | acid precipitation | RP, C18, water-ACN grad., 0.1% (v/v) FA | ESI-QqLIT-MS | [145] |
| 50 | Rat liver enriched membrane fraction | Sonication | (1) SDS (2) SDC | DTT/IAA | trypsin | (1) SDC: acid precipitation (2) SDS: FASP | RP, C18 PepMap, water-ACN grad., 0.1% (v/v) FA | ESI-3D-HC-LIT-MS | [146] |
| 51 | Elaeis guineensis (oil palm) fruit mesocarps | TFA/acetone precipitation | Urea, thiourea, CHAPS, SDC | TCEP/IAA | trypsin | SDC: acid precipitation | RP, C18 PepMap, water-ACN grad., 0.1% (v/v) FA | ESI-Q-Exactive Orbitrap-MS | [147] |
| 52 | Mouse embryonic fibroblasts (STO) cell line | Hypotonic lysis buffer, isolation of cell membranes by centrifugation | (1) PPS Silent (2) AALS I (3) AALS II (4) CALS I (5) CALS II (6) ProteaseMAX (7) RapiGest | TCEP/IAA | trypsin | degradation under acidic conditions | RP, C18-AQ, water-ACN grad., 0.1% (v/v) FA | ESI-LIT(LTQ)-MS | [148] |
| 53 | A375 cell line | Detergent-containing lysis buffer, sonication | (1) ProteaseMAX (2) SDC | DTT/IAA | trypsin | (1) PPS Silent: degradation under acidic conditions (2) SDC: acid precipitation | RP, C18 PepMap, water-ACN grad., 0.1% (v/v) FA | ESI-Q-Orbitrap-MS | [119] |
| 54 | HEK293 kidney cell line | Detergent-containing lysis buffer, sonication | (1) SDC (2) PPS Silent (3) Invitrosol (4) RapiGest | DTT/IAA | trypsin | (1) PPS Silent and RapiGest: degradation under acidic conditions (2) SDC: acid precipitation (3) Invitrosol: none | RP, C18, water-ACN grad., 0.1% (v/v) FA | ESI-Q-Orbitrap-MS | [150] |
| 55 | Escherichia coli | Detergent-containing lysis buffer | SDS | DTT/IAA | trypsin | (1) FASP (2) Protein precipitation in 80% acetone (3) Protein precipitation in TCA with acetone wash (4) KDS precipitation (5) Pierce detergent removal spin columns (6) SCX (7) in-gel digestion | RP, C18, water-ACN grad., 0.1% (v/v) FA | ESI-QqQ-MS | [149] |
| 56 | Mouse pancreatic islets | Centrifugation, detergent-containing lysis buffer, sonication | (1) urea (2) SDS (3) SDC (4) NP-40 (5) RapiGest. | DTT/IAA | trypsin | FASP | RP, C18, water-ACN grad., 0.1% (v/v) FA | ESI-Q-Orbitrap-MS | [153] |
| 57 | Solanum esculentum L. (tomato) roots | Detergent-based extraction | (1) PPS Silent (2) RapiGest (3) GdnCl | TCEP/IAA | trypsin | PPS Silent and RapiGest: degradation under acidic conditions | RP, BEH C18, water-ACN grad., 0.1% (v/v) FA | ESI-QqTOF-MS | [155] |
References
- Wilkins, M.R.; Pasquali, C.; Appel, R.D.; Ou, K.; Golaz, O.; Sanchez, J.C.; Yan, J.X.; Gooley, A.A.; Hughes, G.; Humphery-Smith, I.; et al. From Proteins to Proteomes: Large Scale Protein Identification by Two-Dimensional Electrophoresis and Arnino Acid Analysis. Bio/Technology 1996, 14, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Aslam, B.; Basit, M.; Nisar, M.A.; Khurshid, M.; Rasool, M.H. Proteomics: Technologies and Their Applications. J. Chromatogr. Sci. 2017, 55, 182–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smolikova, G.; Gorbach, D.; Lukasheva, E.; Mavropolo-Stolyarenko, G.; Bilova, T.; Soboleva, A.; Tsarev, A.; Romanovskaya, E.; Podolskaya, E.; Zhukov, V.; et al. Bringing New Methods to the Seed Proteomics Platform: Challenges and Perspectives. Int. J. Mol. Sci. 2020, 21, 9162. [Google Scholar] [CrossRef]
- Iwamoto, N.; Shimada, T. Recent Advances in Mass Spectrometry-Based Approaches for Proteomics and Biologics: Great Contribution for Developing Therapeutic Antibodies. Pharmacol. Ther. 2018, 185, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Frolov, A.; Mamontova, T.; Ihling, C.; Lukasheva, E.; Bankin, M.; Chantseva, V.; Vikhnina, M.; Soboleva, A.; Shumilina, J.; Mavropolo-Stolyarenko, G.; et al. Mining Seed Proteome: From Protein Dynamics to Modification Profiles. Biol. Commun. 2018, 63, 43–58. [Google Scholar] [CrossRef] [Green Version]
- Toby, T.K.; Fornelli, L.; Kelleher, N.L. Progress in Top-Down Proteomics and the Analysis of Proteoforms. Annu. Rev. Anal. Chem. 2016, 9, 499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Catherman, A.D.; Skinner, O.S.; Kelleher, N.L. Top Down Proteomics: Facts and Perspectives. Biochem. Biophys. Res. Commun. 2014, 445, 683. [Google Scholar] [CrossRef] [Green Version]
- Nesvizhskii, A.I.; Aebersold, R. Interpretation of Shotgun Proteomic Data. Mol. Cell. Proteom. 2005, 4, 1419–1440. [Google Scholar] [CrossRef] [Green Version]
- Huang, T.; Wang, J.; Yu, W.; He, Z. Protein Inference: A Review. Brief. Bioinform. 2012, 13, 586–614. [Google Scholar] [CrossRef] [Green Version]
- Dupree, E.J.; Jayathirtha, M.; Yorkey, H.; Mihasan, M.; Petre, B.A.; Darie, C.C. A Critical Review of Bottom-Up Proteomics: The Good, the Bad, and the Future of This Field. Proteomes 2020, 8, 14. [Google Scholar] [CrossRef]
