
Transforming Growth Factorβ1 Overexpression Is Associated with Insulin Resistance and Rapidly Progressive Kidney Fibrosis under Diabetic Conditions



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
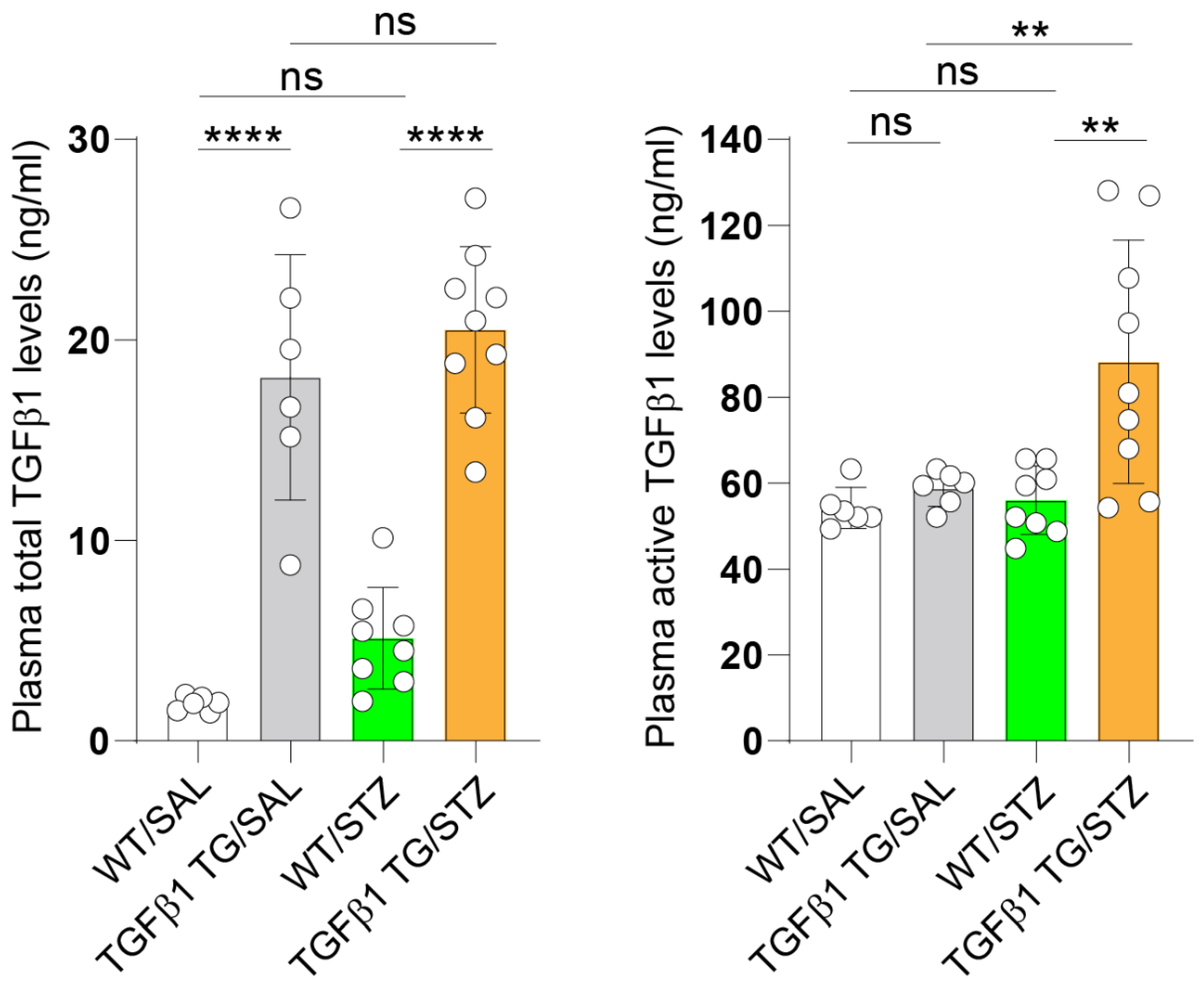
2.1. Increased Circulating Levels of Active TGFβ1 in Diabetic TGFβ1 TG Mice
