
The Interplay between ESIPT and TADF for the 2,2′-Bipyridine-3,3′-diol: A Theoretical Reconsideration


Abstract
:1. Introduction
2. Results and Discussion
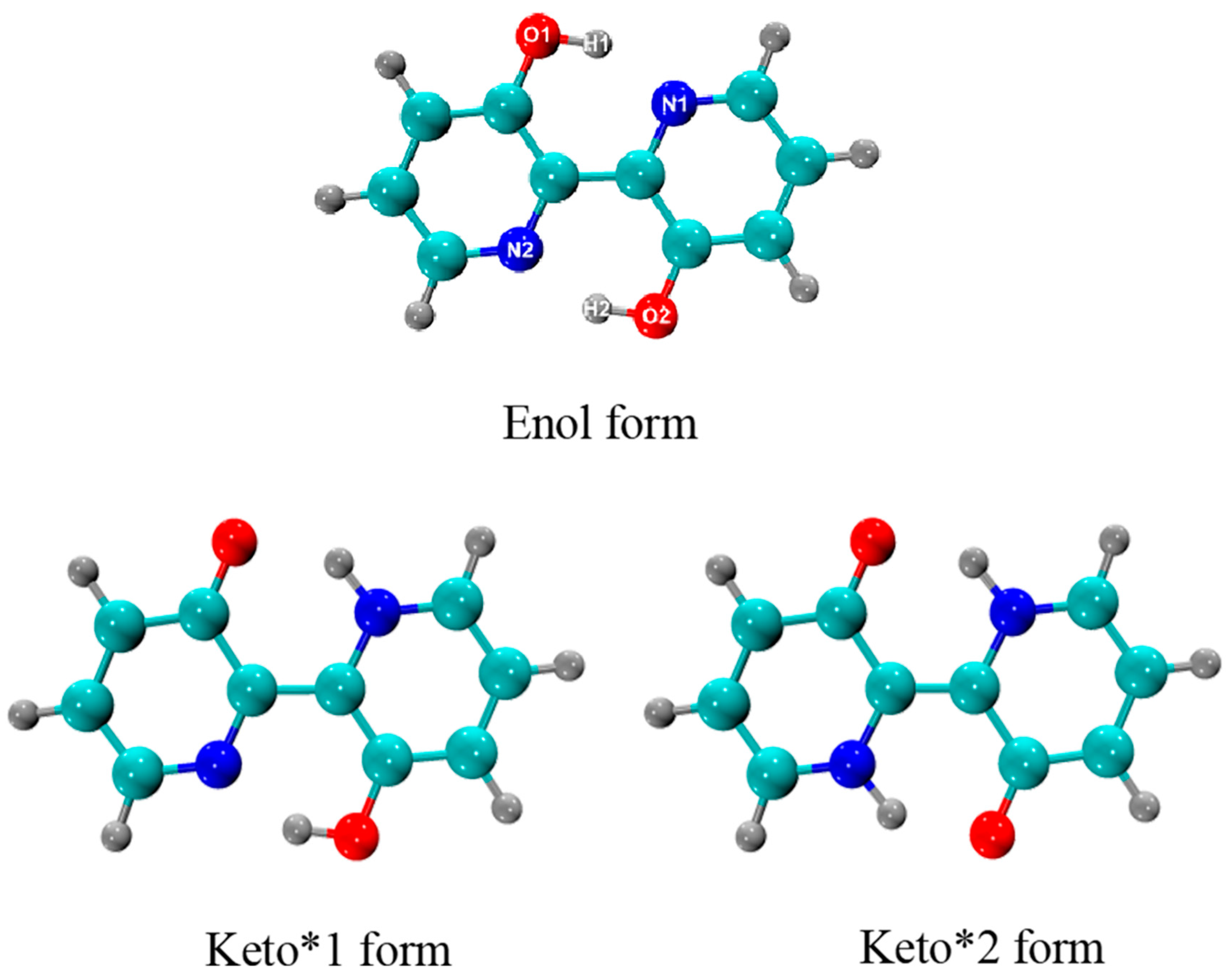
2.1. Structural Analysis and Absorption and Fluorescence Spectra
2.2. Frontier Molecular Orbitals
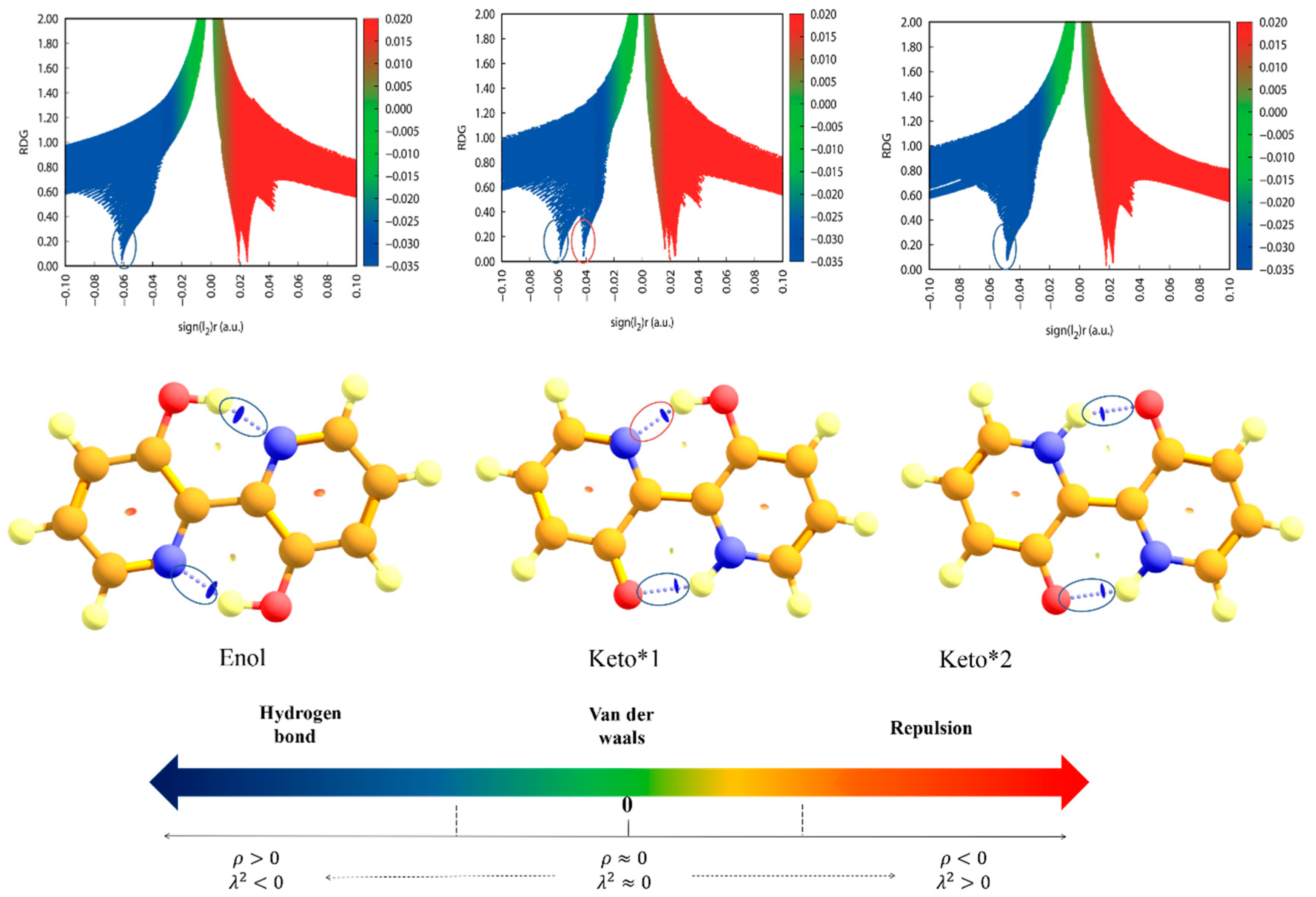
2.3. Reduced Density Gradient Analysis
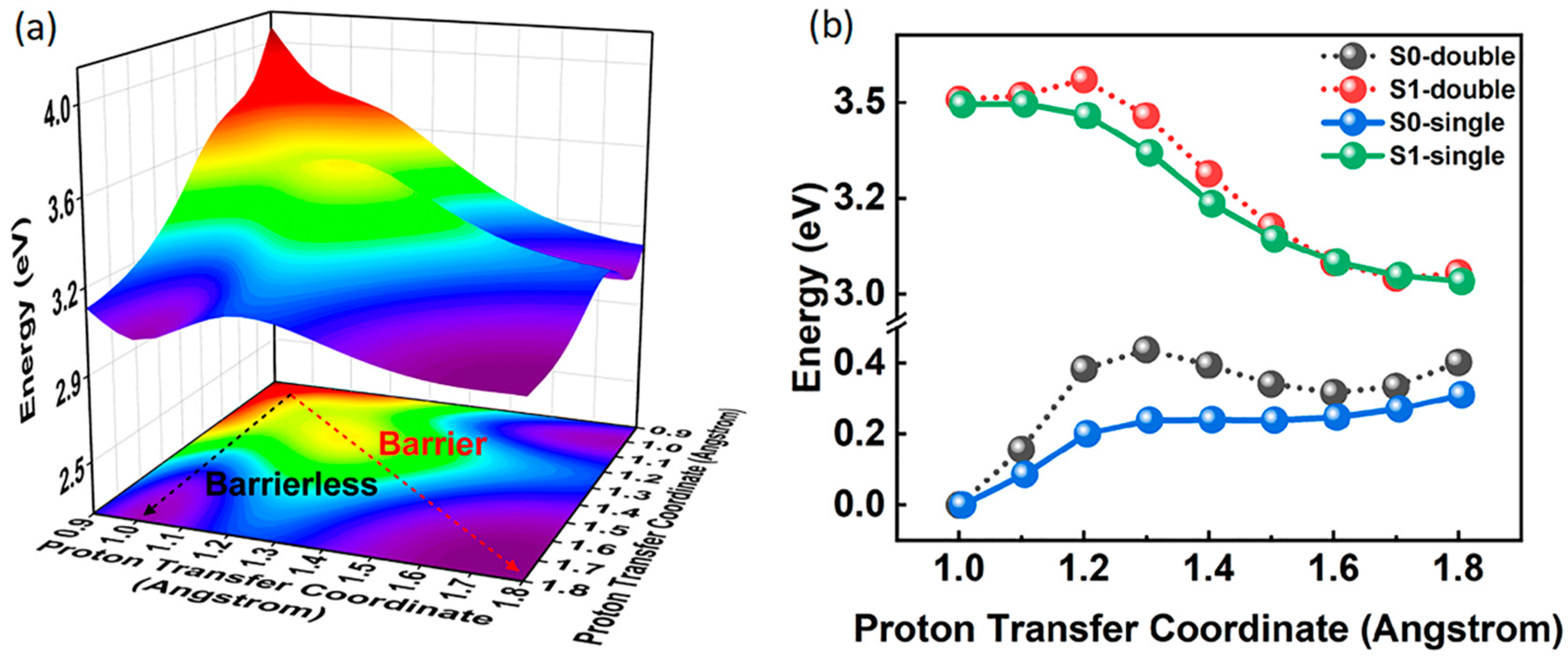
2.4. Potential Energy Surface and Potential Energy Curves
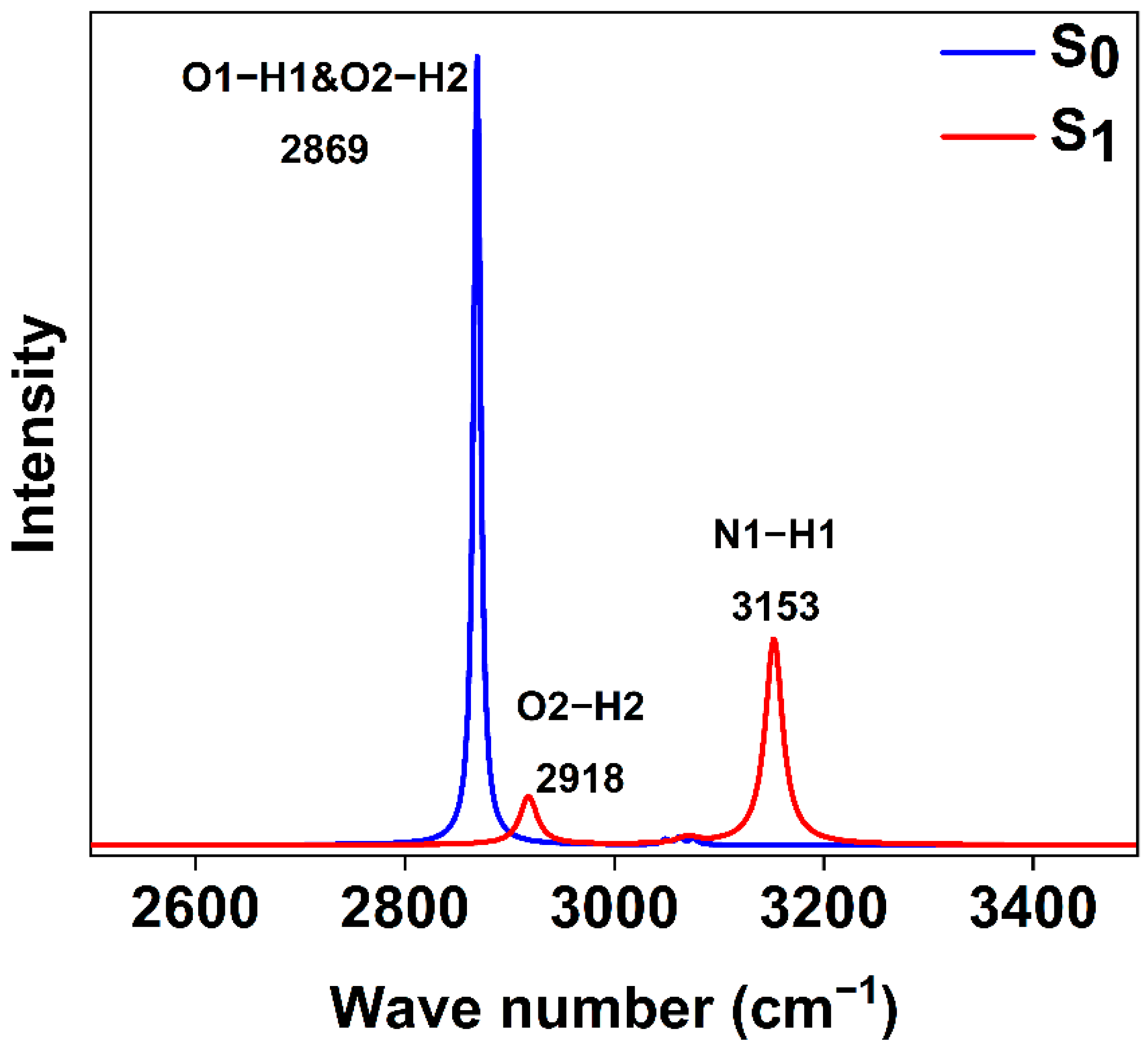
2.5. Infrared Spectra
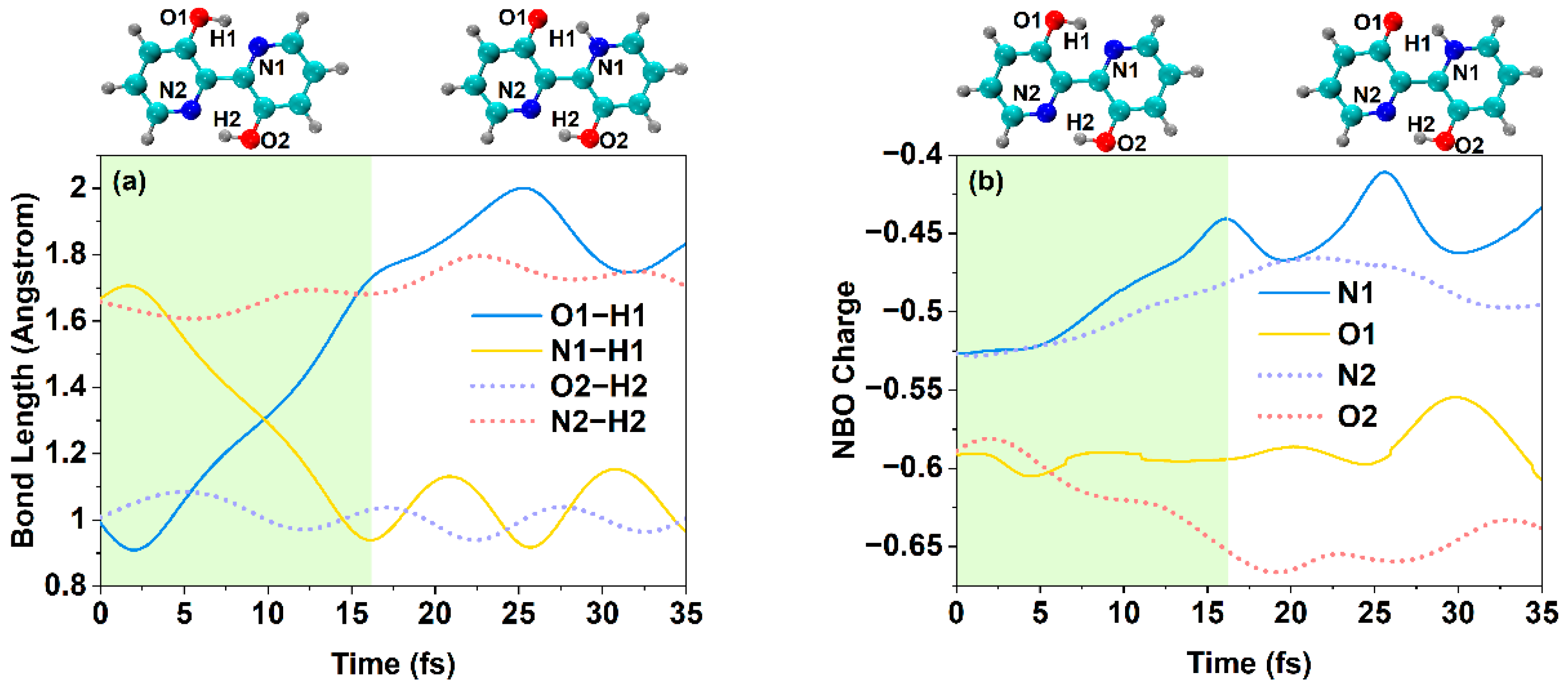
2.6. Excited State Hydrogen Bond Dynamics
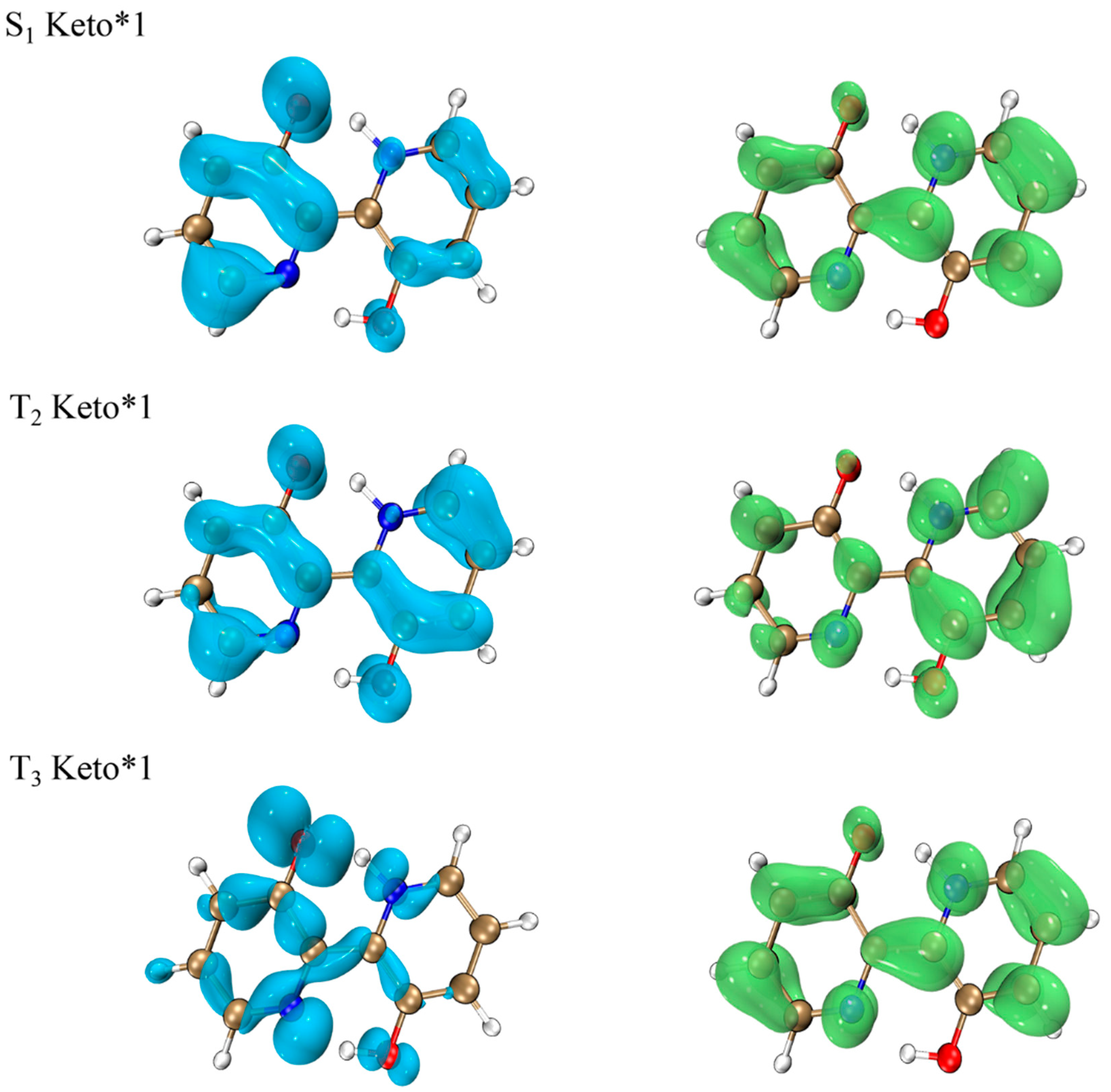
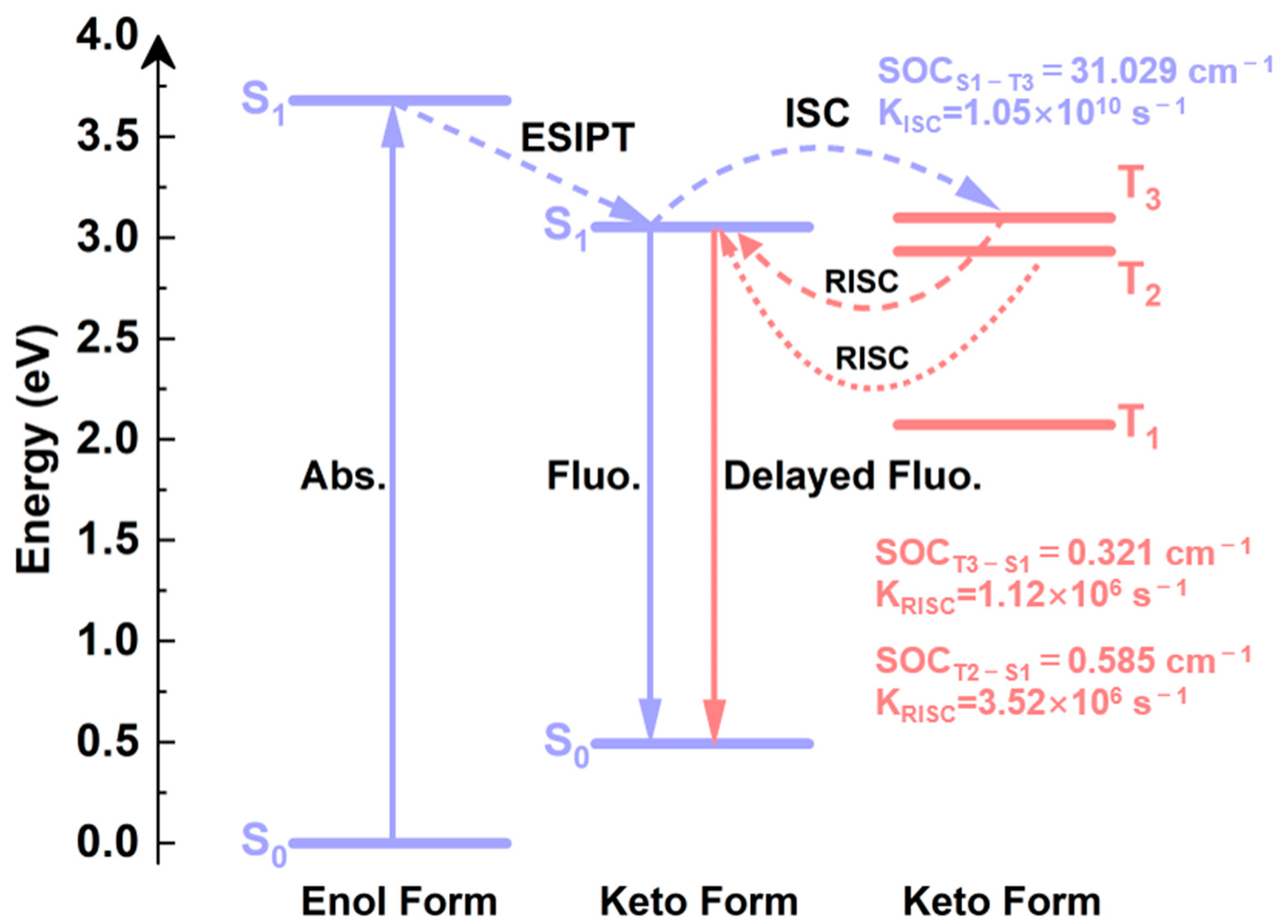
2.7. Spin-Orbit Coupling Interaction
2.8. TADF Mechanism
3. Materials and Methods
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Vazquez, R.J.; Yun, J.H.; Muthike, A.K.; Howell, M.; Kim, H.; Madu, I.K.; Kim, T.; Zimmerman, P.; Lee, J.Y.; Iii, T.G. New Direct Approach for Determining the Reverse Intersystem Crossing Rate in Organic Thermally Activated Delayed Fluorescent (TADF) Emitters. J. Am. Chem. Soc. 2020, 142, 8074–8079. [Google Scholar] [CrossRef]
- Bryden, M.A.; Zysman-Colman, E. Organic thermally activated delayed fluorescence (TADF) compounds used in photocatalysis. Chem. Soc. Rev. 2021, 50, 7587–7680. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.K.; Kim, D.; Bredas, J.L. Thermally Activated Delayed Fluorescence (TADF) Path toward Efficient Electroluminescence in Purely Organic Materials: Molecular Level Insight. Acc. Chem. Res. 2018, 51, 2215–2224. [Google Scholar] [CrossRef] [PubMed]
- Gibson, J.; Penfold, T.J. Nonadiabatic coupling reduces the activation energy in thermally activated delayed fluorescence. Phys. Chem. Chem. Phys. 2017, 19, 8428–8434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheng, R.; Yang, L.; Li, A.; Chen, K.; Zhang, F.; Duan, Y.; Zhao, Y.; Chen, P. Highly efficient orange and white OLEDs based on ultrathin phosphorescent emitters with double reverse intersystem crossing system. J. Lumin. 2022, 246, 118852. [Google Scholar] [CrossRef]
