
The Role of Mitochondrial Enzymes, Succinate-Coupled Signaling Pathways and Mitochondrial Ultrastructure in the Formation of Urgent Adaptation to Acute Hypoxia in the Myocardium

 ,
, 
Abstract
:1. Introduction
2. Results
2.1. Myocardial Mitochondrial Enzymes and Adaptation Factors GPR91, VEGF, HIF-1α under Normoxic Conditions
2.2. Mitochondrial Ultrastructure in Cardiomyocytes under Normoxic Conditions
2.3. Urgent Reaction of Mitochondrial Enzyme Subunits of the Substrate Site of the Myocardial Respiratory Chain (SDHA, NDUFV2) to a Single Hypoxic Exposure In Vivo
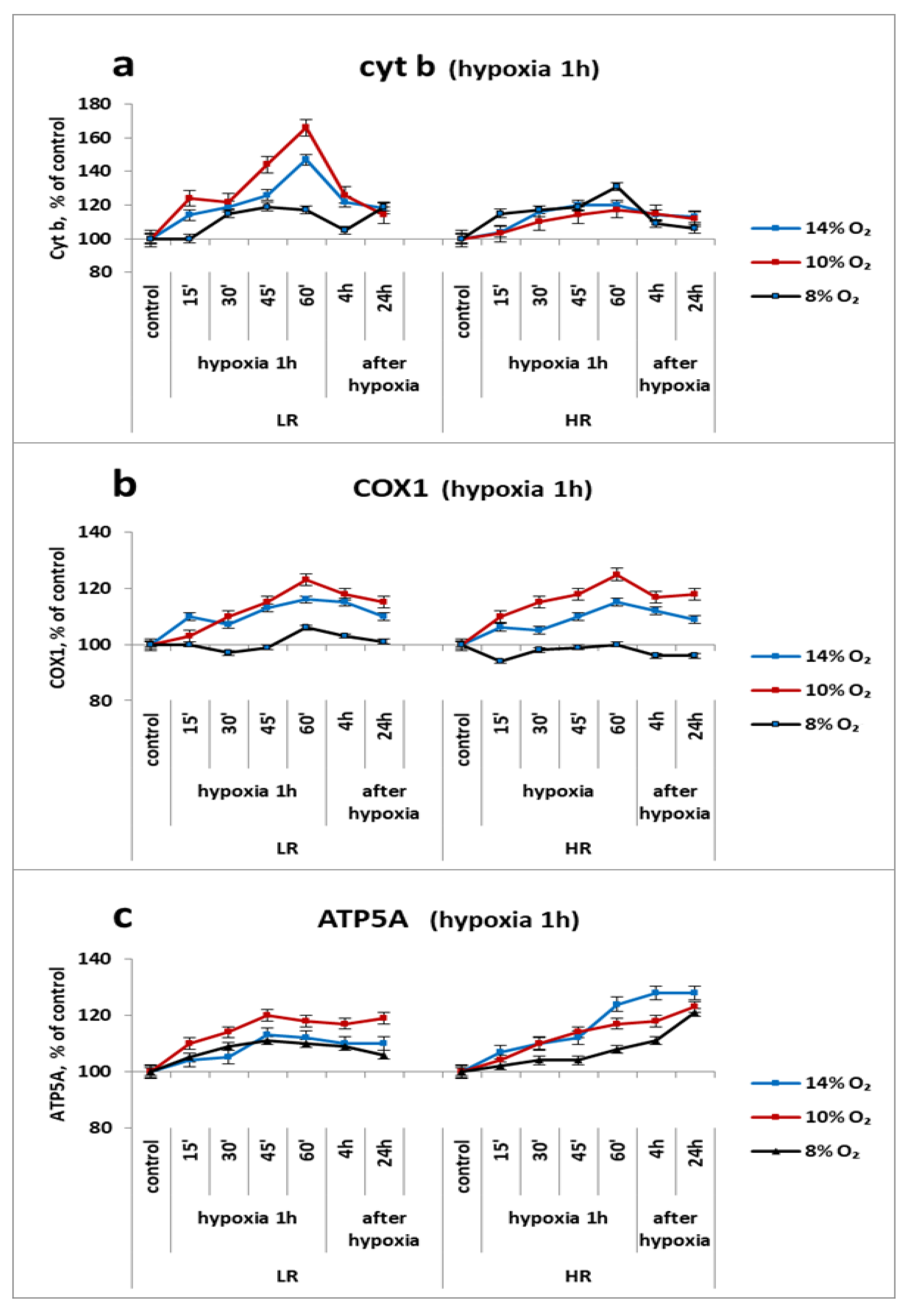
2.4. Urgent Reaction of Mitochondrial Enzymes’ Subunits of the Respiratory Chain Cytochrome Site in the Myocardium (Cyt b, COX2 and ATP5A) to a Single Application of Hypoxia In Vivo
2.5. Urgent Expression of Succinate-Dependent Adaptation Factors (GPR91, VEGF, HIF1-α) in the Myocardium with a Single Hypoxic Exposure of Varying Severity
2.6. Ultrastructure of Three Subpopulations (IFM, SSM, PNM) in the Myocardium of LR and HR Rats after a Single 30-min Different-Severity Hypoxic Exposure
- Effect of hypoxia on the ultrastructure of IFM in the myocardium of HR and LR rats
- b.
- Effect of hypoxia on the ultrastructure of SSM in the myocardium of LR and HR rats
- c.