- Zhang, Y.; Fonslow, B.R.; Shan, B.; Baek, M.C.; Yates, J.R. Protein Analysis by Shotgun/Bottom-up Proteomics. Chem. Rev. 2013, 113, 2343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vercauteren, F.G.G.; Arckens, L.; Quirion, R. Applications and Current Challenges of Proteomic Approaches, Focusing on Two-Dimensional Electrophoresis. Amino Acids 2007, 33, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Magdeldin, S.; Enany, S.; Yoshida, Y.; Xu, B.; Zhang, Y.; Zureena, Z.; Lokamani, I.; Yaoita, E.; Yamamoto, T. Basics and Recent Advances of Two Dimensional-Polyacrylamide Gel Electrophoresis. Clin. Proteom. 2014, 11, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yates, J.R. Mass Spectral Analysis in Proteomics. Annu. Rev. Biophys. Biomol. Struct. 2004, 33, 297–316. [Google Scholar] [CrossRef] [PubMed]
- Aguilar, M.I. Reversed-Phase High-Performance Liquid Chromatography. Methods Mol. Biol. 2004, 251, 9–22. [Google Scholar] [CrossRef]
- Duong, V.A.; Park, J.M.; Lee, H. Review of Three-Dimensional Liquid Chromatography Platforms for Bottom-Up Proteomics. Int. J. Mol. Sci. 2020, 21, 1524. [Google Scholar] [CrossRef] [Green Version]
- Shen, Y.; Zhao, R.; Berger, S.J.; Anderson, G.A.; Rodriguez, N.; Smith, R.D. High-Efficiency Nanoscale Liquid Chromatography Coupled on-Line with Mass Spectrometry Using Nanoelectrospray Ionization for Proteomics. Anal. Chem. 2002, 74, 4235–4249. [Google Scholar] [CrossRef]
- Mitulović, G.; Mechtler, K. HPLC Techniques for Proteomics Analysis--a Short Overview of Latest Developments. Brief. Funct. Genom. Proteomic 2006, 5, 249–260. [Google Scholar] [CrossRef] [Green Version]
- Tao, D.; Zhang, L.; Shan, Y.; Liang, Z.; Zhang, Y. Recent Advances in Micro-Scale and Nano-Scale High-Performance Liquid-Phase Chromatography for Proteome Research. Anal. Bioanal. Chem. 2011, 399, 229–241. [Google Scholar] [CrossRef]
- Krokhin, O.V.; Ens, W.; Standing, K.G. MALDI QqTOF MS Combined with Off-Line HPLC for Characterization of Protein Primary Structure and Post-Translational Modifications. J. Biomol. Tech. 2005, 16, 429. [Google Scholar]
- Soboleva, A.; Schmidt, R.; Vikhnina, M.; Grishina, T.; Frolov, A. Maillard Proteomics: Opening New Pages. Int. J. Mol. Sci. 2017, 18, 2677. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.I.; Cociorva, D.; Norris, J.L.; Yates, J.R. Optimization of Mass Spectrometry-Compatible Surfactants for Shotgun Proteomics. J. Proteome Res. 2007, 6, 2529–2538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loo, R.R.; Dales, N.; Andrews, P.C. The Effect of Detergents on Proteins Analyzed by Electrospray Ionization. Methods Mol. Biol. 1996, 61, 141–160. [Google Scholar] [CrossRef] [PubMed]
- Loo, R.R.O.; Dales, N.; Andrews, P.C. Surfactant Effects on Protein Structure Examined by Electrospray Ionization Mass Spectrometry. Protein Sci. 1994, 3, 1975. [Google Scholar] [CrossRef] [Green Version]
- Rundlett, K.L.; Armstrong, D.W. Mechanism of Signal Suppression by Anionic Surfactants in Capillary Electrophoresis-Electrospray Ionization Mass Spectrometry. Anal. Chem. 1996, 68, 3493–3497. [Google Scholar] [CrossRef]
- Chen, E.I.; McClatchy, D.; Sung, K.P.; Yates, J.R. Comparisons of Mass Spectrometry Compatible Surfactants for Global Analysis of the Mammalian Brain Proteome. Anal. Chem. 2008, 80, 8694–8701. [Google Scholar] [CrossRef] [Green Version]
- Varnavides, G.; Madern, M.; Anrather, D.; Hartl, N.; Reiter, W.; Hartl, M. In Search of a Universal Method: A Comparative Survey of Bottom-Up Proteomics Sample Preparation Methods. J. Proteome Res. 2022, 21, 2397–2411. [Google Scholar] [CrossRef]
- Tsiatsiani, L.; Heck, A.J.R.; Heck, A.J.R.; Bijvoet, P. Proteomics beyond Trypsin. FEBS J. 2015, 282, 2612–2626. [Google Scholar] [CrossRef]
- Vandermarliere, E.; Mueller, M.; Martens, L. Getting Intimate with Trypsin, the Leading Protease in Proteomics. Mass Spectrom. Rev. 2013, 32, 453–465. [Google Scholar] [CrossRef] [Green Version]
- Perutka, Z.; Sebela, M. Pseudotrypsin: A Little-Known Trypsin Proteoform. Chem. Nat. Prod. Chem. 2018, 23, 2637. [Google Scholar] [CrossRef] [Green Version]
- Heissel, S.; Frederiksen, S.J.; Bunkenborg, J.; Højrup, P. Enhanced Trypsin on a Budget: Stabilization, Purification and High-Temperature Application of Inexpensive Commercial Trypsin for Proteomics Applications. PLoS ONE 2019, 14, e0218374. [Google Scholar] [CrossRef]
- Siepen, J.A.; Keevil, E.J.; Knight, D.; Hubbard, S.J. Prediction of Missed Cleavage Sites in Tryptic Peptides Aids Protein Identification in Proteomics. J. Proteome Res. 2007, 6, 399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gershon, P.D. Cleaved and Missed Sites for Trypsin, Lys-C, and Lys-N Can Be Predicted with High Confidence on the Basis of Sequence Context. J. Proteome Res. 2014, 13, 702–709. [Google Scholar] [CrossRef]
- Rodriguez, J.; Gupta, N.; Smith, R.D.; Pevzner, P.A. Does Trypsin Cut before Proline? J. Proteome Res. 2008, 7, 300–305. [Google Scholar] [CrossRef] [PubMed]