- Liu, H.; Liu, Z.; Li, G.; Huang, H.; Zhou, C.; Wang, Z.; Yang, C. Versatile Direct Cyclization Constructs Spiro-acridan Derivatives for Highly Efficient TADF emitters. Angew. Chem. Int. Ed. 2021, 60, 12376–12380. [Google Scholar] [CrossRef]
- Fan, J.; Zhang, Y.; Zhou, Y.; Lin, L.; Wang, C.-K. Excited State Properties of a Thermally Activated Delayed Fluorescence Molecule in Solid Phase Studied by Quantum Mechanics/Molecular Mechanics Method. J. Phys. Chem. C 2018, 122, 2358–2366. [Google Scholar] [CrossRef]
- Hu, Y.-Y.; Luo, W.; Hu, C.-G.; Wang, Y.; Tong, B.-H.; Fung, M.-K.; Tian, Y.-P.; Zhang, Q.-F. The one-pot synthesis of homoleptic phenylphthalazine iridium(III) complexes and their application in high efficiency OLEDs. J. Lumin. 2020, 219, 116846. [Google Scholar] [CrossRef]
- Fan, J.; Zhang, Y.; Ma, Y.; Song, Y.; Lin, L.; Xu, Y.; Wang, C.-K. The role of intermolecular interactions in regulating the thermally activated delayed fluorescence and charge transfer properties: A theoretical perspective. J. Mater. Chem. C 2020, 8, 8601–8612. [Google Scholar] [CrossRef]
- Lin, L.; Wang, Z.; Fan, J.; Wang, C. Theoretical insights on the electroluminescent mechanism of thermally activated delayed fluorescence emitters. Org. Electron. 2017, 41, 17–25. [Google Scholar] [CrossRef]
- Dos Santos, P.L.; Etherington, M.K.; Monkman, A.P. Chemical and conformational control of the energy gaps involved in the thermally activated delayed fluorescence mechanism. J. Mater. Chem. C 2018, 6, 4842–4853. [Google Scholar] [CrossRef]
- Zhou, D.; Liu, D.; Gong, X.; Ma, H.; Qian, G.; Gong, S.; Xie, G.; Zhu, W.; Wang, Y. Solution-Processed Highly Efficient Bluish-Green Thermally Activated Delayed Fluorescence Emitter Bearing an Asymmetric Oxadiazole-Difluoroboron Double Acceptor. ACS Appl. Mater. Interfaces 2019, 11, 24339–24348. [Google Scholar] [CrossRef] [PubMed]