- Effect of hypoxia on the ultrastructure of PNM in the myocardium of LR and HR rats
3. Discussion
4. Materials and Methods
4.1. Evaluation of Animals’ Resistance of to Hypoxia
4.2. Hypoxia Regimens
4.3. Electron Microscopy of the Heart
4.4. Western Blot Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ADP | adenosine diphosphate |
| AMP | adenosine monophosphate |
| ATP5A | subunit of MC V (ATP synthase F1 subunit alpha) |
| AMPK | AMP-activated protein kinase |
| ATP | adenosine triphosphate |
| Cyt b | subunit of MCIII (cytochrome b) |
| COX2 | subunit of MC IV (cytochrome c oxidase subunit 1) |
| FiO2 | oxygen content in inhaled air (%) |
| GPR91 | Succinate G protein-coupled receptor |
| HBH | acute hypobaric hypoxia |
| HIF-1α | hypoxia-inducible factor 1 alpha subunit |
| HR | high resistant rats |
| IFM | interfibrillar mitochondria |
| LR | low resistant rats |
| MC | mitochondrial complex |
| NDUFV2 | a subunit of complex I (MC I, NADH dehydrogenase [ubiquinone] flavoprotein) |
| OXPHOS | oxidative phosphorylation |
| PNM | perinuclear mitochondria |
| SDHA | subunit of succinate dehydrogenase complex (MC II) |
| SSM | subsarcolemmal mitochondria |
| VEGF | vascular endothelial growth factor |
References
- Vasquez-Trincado, C.; Garcia-Carvajal, I.; Pennanen, C.; Parra, V.; Hill, J.A.; Rothermel, B.A.; Lavandero, S. Mitochondrial dynamics, mitophagy and cardiovascular disease. J. Physiol. 2016, 594, 509–525. [Google Scholar] [CrossRef] [PubMed]
- Ventura-Clapier, R.; Garnier, A.; Veksler, V.; Joubert, F. Bioenergetics of the failing heart. Biochim. Biophys. Acta 2011, 1813, 1360–1372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marin, W.; Marin, D.; Ao, X.; Liu, Y. Mitochondria as a therapeutic target for cardiac ischemia-reperfusion injury (Review). Int. J. Mol. Med. 2021, 47, 485–499. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Fernandez-Sanz, C.; Sheu, S.S. Regulation of mitochondrial bioenergetics by the non-canonical roles of mitochondrial dynamics proteins in the heart. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 1991–2001. [Google Scholar] [CrossRef] [PubMed]
- Guzun, R.; Kaambre, T.; Bagur, R.; Grichine, A.; Usson, Y.; Varikmaa, M.; Anmann, T.; Tepp, K.; Timohhina, N.; Shevchuk, I.; et al. Modular organization of cardiac energy metabolism: Energy conversion, transfer and feedback regulation. Acta Physiol. 2015, 213, 84–106. [Google Scholar] [CrossRef] [Green Version]
- Kuznetsov, A.V.; Javadov, S.; Margreiter, R.; Grimm, M.; Hagenbuchner, J.; Ausserlechner, M.J. The role of mitochondria in the mechanisms of cardiac ischemia-reperfusion injury. Antioxidants 2019, 8, 454. [Google Scholar] [CrossRef] [Green Version]
- Kuznetsov, A.V.; Javadov, S.; Grimm, M.; Margreiter, R.; Ausserlechner, M.J.; Hagenbuchner, J. Crosstalk between mitochondria and cytoskeleton in cardiac cells. Cells 2020, 9, 222. [Google Scholar] [CrossRef] [Green Version]
- Lukyanova, L. Molecular, metabolic and functional mechanisms of individual resistance to hypoxia. In Adaptation Biology and Medicine; Sharma, B.K., Takeda, N., Singal, P.K., Eds.; Narosa Publishing House: New Dehli, India, 1997; Volume 1, pp. 261–272. [Google Scholar]
- Lukyanova, L.D.; Kirova, Y.I. Role of mitochondria-dependent receptor GPR-91 in signaling mechanisms of adaptation to hypoxia. In Adaptation Biology and Medicine; Pawan, K., Ed.; Narosa Publishing House Pvt. Ltd.: New Delhi, India, 2015. [Google Scholar]
- Lukyanova, L.D.; Kirova, Y.I. Mitochondria-controlled signaling mechanisms of brain protection in hypoxia. Front. Neurosci. 2015, 9, 320. [Google Scholar] [CrossRef] [Green Version]
- Lukyanova, L.; Germanova, E.; Khmil, N.; Pavlik, L.; Mikheeva, I.; Shigaeva, M.; Mironova, G. Signaling role of mitochondrial enzymes and ultrastructure in the formation of molecular mechanisms of adaptation to hypoxia. Int. J. Mol. Sci. 2021, 22, 8636. [Google Scholar] [CrossRef]
- Krylova, I.B.; Selina, E.N.; Bulion, V.V.; Rodionova, O.M.; Evdokimova, N.R.; Belosludtseva, N.V.; Shigaeva, M.I.; Mironova, G.D. Uridine treatment prevents myocardial injury in rat models of acute ischemia and ischemia/reperfusion by activating the mitochondrial ATP-dependent potassium channel. Sci. Rep. 2021, 11, 16999. [Google Scholar] [CrossRef]
- Hu, J.; Li, T.; Du, S.; Chen, Y.; Wang, S.; Xiong, F.; Wu, Q. The MAPK signaling pathway mediates the GPR91-dependent release of VEGF from RGC-5 cells. Int. J. Mol. Med. 2015, 36, 130–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashmi, S.; Al-Salam, S. Hypoxia-inducible factor-1 alpha in the heart: A double agent? Cardiol. Rev. 2012, 20, 268–273. [Google Scholar] [CrossRef] [PubMed]
- Choudhry, H.; Harris, A.L. Advances in hypoxia-inducible factor biology. Cell Metab. 2018, 27, 281–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lukyanova, L.D.; Germanova, E.L.; Kopaladze, R.A. Development of resistance of an organism under various conditions of hypoxic preconditioning: Role of the hypoxic period and reoxygenation. Bull. Exp. Biol. Med. 2009, 147, 400–404. [Google Scholar] [CrossRef] [PubMed]