- Keil, B. Trypsin. Enzymes 1971, 3, 249–275. [Google Scholar] [CrossRef]
- Šlechtová, T.; Gilar, M.; Kalíková, K.; Tesařová, E. Insight into Trypsin Miscleavage: Comparison of Kinetic Constants of Problematic Peptide Sequences. Anal. Chem. 2015, 87, 7636–7643. [Google Scholar] [CrossRef]
- Proc, J.L.; Kuzyk, M.A.; Hardie, D.B.; Yang, J.; Smith, D.S.; Jackson, A.M.; Parker, C.E.; Borchers, C.H. A Quantitative Study of the Effects of Chaotropic Agents, Surfactants, and Solvents on the Digestion Efficiency of Human Plasma Proteins by Trypsin. J. Proteome Res. 2010, 9, 5422. [Google Scholar] [CrossRef] [Green Version]
- Swaney, D.L.; Wenger, C.D.; Coon, J.J. The Value of Using Multiple Proteases for Large-Scale Mass Spectrometry-Based Proteomics. J. Proteome Res. 2010, 9, 1323. [Google Scholar] [CrossRef] [Green Version]
- Jennings, M.E.; Silveira, J.R.; Treier, K.M.; Tracy, P.B.; Matthews, D.E. Total Retention Liquid Chromatography-Mass Spectrometry to Achieve Maximum Protein Sequence Coverage. Anal. Chem. 2021, 93, 5054. [Google Scholar] [CrossRef]
- Giansanti, P.; Tsiatsiani, L.; Low, T.Y.; Heck, A.J.R. Six Alternative Proteases for Mass Spectrometry-Based Proteomics beyond Trypsin. Nat. Protoc. 2016, 11, 993–1006. [Google Scholar] [CrossRef]
- Darville, L.N.F.; Sokolowski, B.H.A. In-Depth Proteomic Analysis of Mouse Cochlear Sensory Epithelium by Mass Spectrometry. J. Proteome Res. 2013, 12, 3620. [Google Scholar] [CrossRef] [PubMed]
- Priego-Capote, F.; Ramírez-Boo, M.; Finamore, F.; Gluck, F.; Sanchez, J.C. Quantitative Analysis of Glycated Proteins. J. Proteome Res. 2014, 13, 336–347. [Google Scholar] [CrossRef]
- Sonomura, K.; Kuyama, H.; Matsuo, E.; Tsunasawa, S.; Nishimura, O. A Method for Terminus Proteomics: Selective Isolation and Labeling of N-Terminal Peptide from Protein through Transamination Reaction. Bioorg. Med. Chem. Lett. 2009, 19, 6544–6547. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Lu, J.; Yang, L.; Bao, H.; Zhang, L.; Yan, G.; Fang, C.; Lu, H. Rapid and Easy Enrichment Strategy for Naturally Acetylated N Termini Based on LysN Digestion and Amine-Reactive Resin Capture. Anal. Chem. 2020, 92, 8315–8322. [Google Scholar] [CrossRef] [PubMed]
- Miroshnychenko, O.; Chalkley, R.J.; Leib, R.D.; Everts, P.A.; Dragoo, J.L. Proteomic Analysis of Platelet-Rich and Platelet-Poor Plasma. Regen. Ther. 2020, 15, 226–235. [Google Scholar] [CrossRef]
- Wu, Z.; Huang, J.; Huang, J.; Li, Q.; Zhang, X. Lys-C/Arg-C, a More Specific and Efficient Digestion Approach for Proteomics Studies. Anal. Chem. 2018, 90, 9700–9707. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, Q.; Huang, J.; Wu, Z.; Huang, J.; Huang, L.; Li, Y.; Ye, J.; Zhang, X. An Approach to Incorporate Multi-Enzyme Digestion into C-TAILS for C-Terminomics Studies. Proteomics 2018, 18, 1700034. [Google Scholar] [CrossRef]
- Jia, X.; Pan, L.; Zhu, M.; Hu, H.; Zhai, L.; Liu, J.; Hu, M.; Liu, B.; Tan, M. PSILAC Method Coupled with Two Complementary Digestion Approaches Reveals PRPF39 as a New E7070-Dependent DCAF15 Substrate. J. Proteomics 2020, 210, 103545. [Google Scholar] [CrossRef]
- Guo, X.; Trudgian, D.C.; Lemoff, A.; Yadavalli, S.; Mirzaei, H. Confetti: A Multiprotease Map of the HeLa Proteome for Comprehensive Proteomics. Mol. Cell. Proteom. 2014, 13, 1573–1584. [Google Scholar] [CrossRef] [Green Version]
- Kuyama, H.; Nakajima, C.; Tanaka, K. Enriching C-Terminal Peptide from Endopeptidase ArgC Digest for Protein C-Terminal Analysis. Bioorg. Med. Chem. Lett. 2012, 22, 7163–7168. [Google Scholar] [CrossRef]
- Fischer, F.; Wolters, D.; Rögner, M.; Poetsch, A. Toward the Complete Membrane Proteome: High Coverage of Integral Membrane Proteins Through Transmembrane Peptide Detection. Mol. Cell. Proteom. 2006, 5, 444–453. [Google Scholar] [CrossRef] [PubMed]
- Min, L.; Choe, L.H.; Lee, K.H. Improved Protease Digestion Conditions for Membrane Protein Detection. Electrophoresis 2015, 36, 1690. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Huang, Y.; Ferrant, J.; Lyubarskaya, Y.; Zhang, Y.E.; Li, S.P.; Wu, S.L.B. Assessing in Vivo Dynamics of Multiple Quality Attributes from a Therapeutic IgG4 Monoclonal Antibody Circulating in Cynomolgus Monkey. MAbs 2016, 8, 961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiśniewski, J.R. Quantitative Evaluation of Filter Aided Sample Preparation (FASP) and Multienzyme Digestion FASP Protocols. Anal. Chem. 2016, 88, 5438–5443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richards, A.L.; Chen, K.H.; Wilburn, D.B.; Stevenson, E.; Polacco, B.J.; Searle, B.C.; Swaney, D.L. Data-Independent Acquisition Protease-Multiplexing Enables Increased Proteome Sequence Coverage Across Multiple Fragmentation Modes. J. Proteome Res. 2022, 21, 1124–1136. [Google Scholar] [CrossRef] [PubMed]