- Azarias, C.; Budzak, S.; Laurent, A.D.; Ulrich, G.; Jacquemin, D. Tuning ESIPT fluorophores into dual emitters. Chem. Sci. 2016, 7, 3763–3774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Sun, C.; Han, J.; Zhou, Q.; Cao, B.; Yin, H.; Shi, Y. Theoretical investigation of intermolecular hydrogen bond induces fluorescence quenching phenomenon for Coumarin-1. J. Lumin. 2020, 221, 117110. [Google Scholar] [CrossRef]
- Sun, C.; Zhang, X.; Diao, L.; Cao, B.; Yin, H.; Shi, Y. How the atomic electron-accepting ability affect the double ESIPT process of 2,5-bis(benzoxazol-2-yl)thiophene-3,4-diol? J. Lumin. 2020, 225, 117329. [Google Scholar] [CrossRef]
- Yin, H.; Zhang, Y.-M.; Zhao, H.-F.; Yang, G.; Shi, Y.; Zhang, S.X.-A.; Ding, D.-J. Optical anti-counterfeiting of a single molecule by two solvents based on intra- and intermocular excited state proton transfer mechanisms. Dyes Pigments 2018, 159, 506–512. [Google Scholar] [CrossRef]
- Zhao, J.; Dong, H.; Zheng, Y. Elaborating the excited state multiple proton transfer mechanism for 9Hpyrido[3,4-b]indole. J. Lumin. 2018, 195, 228–233. [Google Scholar] [CrossRef]
- Zhu, L.; Zhou, Q.; Cao, B.; Li, B.; Wang, Z.; Zhang, X.; Yin, H.; Shi, Y. Theoretical reconsideration of the mechanism of the excited state proton transfer of indigo carmine in water. J. Mol. Liq. 2022, 347, 118365. [Google Scholar] [CrossRef]
- Yang, Y.; Zhao, J.; Li, Y. Theoretical Study of the ESIPT Process for a New Natural Product Quercetin. Sci. Rep. 2016, 6, 32152. [Google Scholar] [CrossRef] [Green Version]
- Song, P.; Ma, F.-C. Intermolecular hydrogen-bonding effects on photophysics and photochemistry. Int. Rev. Phys. Chem. 2013, 32, 589–609. [Google Scholar] [CrossRef]