- Lukyanova, L.D. Mitochondria signaling in adaptation to hypoxia. J. Physiol. Pathophysiol. 2014, 5, 363–381. [Google Scholar] [CrossRef]
- Mironova, G.D.; Pavlik, L.L.; Kirova, Y.I.; Belosludtseva, N.V.; Mosentsov, A.A.; Khmil, N.V.; Germanova, E.L.; Lukyanova, L.D. Effect of hypoxia on mitochondrial enzymes and ultrastructure in the brain cortex of rats with different tolerance to oxygen shortage. J. Bioenerg. Biomembr. 2019, 51, 329–340. [Google Scholar] [CrossRef]
- Palmer, J.W.; Tandler, B.; Hoppel, C.L. Biochemical properties of subsarcolemmal and interfibrillar mitochondria isolated from rat cardiac muscle. J. Biol. Chem. 1977, 252, 8731–8739. [Google Scholar] [CrossRef]
- Ong, S.B.; Kalkhoran, S.B.; Hernández-Reséndiz, S.; Samangouei, P.; Ong, S.G.; Hausenloy, D.J. Mitochondrial-shaping proteins in cardiac health and disease—The long and the short of it! Cardiovasc. Drugs Ther. 2017, 31, 87–107. [Google Scholar] [CrossRef] [Green Version]
- Riva, A.; Tandler, B.; Loffredo, F.; Vazquez, E.; Hoppel, C. Structural differences in two biochemically defined populations of cardiac mitochondria. Am. J. Physiol. Heart Circ. Physiol. 2005, 289, H868–H872. [Google Scholar] [CrossRef] [Green Version]
- Santel, A.; Fuller, M.T. Control of mitochondrial morphology by a human mitofusin. J. Cell Sci. 2001, 114, 867–874. [Google Scholar] [CrossRef]
- Huang, X.; Sun, L.; Ji, S.; Zhao, T.; Zhang, W.; Xu, J.; Zhang, J.; Wang, Y.; Wang, X.; Franzini-Armstrong, C.; et al. Kissing and nanotunneling mediate intermitochondrial communication in the heart. Proc. Natl. Acad. Sci. USA 2013, 110, 2846–2851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, H.; Dalgard, C.L.; Mohyeldin, A.; McFate, T.; Tait, A.S.; Verma, A. Reversible inactivation of HIF-1 prolyl hydroxylases allows cell metabolism to control basal HIF-1. J. Biol. Chem. 2005, 280, 41928–41939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, Y.; Mansfield, K.D.; Bertozzi, C.C.; Rudenko, V.; Chan, D.A.; Giaccia, A.J.; Simon, M.C. Multiple factors affecting cellular redox status and energy metabolism modulate hypoxia-inducible factor prolyl hydroxylase activity in vivo and in vitro. Mol. Cell Biol. 2007, 27, 912–925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selak, M.A.; Armour, S.M.; MacKenzie, E.D.; Boulahbel, H.; Watson, D.G.; Mansfield, K.D.; Pan, Y.; Simon, M.C.; Thompson, C.B.; Gottlieb, E. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-α prolyl hydroxylase. Cancer Cell 2005, 7, 77–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semenza, G.L. Hydroxylation of HIF-1: Oxygen sensing at the molecular level. Physiology 2004, 19, 176–182. [Google Scholar] [CrossRef] [Green Version]
- Ryan, D.G.; Murphy, M.P.; Frezza, C.; Prag, H.A.; Chouchani, E.T.; O’Neill, L.A.; Mills, E.L. Coupling Krebs cycle metabolites to signaling in immunity and cancer. Nat. Metab. 2019, 1, 16–33. [Google Scholar] [CrossRef]
- Li, L.; Qu, Y.; Li, J.; Xiong, Y.; Mao, M.; Mu, D. Relationship between HIF-1α expression and neuronal apoptosis in neonatal rats with hypoxia–ischemia brain injury. Brain Res. 2007, 1180, 133–139. [Google Scholar] [CrossRef]
- De Castro Fonseca, M.; Aguiar, C.J.; da Rocha Franco, J.A.; Gingold, R.N.; Leite, M.F. GPR91: Expanding the frontiers of Krebs cycle intermediates. Cell Commun. Signal. 2016, 14, 3. [Google Scholar] [CrossRef] [Green Version]
- Fan, W.; Cooper, N.G. Glutamate-induced NFkappaB activation in the retina. Investig. Ophthalmol. Vis. Sci. 2009, 50, 917–925. [Google Scholar] [CrossRef] [Green Version]
- Lukyanova, L.D.; Kirova, Y.I.; Germanova, E.L. Specific features of immediate expression of succinate-dependent receptor GPR91 in tissues during hypoxia. Bull. Exp. Biol. Med. 2016, 160, 742–747. [Google Scholar] [CrossRef]
- Prag, H.A.; Gruszczyk, A.V.; Huang, M.M.; Beach, T.E.; Young, T.; Tronci, L.; Nikitopoulou, E.; Mulvey, J.F.; Ascione, R.; Hadjihambi, A.; et al. Mechanism of succinate efflux upon reperfusion of the ischaemic heart. Cardiovasc. Res. 2021, 117, 1188–1201. [Google Scholar] [CrossRef]
- Osuna-Prieto, F.J.; Martinez-Tellez, B.; Ortiz-Alvarez, L.; Di, X.; Jurado-Fasoli, L.; Xu, H.; Ceperuelo-Mallafre, V.; Nunez-Roa, C.; Kohler, I.; Segura-Carretero, A.; et al. Elevated plasma succinate levels are linked to higher cardiovascular disease risk factors in young adults. Cardiovasc. Diabetol. 2021, 20, 151. [Google Scholar] [CrossRef]
- Zhang, W.; Chen, C.; Wang, J.; Liu, L.; He, Y.; Chen, Q. Mitophagy in cardiomyocytes and in platelets: A major mechanism of cardioprotection against ischemia/reperfusion injury. Physiology 2018, 33, 86–98. [Google Scholar] [CrossRef]
- He, W.; Miao, F.J.P.; Lin, D.C.H.; Schwandner, R.T.; Wang, Z.; Gao, J.; Chen, J.L.; Tian, H.; Ling, L. Citric acid cycle intermediates as ligands for orphan G-protein-coupled receptors. Nature 2004, 429, 188–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ariza, A.C.; Deen, P.M.; Robben, J.H. The succinate receptor as a novel therapeutic target for oxidative and metabolic stress-related conditions. Front. Endocrinol. 2012, 3, 22. [Google Scholar] [CrossRef] [Green Version]