- Morsa, D.; Baiwir, D.; La Rocca, R.; Zimmerman, T.A.; Hanozin, E.; Grifnée, E.; Longuespée, R.; Meuwis, M.A.; Smargiasso, N.; De Pauw, E.; et al. Multi-Enzymatic Limited Digestion: The Next-Generation Sequencing for Proteomics? J. Proteome Res. 2019, 18, 2501–2513. [Google Scholar] [CrossRef]
- Saveliev, S.; Bratz, M.; Zubarev, R.; Szapacs, M.; Budamgunta, H.; Urh, M. Trypsin/Lys-C Protease Mix for Enhanced Protein Mass Spectrometry Analysis. Nat. Methods 2013, 10, i–ii. [Google Scholar] [CrossRef]
- Fossati, A.; Richards, A.L.; Chen, K.H.; Jaganath, D.; Cattamanchi, A.; Ernst, J.D.; Swaney, D.L. Toward Comprehensive Plasma Proteomics by Orthogonal Protease Digestion. J. Proteome Res. 2021, 20, 4031–4040. [Google Scholar] [CrossRef]
- Faktor, J.; Goodlett, D.R.; Dapic, I. Trends in Sample Preparation for Proteome Analysis. In Mass Spectrometry in Life Sciences and Clinical Laboratory; IntechOpen: London, UK, 2021. [Google Scholar] [CrossRef]
- Islam, M.S.; Aryasomayajula, A.; Selvaganapathy, P.R. A Review on Macroscale and Microscale Cell Lysis Methods. Micro Mach. 2017, 8, 83. [Google Scholar] [CrossRef]
- Scheerlinck, E.; Dhaenens, M.; Van Soom, A.; Peelman, L.; De Sutter, P.; Van Steendam, K.; Deforce, D. Minimizing Technical Variation during Sample Preparation Prior to Label-Free Quantitative Mass Spectrometry. Anal. Biochem. 2015, 490, 14–19. [Google Scholar] [CrossRef]
- Linke, D. Detergents: An Overview. Methods Enzymol. 2009, 463, 603–617. [Google Scholar] [CrossRef] [PubMed]
- Anandan, A.; Vrielink, A. Detergents in Membrane Protein Purification and Crystallisation. Adv. Exp. Med. Biol. 2016, 922, 13–28. [Google Scholar] [CrossRef] [PubMed]
- Van Meer, G.; Voelker, D.R.; Feigenson, G.W. Membrane Lipids: Where They Are and How They Behave. Nat. Rev. Mol. Cell Biol. 2008, 9, 112. [Google Scholar] [CrossRef] [PubMed]
- Seddon, A.M.; Curnow, P.; Booth, P.J. Membrane Proteins, Lipids and Detergents: Not Just a Soap Opera. Biochim. Biophys. Acta-Biomembr. 2004, 1666, 105–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, M. Detergents: Triton X-100, Tween-20, and More. Mater. Methods 2013, 3, 163. [Google Scholar] [CrossRef]
- Rabilloud, T. Limits of Proteomics: Protein Solubilisation Issues. eLS 2012, 3. [Google Scholar] [CrossRef]
- Luckey, M. Membrane Structural Biology (with Biochemical and Biophysical Foundations); Cambridge University Press: Cambridge, UK, 2012. [Google Scholar] [CrossRef]
- Le Maire, M.; Champeil, P.; Møller, J.V. Interaction of Membrane Proteins and Lipids with Solubilizing Detergents. Biochim. Biophys. Acta-Biomembr. 2000, 1508, 86–111. [Google Scholar] [CrossRef] [Green Version]
- Kundlacz, T.; Bender, J.; Schmidt, C. Effects of Non-Ionic and Zwitterionic Detergents on Soluble Proteins during Native Mass Spectrometry Experiments. Int. J. Mass Spectrom. 2021, 468, 116652. [Google Scholar] [CrossRef]
- Chamberlain, L.H. Detergents as Tools for the Purification and Classification of Lipid Rafts. FEBS Lett. 2004, 559, 1–5. [Google Scholar] [CrossRef]
- Welling, G.W.; Welling-Wester, S. Integral Membrane Proteins. J. Chromatogr. Libr. 2000, 61, 527–556. [Google Scholar] [CrossRef]
- Dormeyer, W.; Van Hoof, D.; Mummery, C.L.; Krijgsveld, J.; Heck, A.J.R. A Practical Guide for the Identification of Membrane and Plasma Membrane Proteins in Human Embryonic Stem Cells and Human Embryonal Carcinoma Cells. Proteomics 2008, 8, 4036–4053. [Google Scholar] [CrossRef] [PubMed]
- Kopchick, J.J.; List, E.O.; Kohn, D.T.; Keidan, G.M.O.; Qiu, L.; Okada, S. Perspective: Proteomics—See “Spots” Run. Endocrinology 2002, 143, 1990–1994. [Google Scholar] [CrossRef] [PubMed]
- Alfonso-Garrido, J.; Garcia-Calvo, E.; Luque-Garcia, J.L. Sample Preparation Strategies for Improving the Identification of Membrane Proteins by Mass Spectrometry. Anal. Bioanal. Chem. 2015, 407, 4893–4905. [Google Scholar] [CrossRef]
- Feist, P.; Hummon, A.B. Proteomic Challenges: Sample Preparation Techniques for Microgram-Quantity Protein Analysis from Biological Samples. Int. J. Mol. Sci. 2015, 16, 3537–3563. [Google Scholar] [CrossRef] [Green Version]
- Bereman, M.S.; Egertson, J.D.; Maccoss, M.J. Comparison between Procedures Using Sodium Dodecyl Sulfate for Shotgun Proteomic Analyses of Complex Samples. Proteomics 2011, 11, 2931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiśniewski, J.R.; Zougman, A.; Nagaraj, N.; Mann, M. Universal Sample Preparation Method for Proteome Analysis. Nat. Methods 2009, 6, 359–362. [Google Scholar] [CrossRef] [PubMed]
- Botelho, D.; Wall, M.J.; Vieira, D.B.; Fitzsimmons, S.; Liu, F.; Doucette, A. Top-down and Bottom-up Proteomics of Sds-Containing Solutions Following Mass-Based Separation. J. Proteome Res. 2010, 9, 2863–2870. [Google Scholar] [CrossRef]