- Yin, H.; Shi, Y.; Wang, Y. Time-dependent density functional theory study on the excited-state intramolecular proton transfer in salicylaldehyde. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2014, 129, 280–284. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Chen, Y.; Sun, M. Exploring Nonemissive Excited-State Intramolecular Proton Transfer by Plasmon-Enhanced Hyper-Raman Scattering and Two-Photon Excitation Fluorescence. J. Phys. Chem. C 2021, 126, 487–492. [Google Scholar] [CrossRef]
- Yang, Y.; Zhang, Q.; Liu, Y.; Jiang, Z.; Qin, C.; Jiang, K.; Liu, Y. Ultrafast dynamics of dual fluorescence of 2-(2′-hydroxyphenyl) benzothiazole and its derivatives by femtosecond transient absorption spectroscopy. J. Lumin. 2022, 248, 118922. [Google Scholar] [CrossRef]
- Cao, Y.; Eng, J.; Penfold, T.J. Excited State Intramolecular Proton Transfer Dynamics for Triplet Harvesting in Organic Molecules. J. Phys. Chem. A 2019, 123, 2640–2649. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.; Zhang, T.; Wang, Z.; Wang, L.; Zhan, L.; Gong, S.; Zhong, C.; Lu, Z.H.; Zhang, S.; Yang, C. De Novo Design of Excited-State Intramolecular Proton Transfer Emitters via a Thermally Activated Delayed Fluorescence Channel. J. Am. Chem. Soc. 2018, 140, 8877–8886. [Google Scholar] [CrossRef] [PubMed]
- Bulska, H. Intramolecular cooperative double proton transfer in [2,2′-bipyridyl]-3,3′-diol. Chem. Phys. Lett. 1983, 98, 398–402. [Google Scholar] [CrossRef]
- Kunihiro, T.; Mutsuo, K.; Osamu, O. Reverse intersystem crossing from higher triplet to excited singlet in 2,2’-bipyridine-3,3’-diol phototautomer. J. Photochem. Photobiol. A Chem. 1994, 81, 151–158. [Google Scholar] [CrossRef]
- Plasser, F.; Barbatti, M.; Aquino, A.J.A.; Lischka, H. Excited-state diproton transfer in [2,2′-bipyridyl]-3,3′-diol: The mechanism is sequential, not concerted. J. Phys. Chem. A 2009, 113, 8490–8499. [Google Scholar] [CrossRef]