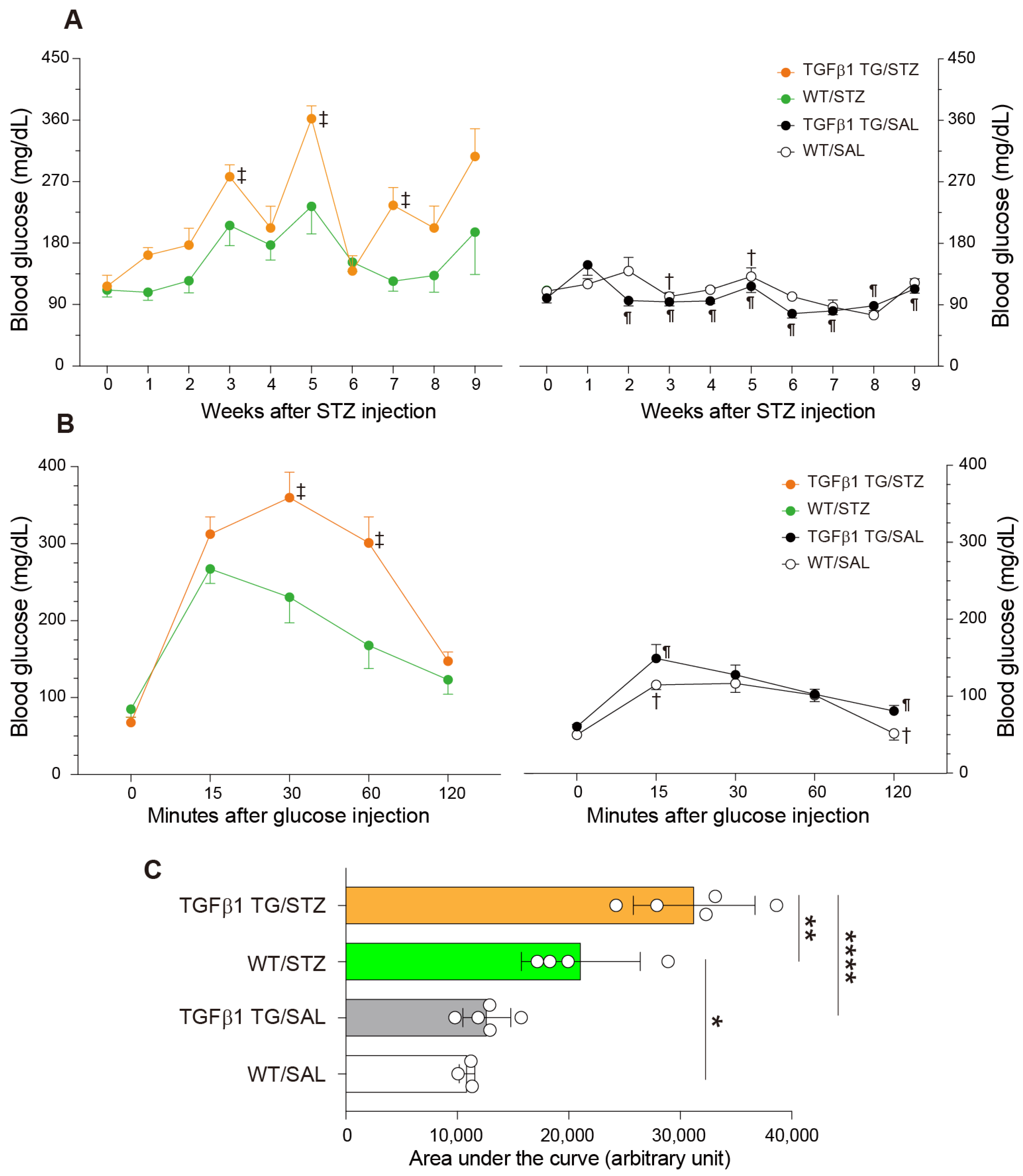
2.2. Increased Blood Glucose Levels and Abnormal Intraperitoneal Glucose Tolerance Test in Diabetic TGFβ1 TG Mice
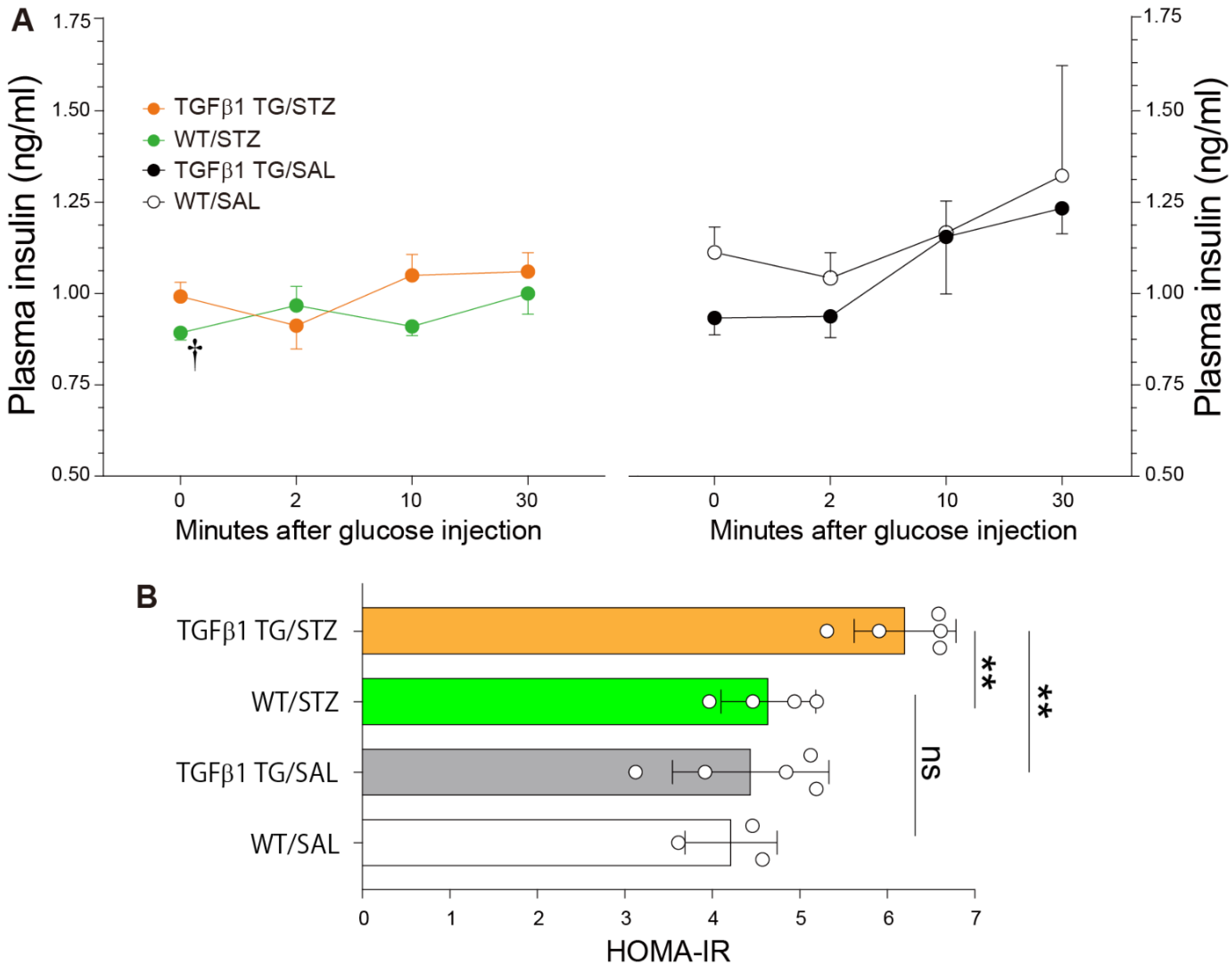