- Zhou, J.Y.; Dann, G.P.; Shi, T.; Wang, L.; Gao, X.; Su, D.; Nicora, C.D.; Shukla, A.K.; Moore, R.J.; Liu, T.; et al. Simple Sodium Dodecyl Sulfate-Assisted Sample Preparation Method for LC-MS-Based Proteomics Applications. Anal. Chem. 2012, 84, 2862–2867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Y.; Jiang, H.; Yan, Y.; Peng, B.; Chen, J.; Lin, H.; Liu, Z. Shotgun Analysis of Membrane Proteomes by an Improved SDS-Assisted Sample Preparation Method Coupled with Liquid Chromatography–Tandem Mass Spectrometry. J. Chromatogr. B 2012, 911, 6–14. [Google Scholar] [CrossRef]
- Sun, D.; Wang, N.; Li, L. Integrated SDS Removal and Peptide Separation by Strong-Cation Exchange Liquid Chromatography for SDS-Assisted Shotgun Proteome Analysis. J. Proteome Res. 2012, 11, 818–828. [Google Scholar] [CrossRef]
- Erde, J.; Loo, R.R.O.; Loo, J.A. Enhanced FASP (EFASP) to Increase Proteome Coverage and Sample Recovery for Quantitative Proteomic Experiments. J. Proteome Res. 2014, 13, 1885–1895. [Google Scholar] [CrossRef] [PubMed]
- Potriquet, J.; Laohaviroj, M.; Bethony, J.M.; Mulvenna, J. A Modified FASP Protocol for High-Throughput Preparation of Protein Samples for Mass Spectrometry. PLoS ONE 2017, 12, e0175967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiśniewski, J.R.; Zielinska, D.F.; Mann, M. Comparison of Ultrafiltration Units for Proteomic and N-Glycoproteomic Analysis by the Filter-Aided Sample Preparation Method. Anal. Biochem. 2011, 410, 307–309. [Google Scholar] [CrossRef] [PubMed]
- Manza, L.L.; Stamer, S.L.; Ham, A.J.L.; Codreanu, S.G.; Liebler, D.C. Sample Preparation and Digestion for Proteomic Analyses Using Spin Filters. Proteomics 2005, 5, 1742–1745. [Google Scholar] [CrossRef]
- Wiśniewski, J.R.; Mann, M. Consecutive Proteolytic Digestion in an Enzyme Reactor Increases Depth of Proteomic and Phosphoproteomic Analysis. Anal. Chem. 2012, 84, 2631–2637. [Google Scholar] [CrossRef]
- Masuda, T.; Tomita, M.; Ishihama, Y. Phase Transfer Surfactant-Aided Trypsin Digestion for Membrane Proteome Analysis. J. Proteome Res. 2008, 7, 731–740. [Google Scholar] [CrossRef]
- Nel, A.J.M.; Garnett, S.; Blackburn, J.M.; Soares, N.C. Comparative Reevaluation of FASP and Enhanced FASP Methods by LC–MS/MS. J. Proteome Res. 2015, 14, 1637–1642. [Google Scholar] [CrossRef]
- Wang, W.Q.; Jensen, O.N.; Møller, I.M.; Hebelstrup, K.H.; Rogowska-Wrzesinska, A. Evaluation of Sample Preparation Methods for Mass Spectrometry-Based Proteomic Analysis of Barley Leaves. Plant Methods 2018, 14, 72. [Google Scholar] [CrossRef]
- Song, G.; Hsu, P.Y.; Walley, J.W. Assessment and Refinement of Sample Preparation Methods for Deep and Quantitative Plant Proteome Profiling. Proteomics 2018, 18, e1800220. [Google Scholar] [CrossRef]
- Vidović, M.; Franchin, C.; Morina, F.; Veljović-Jovanović, S.; Masi, A.; Arrigoni, G. Efficient Protein Extraction for Shotgun Proteomics from Hydrated and Desiccated Leaves of Resurrection Ramonda Serbica Plants. Anal. Bioanal. Chem. 2020, 412, 8299–8312. [Google Scholar] [CrossRef]
- Min, C.W.; Gupta, R.; Agrawal, G.K.; Rakwal, R.; Kim, S.T. Concepts and Strategies of Soybean Seed Proteomics Using the Shotgun Proteomics Approach. Expert Rev. Proteom. 2019, 16, 795–804. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Q.; Li, X.; Niu, F.; Sun, X.; Hu, Z.; Zhang, H. ITRAQ-Based Quantitative Proteomic Analysis of Wheat Roots in Response to Salt Stress. Proteomics 2017, 17, 1600265. [Google Scholar] [CrossRef] [PubMed]
- Szymanski, J.; Levin, Y.; Savidor, A.; Breitel, D.; Chappell-Maor, L.; Heinig, U.; Töpfer, N.; Aharoni, A. Label-Free Deep Shotgun Proteomics Reveals Protein Dynamics during Tomato Fruit Tissues Development. Plant J. 2017, 90, 396–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, R.; Chen, G.; Huang, J. Shot-Gun Proteome and Transcriptome Mapping of the Jujube Floral Organ and Identification of a Pollen-Specific S-Locus F-Box Gene. PeerJ 2017, 5, e3588. [Google Scholar] [CrossRef] [Green Version]
- Leonova, T.; Ihling, C.; Saoud, M.; Rennert, R.; Wessjohann, L.A.; Frolov, A. Does Filter Aided Sample Preparation (FASP) Provide Sufficient Method Linearity for Quantitative Plant Shotgun Proteomics? ChemRxiv 2021. [Google Scholar] [CrossRef]
- Zougman, A.; Selby, P.J.; Banks, R.E. Suspension Trapping (STrap) Sample Preparation Method for Bottom-up Proteomics Analysis. Proteomics 2014, 14, 1006–1010. [Google Scholar] [CrossRef]
- Ludwig, K.R.; Schroll, M.M.; Hummon, A.B. Comparison of In-Solution, FASP, and S-Trap Based Digestion Methods for Bottom-Up Proteomic Studies. J. Proteome Res. 2018, 17, 2480–2490. [Google Scholar] [CrossRef]
- Hughes, C.S.; Foehr, S.; Garfield, D.A.; Furlong, E.E.; Steinmetz, L.M.; Krijgsveld, J. Ultrasensitive Proteome Analysis Using Paramagnetic Bead Technology. Mol. Syst. Biol. 2014, 10, 1. [Google Scholar] [CrossRef]