- Ivanchenko, M.; Tverdokhleb, I.V. The peculiarities of intermitochondrial contacts during ontogenetic formation of mitochondria network in normal and under hypoxic damage of cardiogenesis. I.P. Pavlov. Russ. Med. Biol. Her. 2014, 22, 10–17. (In Russian) [Google Scholar] [CrossRef] [Green Version]
- Kuznetsov, A.V.; Margreiter, R. Heterogeneity of mitochondria and mitochondrial function within cells as another level of mitochondrial complexity. Int. J. Mol. Sci. 2009, 10, 1911–1929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nepomniashchikh, L.M. Regenerative and plastic heart failure: Molecular biological mechanisms and morphological bases. Arkh. Patol. 2007, 69, 3–12. (In Russian) [Google Scholar]
- Nepomnyashchikh, L.M.; Lushnikova, E.L.; Polyakov, L.P.; Molodykh, O.P.; Klinnikova, M.G.; Russkikh, G.S.; Poteryaeva, O.N.; Nepomnyashchikh, R.D.; Pichigin, V.I. Structural changes in the myocardium and serum lipid spectrum in experimental hypercholesterolemia and hypothyroidism. Bull. Exp. Biol. Med. 2013, 155, 692–696. (In Russian) [Google Scholar] [CrossRef]
- Hoppel, C.L.; Tandler, B.; Fujioka, H.; Riva, A. Dynamic organization of mitochondria in human heart and in myocardial disease. Int. J. Biochem. Cell Biol. 2009, 41, 1949–1956. [Google Scholar] [CrossRef] [Green Version]
- Mishra, P.; Chan, D.C. Mitochondrial dynamics and inheritance during cell division, development and disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 634–646. [Google Scholar] [CrossRef] [PubMed]
- Mitra, K.; Wunder, C.; Roysam, B.; Lin, G.; Lippincott-Schwartz, J. A hyperfused mitochondrial state achieved at G1-S regulates cyclin E buildup and entry into S phase. Proc. Natl. Acad. Sci. USA 2009, 106, 11960–11965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tondera, D.; Grandemange, S.; Jourdain, A.; Karbowski, M.; Mattenberger, Y.; Herzig, S.; Da Cruz, S.; Clerc, P.; Raschke, I.; Merkwirth, C.; et al. SLP-2 is required for stress-induced mitochondrial hyperfusion. EMBO J. 2009, 328, 1589–1600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanley, W.C.; Recchia, F.A.; Lopaschuk, G.D. Myocardial substrate metabolism in the normal and failing heart. Physiol. Rev. 2005, 85, 1093–1129. [Google Scholar] [CrossRef] [Green Version]
- Hawkins, B.J.; Levin, M.D.; Doonan, P.J.; Petrenko, N.B.; Davis, C.W.; Patel, V.V.; Madesh, M. Mitochondrial complex II prevents hypoxic but not calcium- and proapoptotic Bcl-2 protein-induced mitochondrial membrane potential loss. J. Biol. Chem. 2010, 285, 494–505. [Google Scholar] [CrossRef] [Green Version]
- Moret, P.R. Myocardial metabolic changes in chronic hypoxia. Cardiology 1971, 56, 161–172. [Google Scholar] [CrossRef]
- Hochachka, P.W.; Mommsen, T.P. Protons and anaerobiosis. Science 1983, 219, 1391–1397. [Google Scholar] [CrossRef]
- Hochachka, P.W. Defense strategies against hypoxia and hypothermia. Science 1986, 231, 234–241. [Google Scholar] [CrossRef]
- Hochachka, P.W.; Buck, L.T.; Doll, C.J.; Land, S.C. Unifying theory of hypoxia tolerance: Molecular/metabolic defense and rescue mechanisms for surviving oxygen lack. Proc. Natl. Acad. Sci. USA 1996, 93, 9493–9498. [Google Scholar] [CrossRef] [Green Version]
- Kondrashova, M.N. The formation and utilization of succinate in mitochondria as a control mechanism of energization and energy state of tissue. In Biological and Biochemical Oscillators; Chance, B., Ed.; Academic Press: New York, NY, USA, 1993; pp. 373–397. [Google Scholar]
- Maklashina, E.; Sher, Y.; Zhou, H.Z.; Gray, M.O.; Karliner, J.S.; Cecchini, G. Effect of anoxia/reperfusion on the reversible active/de-active transition of NADH-ubiquinone oxidoreductase (complex I) in rat heart. Biochim. Biophys. Acta 2002, 1556, 6–12. [Google Scholar] [CrossRef] [Green Version]
- Paddenberg, R.; Ishaq, B.; Goldenberg, A.; Faulhammer, P.; Rose, F.; Weissmann, N.; Braun-Dullaeus, R.C.; Kummer, W. Essential role of complex II of the respiratory chain in hypoxia-induced ROS generation in pulmonary vasculature. Am. J. Physiol. Lung Cell Mol. Physiol. 2003, 284, L710–L719. [Google Scholar] [CrossRef] [PubMed]
- Hems, D.A.; Brosnan, J.T. Effects of ischaemia on content of metabolites in rat liver and kidney in vivo. Biochem. J. 1970, 120, 105–111. [Google Scholar] [CrossRef] [Green Version]
- Komaromy-Hiller, G.; Sundquist, P.D.; Jacobsen, L.J.; Nuttall, K.L. Serum succinate by capillary zone electrophoresis: Marker candidate for hypoxia. Ann. Clin. Lab. Sci. Mar. 1997, 27, 163–168. [Google Scholar]
- Kushnir, M.M.; Komaromy-Hiller, G.; Shushan, B.; Urry, F.M.; Roberts, W.L. Analysis of dicarboxylic acids by tandem mass spectrometry. High-throughput quantitative measurement of methylmalonic acid in serum, plasma, and urine. Clin. Chem. 2001, 47, 1993–2002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pell, V.R.; Chouchani, E.T.; Frezza, C.; Murphy, M.P.; Krieg, T. Succinate metabolism: A new therapeutic target for myocardial reperfusion injury. Cardiovasc. Res. 2016, 111, 134–141. [Google Scholar] [CrossRef] [Green Version]