2.3. Reduced Plasma Insulin Levels after Glucose Injection and Insulin Resistance in Diabetic TGFβ1 TG Mice
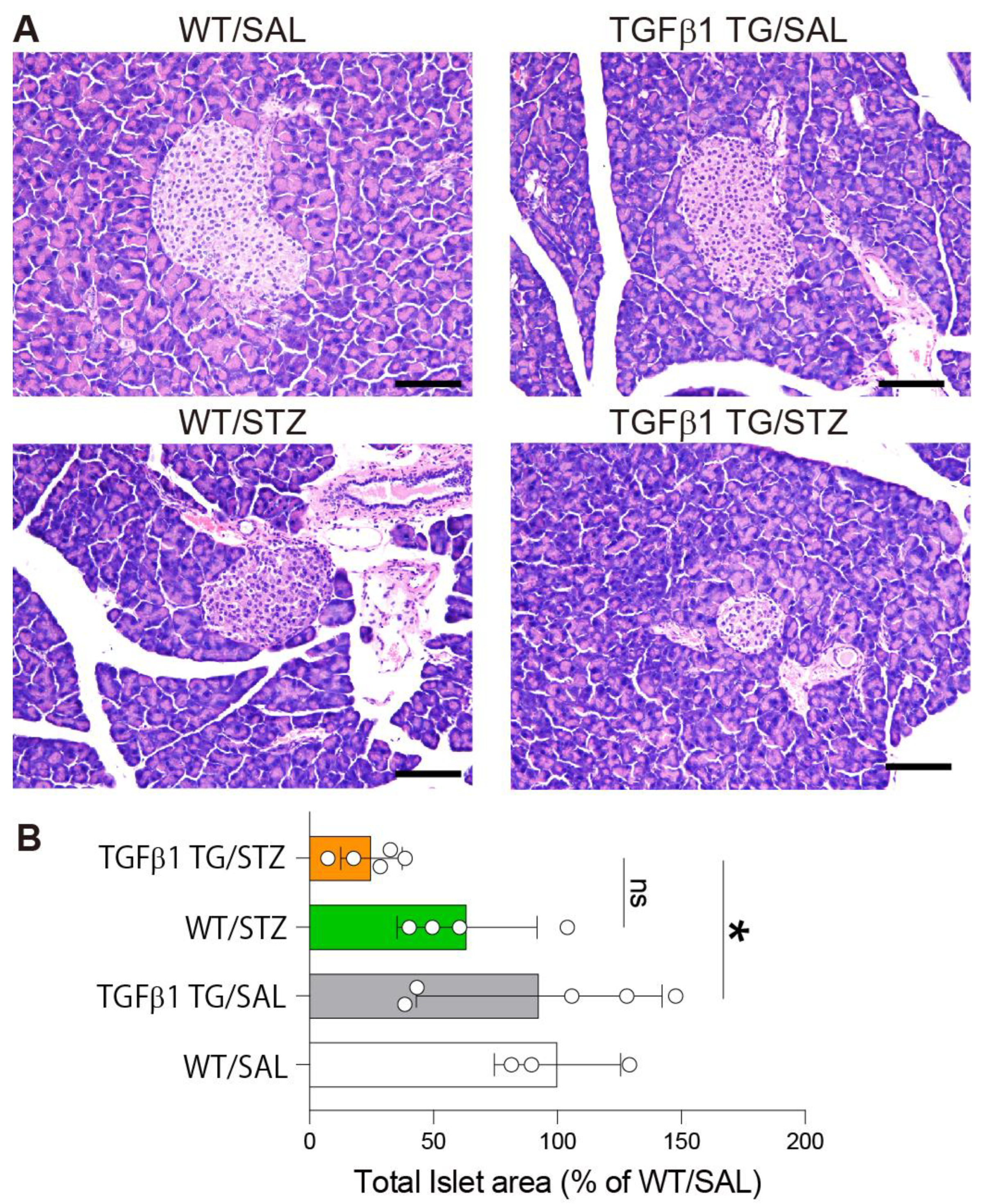