- Sielaff, M.; Kuharev, J.; Bohn, T.; Hahlbrock, J.; Bopp, T.; Tenzer, S.; Distler, U. Evaluation of FASP, SP3, and IST Protocols for Proteomic Sample Preparation in the Low Microgram Range. J. Proteome Res. 2017, 16, 4060–4072. [Google Scholar] [CrossRef]
- Moggridge, S.; Sorensen, P.H.; Morin, G.B.; Hughes, C.S. Extending the Compatibility of the SP3 Paramagnetic Bead Processing Approach for Proteomics. J. Proteome Res. 2018, 17, 1730–1740. [Google Scholar] [CrossRef]
- Hughes, C.S.; Moggridge, S.; Müller, T.; Sorensen, P.H.; Morin, G.B.; Krijgsveld, J. Single-Pot, Solid-Phase-Enhanced Sample Preparation for Proteomics Experiments. Nat. Protoc. 2018, 14, 68–85. [Google Scholar] [CrossRef] [PubMed]
- Neset, L.; Takayidza, G.; Berven, F.S.; Hernandez-Valladares, M. Comparing Efficiency of Lysis Buffer Solutions and Sample Preparation Methods for Liquid Chromatography–Mass Spectrometry Analysis of Human Cells and Plasma. Molecules 2022, 27, 3390. [Google Scholar] [CrossRef] [PubMed]
- Virant-Klun, I.; Leicht, S.; Hughes, C.; Krijgsveld, J. Identification of Maturation-Specific Proteins by Single-Cell Proteomics of Human Oocytes. Mol. Cell. Proteom. 2016, 15, 2616–2627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Höhne, M.; Frese, C.K.; Grahammer, F.; Dafinger, C.; Ciarimboli, G.; Butt, L.; Binz, J.; Hackl, M.J.; Rahmatollahi, M.; Kann, M.; et al. Single-Nephron Proteomes Connect Morphology and Function in Proteinuric Kidney Disease. Kidney Int. 2018, 93, 1308–1319. [Google Scholar] [CrossRef] [PubMed]
- Mikulášek, K.; Konečná, H.; Potěšil, D.; Holánková, R.; Havliš, J.; Zdráhal, Z. SP3 Protocol for Proteomic Plant Sample Preparation Prior LC-MS/MS. Front. Plant Sci. 2021, 12, 369. [Google Scholar] [CrossRef]
- Frolov, A.; Blüher, M.; Hoffmann, R. Glycation Sites of Human Plasma Proteins Are Affected to Different Extents by Hyperglycemic Conditions in Type 2 Diabetes Mellitus. Anal. Bioanal. Chem. 2014, 406, 5755–5763. [Google Scholar] [CrossRef]
- Bilova, T.; Greifenhagen, U.; Paudel, G.; Lukasheva, E.; Brauch, D.; Osmolovskaya, N.; Tarakhovskaya, E.; Balcke, G.U.; Tissier, A.; Vogt, T.; et al. Glycation of Plant Proteins under Environmental Stress—Methodological Approaches, Potential Mechanisms and Biological Role. In Abiotic and Biotic Stress in Plants-Recent Advances and Future Perspectives; IntechOpen: London, UK, 2016. [Google Scholar] [CrossRef] [Green Version]
- Soboleva, A.; Modzel, M.; Didio, A.; Płóciennik, H.; Kijewska, M.; Grischina, T.; Karonova, T.; Bilova, T.; Stefanov, V.; Stefanowicz, P.; et al. Quantification of Prospective Type 2 Diabetes Mellitus Biomarkers by Stable Isotope Dilution with Bi-Labeled Standard Glycated Peptides. Anal. Methods 2017, 9, 409–418. [Google Scholar] [CrossRef]
- Soboleva, A.; Mavropulo-Stolyarenko, G.; Karonova, T.; Thieme, D.; Hoehenwarter, W.; Ihling, C.; Stefanov, V.; Grishina, T.; Frolov, A. Multiple Glycation Sites in Blood Plasma Proteins as an Integrated Biomarker of Type 2 Diabetes Mellitus. Int. J. Mol. Sci. 2019, 20, 2329. [Google Scholar] [CrossRef]
- Manufacturer Protocol for PPS Silent® Surfactant. Available online: Chrome-extension://efaidnbmnnnibpcajpcglclefindmkaj/https://proteomicsresource.washington.edu/docs/protocols03/expedeon_PPS_SilentSurfactant.pdf (accessed on 4 May 2022).
- Waanders, L.F.; Chwalek, K.; Monetti, M.; Kumar, C.; Lammert, E.; Mann, M. Quantitative Proteomic Analysis of Single Pancreatic Islets. Proc. Natl. Acad. Sci. USA 2009, 106, 18902–18907. [Google Scholar] [CrossRef] [Green Version]
- Chooneea, D.; Karlsson, R.; Encheva, V.; Arnold, C.; Appleton, H.; Shah, H. Elucidation of the Outer Membrane Proteome of Salmonella Enterica Serovar Typhimurium Utilising a Lipid-Based Protein Immobilization Technique. BMC Microbiol. 2010, 10, 44. [Google Scholar] [CrossRef] [Green Version]
- Ting, Y.S.; Egertson, J.D.; Bollinger, J.G.; Searle, B.C.; Payne, S.H.; Noble, W.S.; MacCoss, M.J. PECAN: Library-Free Peptide Detection for Data-Independent Acquisition Tandem Mass Spectrometry Data. Nat. Methods 2017, 14, 903–908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aryal, U.K.; Krochko, J.E.; Ross, A.R.S. Identification of Phosphoproteins in Arabidopsis Thaliana Leaves Using Polyethylene Glycol Fractionation, Immobilized Metal-Ion Affinity Chromatography, Two-Dimensional Gel Electrophoresis and Mass Spectrometry. J. Proteome Res. 2012, 11, 425–437. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.; Liu, M.; Bachschmid, M.M.; Costello, C.E.; Lin, C. Surfactant-Induced Artifacts during Proteomic Sample Preparation. Anal. Chem. 2015, 87, 5500–5504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manufacturer Protocol for ProteaseMAXTM. Available online: https://www.promega.com/-/media/files/resources/protocols/technical-bulletins/101/proteasemax-surfactant-trypsin-enhancer.pdf (accessed on 4 May 2022).