- Lopaschuk, G.D.; Ussher, J.R.; Folmes, C.D.; Jaswal, J.S.; Stanley, W.C. Myocardial fatty acid metabolism in health and disease. Physiol. Rev. 2010, 90, 207–258. [Google Scholar] [CrossRef]
- Guzy, R.D.; Sharma, B.; Bell, E.; Chandel, N.S.; Schumacker, P.T. Loss of the SdhB, but not the SdhA, subunit of complex II triggers reactive oxygen species-dependent hypoxia-inducible factor activation and tumorigenesis. Mol. Cell Biol. 2008, 28, 718–731. [Google Scholar] [CrossRef] [Green Version]
- Chouchani, E.T.; Pell, V.R.; Gaude, E.; Aksentijević, D.; Sundier, S.Y.; Robb, E.L.; Logan, A.; Nadtochiy, S.M.; Ord, E.N.J.; Smith, A.C.; et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 2014, 515, 431–435. [Google Scholar] [CrossRef] [Green Version]
- Chance, B. Reaction of oxygen with respiratory chain in cells and tissues. J. Gen. Physiol. 1965, 49, 163–188. [Google Scholar] [CrossRef] [Green Version]
- Lisa, F.D.; Menabò, R.; Canton, M.; Petronilli, V. The role of mitochondria in the salvage and the injury of the ischemic myocardium. Biochim. Biophys. Acta 1998, 1366, 69–78. [Google Scholar] [CrossRef] [Green Version]
- Weiss, R.G.; Gerstenblith, G.; Bottomle, P.A. ATP flux through creatine kinase in the normal, stressed, and failing human heart. Proc. Natl. Acad. Sci. USA 2005, 102, 808–813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiss, J.N.; Venkatesh, N. Metabolic regulation of cardiac ATP-sensitive K+ channels. Cardiovasc. Drugs Ther. 1993, 7 (Suppl. 3), 499–505; [Google Scholar] [CrossRef] [PubMed]
- Schwenk, R.W.; Luiken, J.J.; Bonen, A.; Glatz, J.F. Regulation of sarcolemmal glucose and fatty acid transporters in cardiac disease. Cardiovasc. Res. 2008, 79, 249–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taegtmeyer, H.; Razeghi, P.; Young, M.E. Mitochondrial proteins in hypertrophy and atrophy: A transcript analysis in rat heart. Clin. Exp. Pharmacol. Physiol. 2002, 29, 346–350. [Google Scholar] [CrossRef] [PubMed]
- King, A.; Selak, M.A.; Gottlieb, E. Succinate dehydrogenase and fumarate hydratase: Linking mitochondrial dysfunction and cancer. Oncogene 2006, 25, 4675–4682. [Google Scholar] [CrossRef] [Green Version]
- Sadagopan, N.; Li, W.; Roberds, S.L.; Major, T.; Preston, G.M.; Yu, Y.; Tones, M.A. Circulating succinate is elevated in rodent models of hypertension and metabolic disease. Am. J. Hypertens. 2007, 20, 1209–1215. [Google Scholar] [CrossRef]
- Lukyanova, L.D.; Kirova, Y.I.; Germanova, E.L. Peculiarities of immediate response of respiratory chain enzymes in rat cerebral cortex to hypoxia. Bull. Exp. Biol. Med. 2019, 166, 426–431. [Google Scholar] [CrossRef]
- Kumar, A.; Noda, K.; Philips, B.; Velayutham, M.; Stolz, D.B.; Gladwin, M.T.; Shiva, S.; D’Cunha, J. Nitrite attenuates mitochondrial impairment and vascular permeability induced by ischemia-reperfusion injury in the lung. Am. J. Physiol. Lung Cell Mol. Physiol. 2020, 318, 580–591. [Google Scholar] [CrossRef]
- Arrieta, A.; Blackwood, E.A.; Stauffer, W.T.; Glembotski, C.C. Integrating ER and mitochondrial proteostasis in the healthy and diseased heart. Front. Cardiovasc. Med. 2020, 6, 193. [Google Scholar] [CrossRef]
- Feldkamp, T.; Kribben, A.; Weinberg, J.M. Assessment of mitochondrial membrane potential in proximal tubules after hypoxia-reoxygenation. Am. J. Physiol. Renal Physiol. 2005, 288, F1092–F1102. [Google Scholar] [CrossRef] [Green Version]
- Hochachka, P.W. Mechanism and evolution of hypoxia-tolerance in humans. J. Exp. Biol. 1998, 201, 1243–1254. [Google Scholar] [CrossRef] [PubMed]
- Ostadal, B.; Ostadalova, I.; Dhalla, N.S. Development of cardiac sensitivity to oxygen deficiency: Comparative and ontogenetic aspects. Physiol. Rev. 1999, 79, 635–659. [Google Scholar] [CrossRef] [PubMed]
- Kondrashova, M.N.; Doliba, N.M. Polarografiphic observation of substrate-level phosphorylation and its stimulation by acetylcholine. FEBS Lett. 1989, 243, 153–155. [Google Scholar] [CrossRef] [Green Version]
- Zakharchenko, M.V.; Zakharchenko, A.V.; Khunderyakova, N.V.; Tutukina, M.N.; Simonova, M.A.; Vasilieva, A.A.; Romanova, O.I.; Fedotcheva, N.I.; Litvinova, E.G.; Maevsky, E.I.; et al. Burst of succinate dehydrogenase and α-ketoglutarate dehydrogenase activity in concert with the expression of genes coding for respiratory chain proteins underlies short-term beneficial physiological stress in mitochondria. Int. J. Biochem. Cell Biol. 2013, 45, 190–200. [Google Scholar] [CrossRef] [PubMed]
- Osellame, L.D.; Blacker, T.S.; Duchen, M.R. Cellular and molecular mechanisms of mitochondrial function. Best Pract. Res. Clin. Endocrinol. Metab. 2012, 26, 711–723. [Google Scholar] [CrossRef] [Green Version]
- Lynch, G.S.; Schertzer, J.D.; Ryall, J.G. Therapeutic approaches for muscle wasting disorders. Pharmacol. Ther. 2007, 113, 461–487. [Google Scholar] [CrossRef]