- Zhao, J.; Liu, X.; Zheng, Y. Controlling Excited State Single versus Double Proton Transfer for 2,2’-Bipyridyl-3,3′-diol: Solvent Effect. J. Phys. Chem. A 2017, 121, 4002–4008. [Google Scholar] [CrossRef]
- Chen, K.; Kurganskii, I.V.; Zhang, X.; Elmali, A.; Zhao, J.; Karatay, A.; Fedin, M.V. Intersystem Crossing and Electron Spin Selectivity in Anthracene-Naphthalimide Compact Electron Donor-Acceptor Dyads Showing Different Geometry and Electronic Coupling Magnitudes. Chem. Eur. J. 2021, 27, 7572–7587. [Google Scholar] [CrossRef]
- Xu, C.; Yu, L.; Zhu, C.; Yu, J.; Cao, Z. Intersystem crossing-branched excited-state intramolecular proton transfer for o-nitrophenol: An ab initio on-the-fly nonadiabatic molecular dynamic simulation. Sci. Rep. 2016, 6, 26768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Marshall, M.; Collins, E.; Marquez, S.; Mu, C.; Bowen, K.H.; Zhang, X. Intramolecular electron-induced proton transfer and its correlation with excited-state intramolecular proton transfer. Nat. Commun. 2019, 10, 1170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Yu, T.; Ubba, E.; Xie, Z.; Yang, Z.; Zhang, Y.; Liu, S.; Xu, J.; Aldred, M.P.; Chi, Z. Achieving Dual-Emissive and Time-Dependent Evolutive Organic Afterglow by Bridging Molecules with Weak Intermolecular Hydrogen Bonding. Adv. Opt. Mater. 2019, 7, 1801593. [Google Scholar] [CrossRef]
- Samant, V.; Singh, A.K.; Ramakrishna, G.; Ghosh, H.N.; Ghanty, T.K.; Palit, D.K. Ultrafast Intermolecular Hydrogen Bond Dynamics in the Excited State of Fluorenone. J. Phys. Chem. A 2005, 109, 8693–8704. [Google Scholar] [CrossRef] [PubMed]
- Dommett, M.; Crespo-Otero, R. Excited state proton transfer in 2′-hydroxychalcone derivatives. Phys. Chem. Chem. Phys. 2017, 19, 2409–2416. [Google Scholar] [CrossRef]
- Lawetz, V.; Orlandi, G.; Siebrand, W. Theory of Intersystem Crossing in Aromatic Hydrocarbons. J. Chem. Phys. 1972, 56, 4058–4072. [Google Scholar] [CrossRef]