- Pirmoradian, M.; Budamgunta, H.; Chingin, K.; Zhang, B.; Astorga-Wells, J.; Zubarev, R.A. Rapid and Deep Human Proteome Analysis by Single-Dimension Shotgun Proteomics. Mol. Cell. Proteom. 2013, 12, 3330–3338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, S.; Zweyer, M.; Henry, M.; Meleady, P.; Mundegar, R.R.; Swandulla, D.; Ohlendieck, K. Proteomic Profiling of Liver Tissue from the Mdx-4cv Mouse Model of Duchenne Muscular Dystrophy. Clin. Proteom. 2018, 15, 34. [Google Scholar] [CrossRef]
- Delcourt, V.; Franck, J.; Quanico, J.; Gimeno, J.P.; Wisztorski, M.; Raffo-Romero, A.; Kobeissy, F.; Roucou, X.; Salzet, M.; Fournier, I. Spatially-Resolved Top-down Proteomics Bridged to MALDI MS Imaging Reveals the Molecular Physiome of Brain Regions. Mol. Cell. Proteom. 2018, 17, 357–372. [Google Scholar] [CrossRef] [Green Version]
- Miyauchi, E.; Furuta, T.; Ohtsuki, S.; Tachikawa, M.; Uchida, Y.; Sabit, H.; Obuchi, W.; Baba, T.; Watanabe, M.; Terasaki, T.; et al. Identification of Blood Biomarkers in Glioblastoma by SWATH Mass Spectrometry and Quantitative Targeted Absolute Proteomics. PLoS ONE 2018, 13, e0193799. [Google Scholar] [CrossRef] [Green Version]
- Deblasio, S.L.; Johnson, R.S.; Maccoss, M.J.; Gray, S.M.; Cilia, M. Model System-Guided Protein Interaction Mapping for Virus Isolated from Phloem Tissue. J. Proteome Res. 2016, 15, 4601–4611. [Google Scholar] [CrossRef]
- Robison, F.M.; Heuberger, A.L.; Brick, M.A.; Prenni, J.E. Proteome Characterization of Leaves in Common Bean. Proteomes 2015, 3, 236. [Google Scholar] [CrossRef]
- Li, M.; Powell, M.J.; Razunguzwa, T.T.; O’doherty, G.A. A General Approach to Anionic Acid-Labile Surfactants with Tunable Properties. J. Org. Chem. 2010, 75, 6149–6153. [Google Scholar] [CrossRef]
- Manufacturer Protocol for AALS II. Available online: https://us.vwr.com/store/product/14450751/progentatm-anionic-acid-labile-surfactant-ii-aals-ii-protea (accessed on 30 August 2022).
- Zeller, M.; Brown, E.K.; Bouvier, E.S.P.; König, S. Use of an Acid-Labile Surfactant as an SDS Substitute for Gel Electrophoresis and Proteomic Analysis. J. Biomol. Tech. 2002, 13, 1. [Google Scholar] [PubMed]
- Frolov, A.; Didio, A.; Ihling, C.; Chantzeva, V.; Grishina, T.; Hoehenwarter, W.; Sinz, A.; Smolikova, G.; Bilova, T.; Medvedev, S.; et al. The Effect of Simulated Microgravity on the Brassica Napus Seedling Proteome. Funct. Plant Biol. 2017, 45, 440–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mamontova, T.; Lukasheva, E.; Mavropolo-Stolyarenko, G.; Proksch, C.; Bilova, T.; Kim, A.; Babakov, V.; Grishina, T.; Hoehenwarter, W.; Medvedev, S.; et al. Proteome Map of Pea (Pisum sativum L.) Embryos Containing Different Amounts of Residual Chlorophylls. Int. J. Mol. Sci. 2018, 19, 4066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mamontova, T.; Afonin, A.M.; Ihling, C.; Soboleva, A.; Lukasheva, E.; Sulima, A.S.; Shtark, O.Y.; Akhtemova, G.A.; Povydysh, M.N.; Sinz, A.; et al. Profiling of Seed Proteome in Pea (Pisum sativum L.) Lines Characterized with High and Low Responsivity to Combined Inoculation with Nodule Bacteria and Arbuscular Mycorrhizal Fungi. Molecules 2019, 24, 1603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matamoros, M.A.; Kim, A.; Peñuelas, M.; Ihling, C.; Griesser, E.; Hoffmann, R.; Fedorova, M.; Frolov, A.; Becana, M. Protein Carbonylation and Glycation in Legume Nodules. Plant Physiol. 2018, 177, 1510–1528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osawa, S.; Jo, R.; Xiong, Y.; Reidel, B.; Tserentsoodol, N.; Arshavsky, V.Y.; Iuvone, P.M.; Weiss, E.R. Phosphorylation of G Protein-Coupled Receptor Kinase 1 (GRK1) Is Regulated by Light but Independent of Phototransduction in Rod Photoreceptors. J. Biol. Chem. 2011, 286, 20923–20929. [Google Scholar] [CrossRef] [Green Version]
- Van Itallie, C.M.; Tietgens, A.J.; LoGrande, K.; Aponte, A.; Gucek, M.; Anderson, J.M. Phosphorylation of Claudin-2 on Serine 208 Promotes Membrane Retention and Reduces Trafficking to Lysosomes. J. Cell Sci. 2012, 125, 4902–4912. [Google Scholar] [CrossRef] [Green Version]
- Waters Corporation Manufacturer Protocol for RapiGest. Available online: https://www.waters.com/nextgen/us/en/products/standards-and-reagents/rapigest-sf-surfactant.html (accessed on 30 August 2022).
- Wu, F.; Sun, D.; Wang, N.; Gong, Y.; Li, L. Comparison of Surfactant-Assisted Shotgun Methods Using Acid-Labile Surfactants and Sodium Dodecyl Sulfate for Membrane Proteome Analysis. Anal. Chim. Acta 2011, 698, 36–43. [Google Scholar] [CrossRef]
- Kramer, G.; Woolerton, Y.; Van Straalen, J.P.; Vissers, J.P.C.; Dekker, N.; Langridge, J.I.; Beynon, R.J.; Speijer, D.; Sturk, A.; Aerts, J.M.F.G. Accuracy and Reproducibility in Quantification of Plasma Protein Concentrations by Mass Spectrometry without the Use of Isotopic Standards. PLoS ONE 2015, 10, e0140097. [Google Scholar] [CrossRef]
- Frei, A.P.; Jeon, O.Y.; Kilcher, S.; Moest, H.; Henning, L.M.; Jost, C.; Plückthun, A.; Mercer, J.; Aebersold, R.; Carreira, E.M.; et al. Direct Identification of Ligand-Receptor Interactions on Living Cells and Tissues. Nat. Biotechnol. 2012, 30, 997–1001. [Google Scholar] [CrossRef]
- Schoor, C.; Brocke-Ahmadinejad, N.; Gieselmann, V.; Winter, D. Investigation of Oligodendrocyte Precursor Cell Differentiation by Quantitative Proteomics. Proteomics 2019, 19, 1900057. [Google Scholar] [CrossRef] [PubMed]