- Cairns, S.P.; Borrani, F. β-Adrenergic modulation of skeletal muscle contraction: Key role of excitation-contraction coupling. J. Physiol. 2015, 593, 4713–4727. [Google Scholar] [CrossRef] [Green Version]
- Acin-Perez, R.; Salazar, E.; Kamenetsky, M.; Buck, J.; Levin, L.R.; Manfredi, G. Cyclic AMP produced inside mitochondria regulates oxidative phosphorylation. Cell Metab. 2009, 9, 265–276. [Google Scholar] [CrossRef] [Green Version]
- Azevedo, R.D.; Falcão, K.V.; Assis, C.R.; Martins, R.M.; Araújo, M.C.; Yogui, G.T.; Neves, J.L.; Seabra, G.M.; Maia, M.B.; Amaral, I.P.; et al. Effects of pyriproxyfen on zebrafish brain mitochondria and acetylcholinesterase. Chemosphere 2021, 263, 12802. [Google Scholar] [CrossRef]
- Feliciello, A.; Gottesman, M.E.; Avvedimento, E.V. cAMP-PKA signaling to the mitochondria: Protein scaffolds, mRNA and phosphatases. Cell Signal. 2005, 17, 279–287. [Google Scholar] [CrossRef]
- Lark, D.S.; Reese, L.R.; Ryan, T.E.; Torres, M.J.; Smith, C.D.; Lin, C.T.; Neufer, P.D. Protein kinase A governs oxidative phosphorylation kinetics and oxidant emitting ootential at complex I. Front. Physiol. 2015, 6, 332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wettschureck, N.; Offermanns, S. Mammalian G proteins and their cell type specific functions. Physiol. Rev. 2005, 85, 1159–1204. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Yuan, Y.; Luo, P.; Yang, J.; Zhou, J.; Zhu, C.; Jiang, Q.; Shu, G. Acute succinate administration increases oxidative phosphorylation and skeletal muscle explosive strength via SUCNR1. Front. Vet. Sci. 2021, 8, 808863. [Google Scholar] [CrossRef]
- Tannahill, G.M.; Curtis, A.M.; Adamik, J.; Palsson-McDermott, E.M.; McGettrick, A.F.; Goel, G.; Frezza, C.; Bernard, N.J.; Kelly, B.; Foley, N.H.; et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature 2013, 496, 238–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luk’yanova, L.D.; Dudchenko, A.M. Parameters of adenylate pool as predictors of energy metabolism disturbances in hepatocytes during hypoxia. Bull. Exp. Biol. Med. 2003, 136, 34–37. [Google Scholar] [CrossRef] [PubMed]
- Dyck, J.R.; Lopaschuk, G.D. AMPK alterations in cardiac physiology and pathology: Enemy or ally? J. Physiol. 2006, 574, 95–112. [Google Scholar] [CrossRef]
- Gruzman, A.; Babai, G.; Sasson, S. Adenosine monophosphate-activated protein kinase (AMPK) as a new target for antidiabetic drugs: A review on metabolic, pharmacological and chemical considerations. Rev. Diabet. Stud. 2009, 6, 13–36. [Google Scholar] [CrossRef] [Green Version]
- Viollet, B.; Athea, Y.; Mounier, R.; Guigas, B.; Zarrinpashneh, E.; Horman, S.; Lantier, L.; Hebrard, S.; Devin-Leclerc, J.; Beauloye, C.; et al. AMPK: Lessons from transgenic and knockout animals. Front. Biosci. 2009, 14, 19–44. [Google Scholar] [CrossRef] [Green Version]
- Solaini, G.; Baracca, A.; Lenaz, G.; Sgarbi, G. Hypoxia and mitochondrial oxidative metabolism. Biochim. Biophys. Acta 2010, 1797, 1171–1177. [Google Scholar] [CrossRef] [Green Version]
- Moussa, A.; Li, J. AMPK in myocardial infarction and diabetes: The yin/yang effect. Acta Pharm. Sin. B 2012, 2, 368–378. [Google Scholar] [CrossRef] [Green Version]
- Dzeja, P.P.; Zelenznikar, R.J.; Goldberg, N.D. Adenylate kinase: Kinetic behavior in intact cells indicates it is integral to multiple cellular processes. Mol. Cell Biochem. 1998, 184, 169–182. [Google Scholar] [CrossRef] [PubMed]
- Dzeja, P.P.; Terzic, A. Phosphotransfer networks and cellular energetics. J. Exp. Biol. 2003, 206, 2039–2047. [Google Scholar] [CrossRef] [PubMed]
- Dzeja, P.; Terzic, A. Adenylate kinase and AMP signaling networks: Metabolic monitoring, signal communication and body energy sensing. Int. J. Mol. Sci. 2009, 10, 1729–1772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardie, D.G. The AMP-activated protein kinase pathway–new players upstream and downstream. J. Cell Sci. 2004, 117, 5479–5487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saks, V.; Dzeja, P.; Schlattner, U.; Vendelin, M.; Terzic, A.; Wallimann, T. Cardiac system bioenergetics: Metabolic basis of Frank-Starling law. J. Physiol. 2006, 571, 253–273. [Google Scholar] [CrossRef]
- Brunk, U.T.; Terman, A. The mitochondrial-lysosomal axis theory of aging: Accumulation of damaged mitochondria as a result of imperfect autophagocytosis. Eur. J. Biochem. 2002, 269, 1996–2002. [Google Scholar] [CrossRef]
- Butow, R.A.; Avadhani, N.G. Mitochondrial signaling: The retrograde response. Mol. Cell 2004, 14, 1–15. [Google Scholar] [CrossRef]
- Chandel, N.S.; Schumacker, P.T. Cells depleted of mitochondrial DNA (rho0) yield insight into physiological mechanisms. FEBS Lett. 1999, 454, 173–176. [Google Scholar] [CrossRef]
- Chandel, N.S.; Schumacker, P.T. Cellular oxygen sensing by mitochondria: Old questions, new insight. J. Appl. Physiol. (1985) 2000, 88, 1880–1889. [Google Scholar] [CrossRef] [Green Version]