- Jiang, G.; Li, F.; Fan, J.; Song, Y.; Wang, C.-K.; Lin, L. Theoretical perspective for luminescent mechanism of thermally activated delayed fluorescence emitter with excited-state intramolecular proton transfer. J. Mater. Chem. C 2020, 8, 98–108. [Google Scholar] [CrossRef]
- Samanta, P.K.; Kim, D.; Coropceanu, V.; Bredas, J.L. Up-Conversion Intersystem Crossing Rates in Organic Emitters for Thermally Activated Delayed Fluorescence: Impact of the Nature of Singlet vs Triplet Excited States. J. Am. Chem. Soc. 2017, 139, 4042–4051. [Google Scholar] [CrossRef]
- Zhang, M.; Ren, B.; Wang, Y.; Zhao, C. A DFT/TDDFT study on the excited-state hydrogen bonding dynamics of 6-aminocoumarin in water solution. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2013, 101, 191–195. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Lanke, S.K.; Sekar, N. Pyrazole based NLOphores: Synthesis, photophysical, DFT, TDDFT studies. Dyes Pigments 2016, 127, 116–127. [Google Scholar] [CrossRef]
- Salomon, O.; Reiher, M.; Hess, B.A. Assertion and validation of the performance of the B3LYP⋆ functional for the first transition metal row and the G2 test set. J. Chem. Phys. 2002, 117, 4729–4737. [Google Scholar] [CrossRef]
- Navarro, L.; Rodriguez, F.; Cirera, J. Controlling the spin-crossover behavior of the [Cr(indenyl)2] family via ligand functionalization. Dalton Trans. 2021, 50, 8704–8710. [Google Scholar] [CrossRef] [PubMed]
- Chrayteh, M.; Huet, T.R.; Drean, P. Microsolvation of myrtenal studied by microwave spectroscopy highlights the role of quasi-hydrogen bonds in the stabilization of its hydrates. J. Chem. Phys. 2020, 153, 104304. [Google Scholar] [CrossRef] [PubMed]
- Mennucci, B.; Cancès, E.; Tomasi, J. Evaluation of Solvent Effects in Isotropic and Anisotropic Dielectrics and in Ionic Solutions with a Unified Integral Equation Method: Theoretical Bases, Computational Implementation, and Numerical Applications. J. Phys. Chem. B 1997, 101, 10506–10517. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision B 01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Nguyen, P.D.; Ding, F.; Fischer, S.A.; Liang, W.; Li, X. Solvated First-Principles Excited-State Charge-Transfer Dynamics with Time-Dependent Polarizable Continuum Model and Solvent Dielectric Relaxation. J. Phys. Chem. Lett. 2012, 3, 2898–2904. [Google Scholar] [CrossRef]
- Yin, H.; Li, B.; Zhao, X.; Liu, Y.; Shi, Y.; Ding, D. Restriction of intramolecular torsion induces abnormal blue-shifted fluorescence in the aggregate state. Dyes Pigments 2022, 201, 110192. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Gao, X.; Bai, S.; Fazzi, D.; Niehaus, T.; Barbatti, M.; Thiel, W. Evaluation of Spin-Orbit Couplings with Linear-Response Time-Dependent Density Functional Methods. J. Chem. Theory Comput. 2017, 13, 515–524. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Wan, Y.; Zhou, Q.; Li, Y.; Li, B.; Zhu, L.; Wan, Y.; Yin, H.; Shi, Y. Exploring the effect of nitrile substituent position on fluorescence quantum yield of ESIPT-based oxazoline derivatives: A TDDFT investigation. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2022, 272, 120953–120959. [Google Scholar] [CrossRef]
- Niu, Y.; Li, W.; Peng, Q.; Geng, H.; Yi, Y.; Wang, L.; Nan, G.; Wang, D.; Shuai, Z. MOlecular MAterials Property Prediction Package (MOMAP) 1.0: A software package for predicting the luminescent properties and mobility of organic functional materials. Mol. Phys. 2018, 116, 1078–1090. [Google Scholar] [CrossRef]
- Peng, Q.; Yi, Y.; Shuai, Z.; Shao, J. Toward Quantitative Prediction of Molecular Fluorescence Quantum Efficiency: Role of Duschinsky Rotation. J. Am. Chem. Soc. 2007, 129, 9333–9339. [Google Scholar] [CrossRef] [PubMed]
- Niu, Y.; Peng, Q.; Shuai, Z. Promoting-mode free formalism for excited state radiationless decay process with Duschinsky rotation effect. Sci. China Chem. 2008, 51, 1153–1158. [Google Scholar] [CrossRef]