- Kaspar-Schoenefeld, S.; Merx, K.; Jozefowicz, A.M.; Hartmann, A.; Seiffert, U.; Weschke, W.; Matros, A.; Mock, H.P. Label-Free Proteome Profiling Reveals Developmental-Dependent Patterns in Young Barley Grains. J. Proteom. 2016, 143, 106–121. [Google Scholar] [CrossRef] [PubMed]
- Maldonado-Valderrama, J.; Wilde, P.; MacIerzanka, A.; MacKie, A. The Role of Bile Salts in Digestion. Adv. Colloid Interface Sci. 2011, 165, 36–46. [Google Scholar] [CrossRef]
- Mukhopadhyay, S.; Maitra, U. Chemistry and Biology of Bile Acids. Curr. Sci. 2004, 87, 1666–1683. [Google Scholar]
- Lin, Y.; Liu, Y.; Li, J.; Zhao, Y.; He, Q.; Han, W.; Chen, P.; Wang, X.; Liang, S. Evaluation and Optimization of Removal of an Acid-Insoluble Surfactant for Shotgun Analysis of Membrane Proteome. Electrophoresis 2010, 31, 2705–2713. [Google Scholar] [CrossRef]
- Serra, A.; Zhu, H.; Gallart-Palau, X.; Park, J.E.; Ho, H.H.; Tam, J.P.; Sze, S.K. Plasma Proteome Coverage Is Increased by Unique Peptide Recovery from Sodium Deoxycholate Precipitate. Anal. Bioanal. Chem. 2016, 408, 1963–1973. [Google Scholar] [CrossRef]
- Greifenhagen, U.; Frolov, A.; Blüher, M.; Hoffmann, R. Plasma Proteins Modified by Advanced Glycation End Products (AGEs) Reveal Site-Specific Susceptibilities to Glycemic Control in Patients with Type 2 Diabetes. J. Biol. Chem. 2016, 291, 9610–9616. [Google Scholar] [CrossRef] [Green Version]
- Spiller, S.; Frolov, A.; Hoffmann, R. Quantification of Specific Glycation Sites in Human Serum Albumin as Prospective Type 2 Diabetes Mellitus Biomarkers. Protein Pept. Lett. 2017, 24, 887–896. [Google Scholar] [CrossRef]
- Lin, Y.; Lin, H.; Liu, Z.; Wang, K.; Yan, Y. Improvement of a Sample Preparation Method Assisted by Sodium Deoxycholate for Mass-Spectrometry-Based Shotgun Membrane Proteomics. J. Sep. Sci. 2014, 37, 3321–3329. [Google Scholar] [CrossRef]
- Chung Lau, B.Y.; Othman, A. Evaluation of Sodium Deoxycholate as Solubilization Buffer for Oil Palm Proteomics Analysis. PLoS ONE 2019, 14, e0221052. [Google Scholar] [CrossRef]
- Waas, M.; Bhattacharya, S.; Chuppa, S.; Wu, X.; Jensen, D.R.; Omasits, U.; Wollscheid, B.; Volkman, B.F.; Noon, K.R.; Gundry, R.L. Combine and Conquer: Surfactants, Solvents, and Chaotropes for Robust Mass Spectrometry Based Analyses of Membrane Proteins. Anal. Chem. 2014, 86, 1551–1559. [Google Scholar] [CrossRef] [PubMed]
- Kachuk, C.; Stephen, K.; Doucette, A. Comparison of Sodium Dodecyl Sulfate Depletion Techniques for Proteome Analysis by Mass Spectrometry. J. Chromatogr. A 2015, 1418, 158–166. [Google Scholar] [CrossRef] [PubMed]
- Porter, C.J. Comparison of Commercial LC MS/MS Compatible Detergents with Sodium Deoxycholate for Shotgun Proteomics. J. Proteins Proteom. 2014, 5, 151–161. [Google Scholar]
- Ishihama, Y. Analytical Platforms for Mass Spectrometry-Based Proteomics. Chromatography 2019, 40, 89–97. [Google Scholar] [CrossRef] [Green Version]
- Baniasad, M.; Reed, A.J.; Lai, S.M.; Zhang, L.; Schulte, K.Q.; Smith, A.R.; LeSassier, D.S.; Weber, K.L.; Hewitt, F.C.; Woerner, A.E.; et al. Optimization of Proteomics Sample Preparation for Forensic Analysis of Skin Samples. J. Proteom. 2021, 249, 104360. [Google Scholar] [CrossRef] [PubMed]
- Schmudlach, A.; Felton, J.; Cipolla, C.; Sun, L.; Kennedy, R.T.; Dovichi, N.J. Sample Preparation Protocol for Bottom-up Proteomic Analysis of the Secretome of the Islets of Langerhans. Analyst 2016, 141, 1700. [Google Scholar] [CrossRef] [Green Version]
- Jez, J.M.; Topp, C.N.; Smythers, A.L.; Hicks, L.M. Mapping the Plant Proteome: Tools for Surveying Coordinating Pathways. Emerg. Top. Life Sci. 2021, 5, 203–220. [Google Scholar] [CrossRef]
- Mbeunkui, F.; Goshe, M.B. Investigation of Solubilization and Digestion Methods for Microsomal Membrane Proteome Analysis Using Data-Independent LC-MSE. Proteomics 2011, 11, 898–911. [Google Scholar] [CrossRef]
- Musunuri, S.; Shevchenko, G.; Bergquist, J. Neuroproteomic Profiling of Human Brain Tissue Using Multidimensional Separation Techniques and Selective Enrichment of Membrane Proteins. Electrophoresis 2012, 33, 3779–3785. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Danko, K.; Lukasheva, E.; Zhukov, V.A.; Zgoda, V.; Frolov, A. Detergent-Assisted Protein Digestion—On the Way to Avoid the Key Bottleneck of Shotgun Bottom-Up Proteomics. Int. J. Mol. Sci. 2022, 23, 13903. https://doi.org/10.3390/ijms232213903
Danko K, Lukasheva E, Zhukov VA, Zgoda V, Frolov A. Detergent-Assisted Protein Digestion—On the Way to Avoid the Key Bottleneck of Shotgun Bottom-Up Proteomics. International Journal of Molecular Sciences. 2022; 23(22):13903. https://doi.org/10.3390/ijms232213903
Chicago/Turabian StyleDanko, Katerina, Elena Lukasheva, Vladimir A. Zhukov, Viktor Zgoda, and Andrej Frolov. 2022. "Detergent-Assisted Protein Digestion—On the Way to Avoid the Key Bottleneck of Shotgun Bottom-Up Proteomics" International Journal of Molecular Sciences 23, no. 22: 13903. https://doi.org/10.3390/ijms232213903
APA StyleDanko, K., Lukasheva, E., Zhukov, V. A., Zgoda, V., & Frolov, A. (2022). Detergent-Assisted Protein Digestion—On the Way to Avoid the Key Bottleneck of Shotgun Bottom-Up Proteomics. International Journal of Molecular Sciences, 23(22), 13903. https://doi.org/10.3390/ijms232213903