- Felty, Q.; Roy, D. Mitochondrial signals to nucleus regulate estrogen-induced cell growth. Med. Hypotheses 2005, 64, 133–141. [Google Scholar] [CrossRef]
- Kuznetsov, A.V.; Schneeberger, S.; Seiler, R.; Brandacher, G.; Mark, W.; Steurer, W.; Saks, V.; Usson, Y.; Margreiter, R.; Gnaiger, E. Mitochondrial defects and heterogeneous cytochrome c release after cardiac cold ischemia and reperfusion. Am. J. Physiol. Heart Circ. Physiol. 2004, 286, H1633–H1641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McBride, H.M.; Neuspiel, M.; Wasiak, S. Mitochondria: More than just a powerhouse. Curr. Biol. 2006, 16, R551–R560. [Google Scholar] [CrossRef] [PubMed]
- McDonald, E.S.; Windebank, A.J. Cisplatin-induced apoptosis of DRG neurons involves bax redistribution and cytochrome c release but not fas receptor signaling. Neurobiol. Dis. 2002, 9, 220–233. [Google Scholar] [CrossRef] [Green Version]
- Michiels, C. Physiological and pathological responses to hypoxia. Am. J. Pathol. 2004, 164, 1875–1882. [Google Scholar] [CrossRef] [Green Version]
- Nicholls, D.G.; Budd, S.L. Mitochondria and neuronal survival. Physiol. Rev. 2000, 80, 315–360. [Google Scholar] [CrossRef] [Green Version]
- Nishimura, G.; Proske, R.J.; Doyama, H.; Higuchi, M. Regulation of apoptosis by respiration: Cytochrome c release by respiratory substrates. FEBS Lett. 2001, 505, 399–404. [Google Scholar] [CrossRef] [Green Version]
- Tait, S.W.G.; Green, D.R. Mitochondria and cell signaling. J. Cell Sci. 2012, 125, 807–815. [Google Scholar] [CrossRef] [Green Version]
- Zhu, H.; Bunn, H.F. Oxygen sensing and signaling: Impact on the regulation of physiologically important genes. Respir. Physiol. 1999, 115, 239–247. [Google Scholar] [CrossRef] [Green Version]
- Semenza, G.L.; Jiang, B.H.; Leung, S.W.; Passantino, R.; Concordet, J.P.; Maire, P.; Giallongo, A. Hypoxia response elements in the aldolase A, enolase 1, and lactate dehydrogenase A gene promoters contain essential binding sites for hypoxia-inducible factor 1. J. Biol. Chem. 1996, 20271, 32529–32537. [Google Scholar] [CrossRef] [Green Version]
- Wenger, R.H. Cellular adaptation to hypoxia: O2-sensing protein hydroxylases, hypoxia-inducible transcription factors, and O2-regulated gene expression. FASEB J. 2002, 16, 1151–1162. [Google Scholar] [CrossRef] [Green Version]
- Boengler, K.; Lochnit, G.; Schulz, R. Mitochondria “THE” target of myocardial conditioning. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H1215–H1231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geng, Y.; Hu, Y.; Wang, H.; Shi, S.; Shi, J.; Qiu, Z. Deficiency of interfibrillar mitochondria in post-acute myocardial infarction heart failure. Pak. J. Pharm. Sci. 2017, 30, 1089–1094. [Google Scholar] [PubMed]
- Lombardi, A.; Damon, M.; Vincent, A.; Goglia, F.; Herpin, P. Characterisation of oxidative phosphorylation in skeletal muscle mitochondria subpopulations in pig: A study using top-down elasticity analysis. FEBS Lett. 2000, 475, 84–88. [Google Scholar] [CrossRef] [Green Version]
- Carafoli, E. Mitochondria, Ca2+ transport and the regulation of heart contraction and metabolism. J. Mol. Cell. Cardiol. 1975, 7, 83–87. [Google Scholar] [CrossRef]
- Palmer, J.W.; Tandler, B.; Hoppel, C.L. Heterogeneous response of subsarcolemmal heart mitochondria to calcium. Am. J. Physiol. 1986, 250, H741–H748. [Google Scholar] [CrossRef]
- Kuznetsov, A.V.; Javadov, S.; Guzun, R.; Grimm, M.; Saks, V. Cytoskeleton and regulation of mitochondrial function: The role of beta-tubulin II. Front. Physiol. 2013, 4, 82. [Google Scholar] [CrossRef] [Green Version]
- Woods, D.C. Mitochondrial heterogeneity: Evaluating mitochondrial subpopulation dynamics in stem cells. Stem Cells Int. 2017, 2017, 7068567. [Google Scholar] [CrossRef] [Green Version]
- Al-Mehdi, A.B.; Pastukh, V.M.; Swiger, B.M.; Reed, D.J.; Patel, M.R.; Bardwell, G.C.; Pastukh, V.V.; Alexeyev, M.F.; Gillespie, M.N. Perinuclear mitochondrial clustering creates an oxidant-rich nuclear domain required for hypoxia-induced transcription. Sci. Signal. 2012, 35, ra47. [Google Scholar] [CrossRef] [Green Version]
- Saks, V.A.; Kaambre, T.; Sikk, P.; Eimre, M.; Orlova, E.; Paju, K.; Piirsoo, A.; Appaix, F.; Kay, L.; Regitz-Zagrosek, V.; et al. Intracellular energetic units in red muscle cells. Biochem. J. 2001, 356, 643–657. [Google Scholar] [CrossRef]
- Collins, T.J.; Bootman, M.D. Mitochondria are morphologically heterogeneous within cells. J. Exp. Biol. 2003, 206, 1993–2000. [Google Scholar] [CrossRef] [Green Version]
- Dzeja, P.P.; Bortolon, R.; Perez-Terzic, C.; Holmuhamedov, E.L.; Terzic, A. Energetic communication between mitochondria and nucleus directed by catalyzed phosphotransfer. Proc. Natl. Acad. Sci. USA 2002, 99, 10156–10161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weakley, B.S. A Beginners Handbook in Biological Electron Microscopy; Churchill Livingstone: Edinburg, TX, USA; London, UK, 1972; p. 228. [Google Scholar]