- Shuai, Z. Thermal Vibration Correlation Function Formalism for Molecular Excited State Decay Rates. Chin. J. Chem. 2020, 38, 1223–1232. [Google Scholar] [CrossRef]









{kind=link}
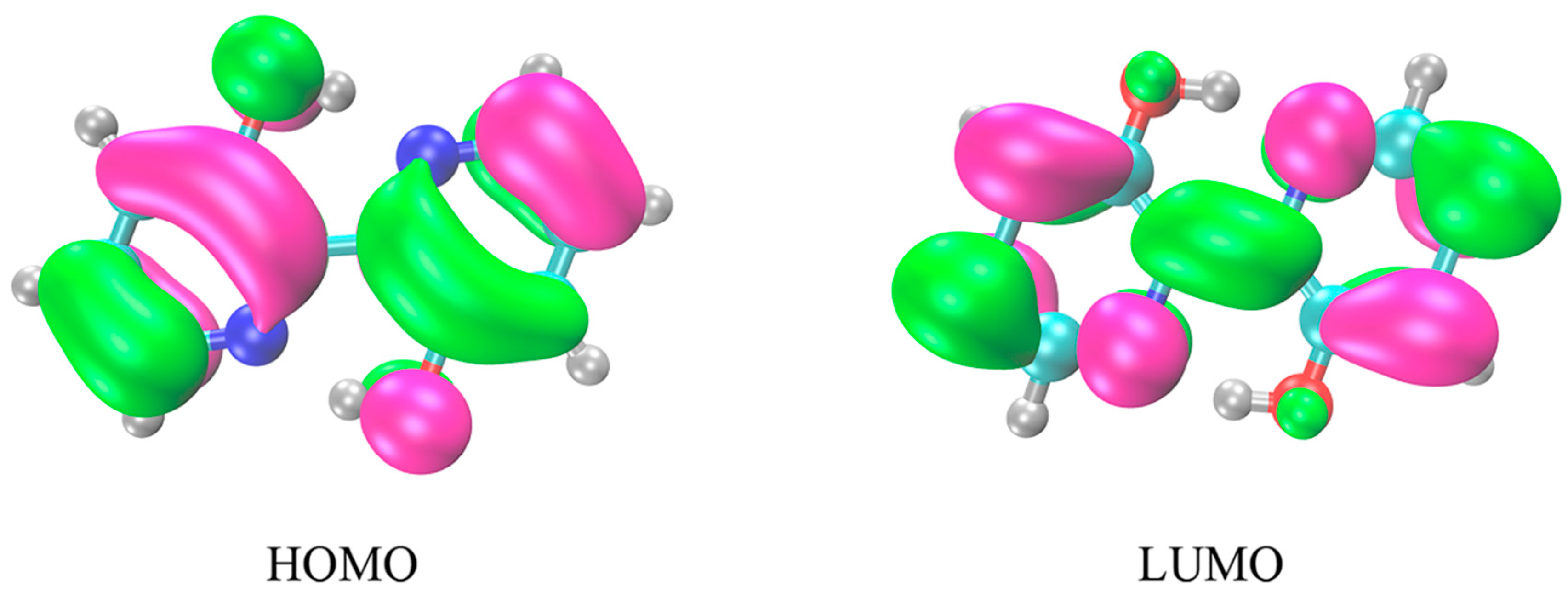{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Unit Å | Enol | Keto*1 | Keto*2 |
|---|---|---|---|
| N1-H1 (Å) | 1.659 | 1.026 | 1.034 |
| O1-H1 (Å) | 1.005 | 1.788 | 1.725 |
| N2-H2 (Å) | 1.659 | 1.686 | 1.034 |
| O2-H2 (Å) | 1.000 | 1.000 | 1.724 |
| Abs. (nm) | Fluo. (nm) | ||
|---|---|---|---|
| Exp. | 345 | 499 | |
| Theor. | 337(Enol) | 484(Keto*1) | 479(Keto*2) |
| S1-T1 | S1-T2 | S1-T3 | |
|---|---|---|---|
| ΔE (eV) | 0.980 | 0.120 | −0.046 |
| SOCISC (cm−1) | 0.152 | 0.411 | 31.029 |
| SOCRISC (cm−1) | 0.095 | 0.585 | 0.321 |
| KISC (s−1) | 1.40 × 106 | 1.73 × 106 | 1.05 × 1010 |
| KRISC (s−1) | 5.48 × 105 | 3.52 × 106 | 1.12 × 106 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, X.; Zhu, L.; Li, Q.; Yin, H.; Shi, Y. The Interplay between ESIPT and TADF for the 2,2′-Bipyridine-3,3′-diol: A Theoretical Reconsideration. Int. J. Mol. Sci. 2022, 23, 13969. https://doi.org/10.3390/ijms232213969
Zhao X, Zhu L, Li Q, Yin H, Shi Y. The Interplay between ESIPT and TADF for the 2,2′-Bipyridine-3,3′-diol: A Theoretical Reconsideration. International Journal of Molecular Sciences. 2022; 23(22):13969. https://doi.org/10.3390/ijms232213969
Chicago/Turabian StyleZhao, Xin, Lixia Zhu, Qi Li, Hang Yin, and Ying Shi. 2022. "The Interplay between ESIPT and TADF for the 2,2′-Bipyridine-3,3′-diol: A Theoretical Reconsideration" International Journal of Molecular Sciences 23, no. 22: 13969. https://doi.org/10.3390/ijms232213969
APA StyleZhao, X., Zhu, L., Li, Q., Yin, H., & Shi, Y. (2022). The Interplay between ESIPT and TADF for the 2,2′-Bipyridine-3,3′-diol: A Theoretical Reconsideration. International Journal of Molecular Sciences, 23(22), 13969. https://doi.org/10.3390/ijms232213969