- Chilov, D.; Camenisch, G.; Kvietikova, I.; Ziegler, U.; Gassmann, M.; Wenger, R.H. Induction and nuclear translocation of hypoxia-inducible factor-1 (HIF-1): Heterodimerization with ARNT is not necessary for nuclear accumulation of HIF-1a. J. Cell Sci. 1999, 112, 1203–1212. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Huang, C.J.; Nazarian, R.; Ritchie, T.; de Vellis, J.S.; Noble, E.P. Isolation of nuclear protein from human brains. Biothechniques 1997, 22, 848–850. [Google Scholar] [CrossRef] [PubMed]
- Pugh, C.W.; Ratcliffe, P.J. Regulation of angiogenesis by hypoxia: Role of the HIF system. Nat. Med. 2003, 9, 677–684. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of Mitochondria | LR, Units/10 µm2 | HR, Units/10 µm2 | ||
|---|---|---|---|---|
| Avg. Number of Small mt (perimeter ≤ 0.25 µm) | Avg. Number of Medium and Large mt | Avg. Number of Small mt (Perimeter ≤ 0.25 µm) | Avg. Number of Medium and Large mt | |
| IFM | 1.41 ± 0.97, 12% | 9.89 ± 0.211, 88% | 2.48 ± 1.47, 15% | 13.56 ± 2.84, 85% |
| SSM | 1.92 ± 0.86, 18% | 9.02 ± 0.189, 82% | 1.82 ± 0.69, 16% | 9.84 ± 1.79, 84% |
| PNM | 6.60 ± 1.67, 37% | 11.40 ± 2.85, 63% | 15.92 ± 3.96 *, 55% | 12.88 ± 2.96, 45% |
| FiO2 | Average Area of IFM, µm2 | Average Area of SSM, µm2 | Average Area of PNM, µm2 | |||
|---|---|---|---|---|---|---|
| LR Rats | HR Rats | LR Rats | HR Rats | LR Rats | HR Rats | |
| Control | 0.673 ± 0.430 | 0.685 ± 0.460 | 0.485 ± 0.291 | 0.535 ± 0.260 | 0.453 ± 0.046 | 2.556 ± 0.154 |
| 14% | 0.508 ± 0.360 ** | 0.543 ± 0.394 * | 0.496 ± 0.252 | 0.443 ± 0.354 | 0.319 ± 0.030 | 2.143 ± 0.192 |
| 10% | 0.745 ± 0.586 | 0.844 ± 0.740 ** | 0.564 ± 0.230 | 0.644 ± 0.340 | 0.586 ± 0.084 | 2.705 ± 0.222 |
| 8% | 0.512 ± 0.353 ** | 0.660 ± 0.527 | 0.532 ± 0.327 | 0.460 ± 0.227 | 0.241 ± 0.019 * | 1.827 ± 0.081 * |
| FiO2 | LR, Units/10 µm2 | HR, Units/10 µm2 | ||
|---|---|---|---|---|
| Total Number of IFM | Average Number of Small IFM | Total Number of IFM | Average Number of Small IFM | |
| Control | 11.30 ± 2.85 (n = 27, mt = 305) | 1.41 ± 0.97 | 16.04 ± 4.65 (n = 23, mt = 369) | 2.48 ± 1.47 |
| 14% | 15.56 ± 4.08 * (n = 16, mt = 249) | 4.44 ± 1.63 * | 13.89 ± 3.86 (n = 9, mt = 125) | 3.56 ± 2.60 |
| 10% | 11.67 ± 4.20 (n = 15, mt = 175) | 2.47 ± 1.25 * | 8.96 ± 3.41 * (n = 27, mt = 242) | 1.44 ± 1.42 |
| 8% | 17.00 ± 3.79 * (n = 16, mt = 272) | 3.88 ± 2.19 * | 13.93 ± 5.43 (n = 14, mt = 195) | 3.50 ± 2.14 |
| FiO2 | LR, Units/10 µm2 | HR, Units/10 µm2 | ||
|---|---|---|---|---|
| Total Number of SSM | Average Number of Small SSM | Total Number of SSM | Average Number of Small SSM | |
| Control | 10.94 ± 2.25 (n = 17, mt = 186) | 1.92 ± 0.86 | 11.65 ± 3.14 (n = 23, mt = 199) | 1.84 ± 0.69 |
| 14% | 8.58 ± 2.75 (n = 12, mt = 103) | 1.56 ± 0.90 | 7.41 ± 1.86 (n = 17, mt = 126) | 0.84 ± 0.63 |
| 10% | 6.86 ± 3.86 (n = 14, mt = 96) | 0.54 ± 0.62 | 6.89 ± 2.67 (n = 19, mt = 131) | 1.47 ± 1.25 |
| 8% | 8.78 ± 2.51 (n = 9, mt = 79) | 1.50 ± 0.84 | 10.13 ± 3.26 (n = 8, mt = 81) | 1.88 ± 0.91 |
| FiO2 | LR, Units/10 µm2 | HR, Units/10 µm2 | ||
|---|---|---|---|---|
| Total Number of PNM | Average Number of Small PNM | Total Number of PNM | Average Number of Small PNM | |
| Control | 18.00 ± 3.31 (n = 18, mt = 324) | 6.60 ± 1.67 | 28.80 ± 5.82 $ (n = 15, mt = 432) | 15.92 ± 3.96 $ |
| 14% | 16.10 + 2.80 (n = 20, mt = 322) | 7.24 ± 2.56 | 23.46 ± 3.67 (n = 13, MX = 305) | 13.66 ± 2.57 $ |
| 10% | 18.79 + 3.04 (n = 14, mt = 263) | 12.16 ± 3.85 | 21.50 ± 5.79 (n = 16, MX = 344) | 11.84 ± 4.18 |
| 8% | 41.87 ± 6.00 * (n = 15, mt = 628) | 20.50 ± 5.72 * | 37.80 ± 3.08 * (n = 15, MX = 567) | 9.86 ± 2.13 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Germanova, E.; Khmil, N.; Pavlik, L.; Mikheeva, I.; Mironova, G.; Lukyanova, L. The Role of Mitochondrial Enzymes, Succinate-Coupled Signaling Pathways and Mitochondrial Ultrastructure in the Formation of Urgent Adaptation to Acute Hypoxia in the Myocardium. Int. J. Mol. Sci. 2022, 23, 14248. https://doi.org/10.3390/ijms232214248
Germanova E, Khmil N, Pavlik L, Mikheeva I, Mironova G, Lukyanova L. The Role of Mitochondrial Enzymes, Succinate-Coupled Signaling Pathways and Mitochondrial Ultrastructure in the Formation of Urgent Adaptation to Acute Hypoxia in the Myocardium. International Journal of Molecular Sciences. 2022; 23(22):14248. https://doi.org/10.3390/ijms232214248
Chicago/Turabian StyleGermanova, Elita, Natalya Khmil, Lyubov Pavlik, Irina Mikheeva, Galina Mironova, and Ludmila Lukyanova. 2022. "The Role of Mitochondrial Enzymes, Succinate-Coupled Signaling Pathways and Mitochondrial Ultrastructure in the Formation of Urgent Adaptation to Acute Hypoxia in the Myocardium" International Journal of Molecular Sciences 23, no. 22: 14248. https://doi.org/10.3390/ijms232214248
APA StyleGermanova, E., Khmil, N., Pavlik, L., Mikheeva, I., Mironova, G., & Lukyanova, L. (2022). The Role of Mitochondrial Enzymes, Succinate-Coupled Signaling Pathways and Mitochondrial Ultrastructure in the Formation of Urgent Adaptation to Acute Hypoxia in the Myocardium. International Journal of Molecular Sciences, 23(22), 14248. https://doi.org/10.3390/ijms232214248