2.4. Reduced Area of Pancreatic Islets in Diabetic TGFβ1 TG Mice
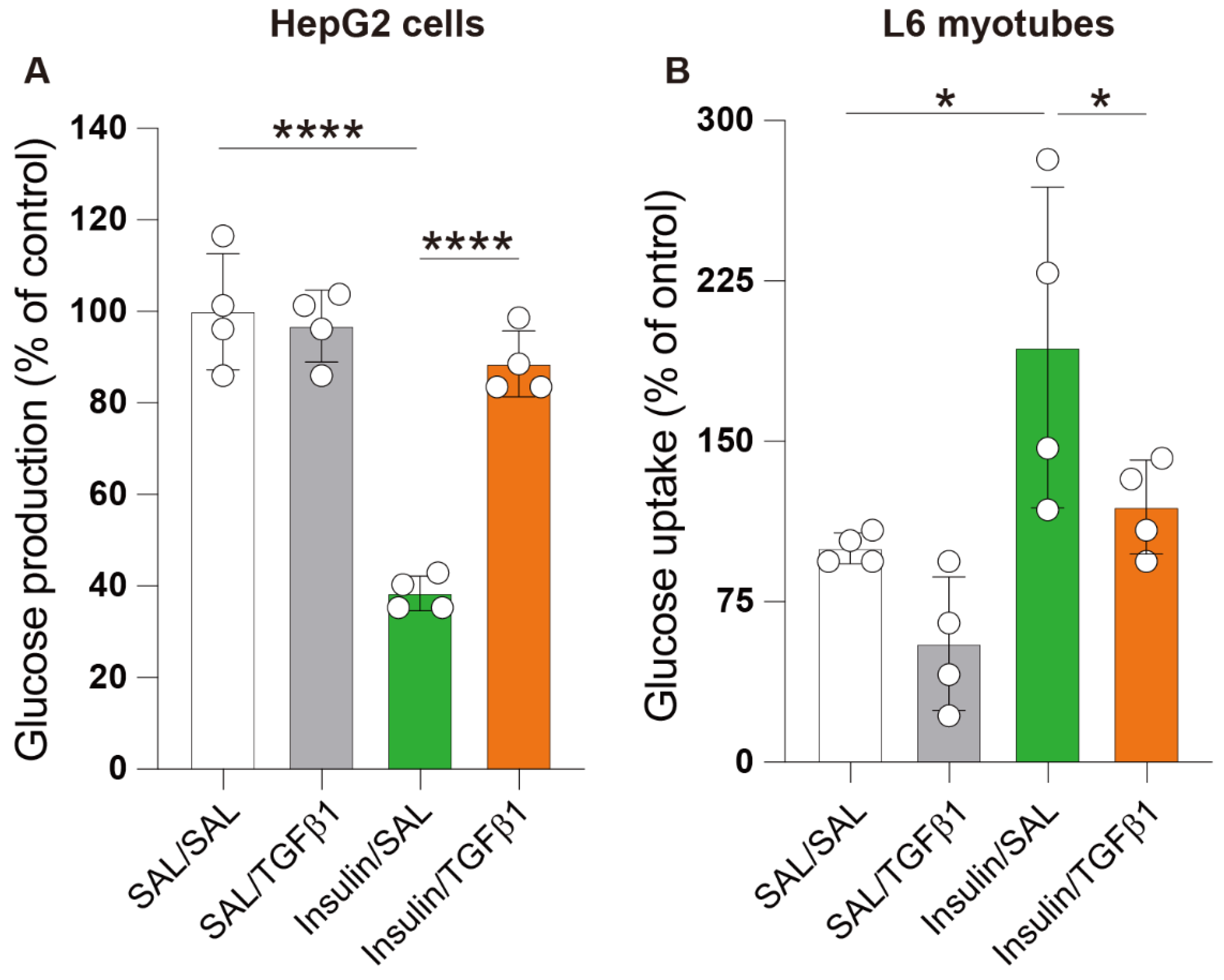
2.5. TGFβ1 Impaired Insulin Action in the Hepatocyte and Skeletal Muscle Cell Lines
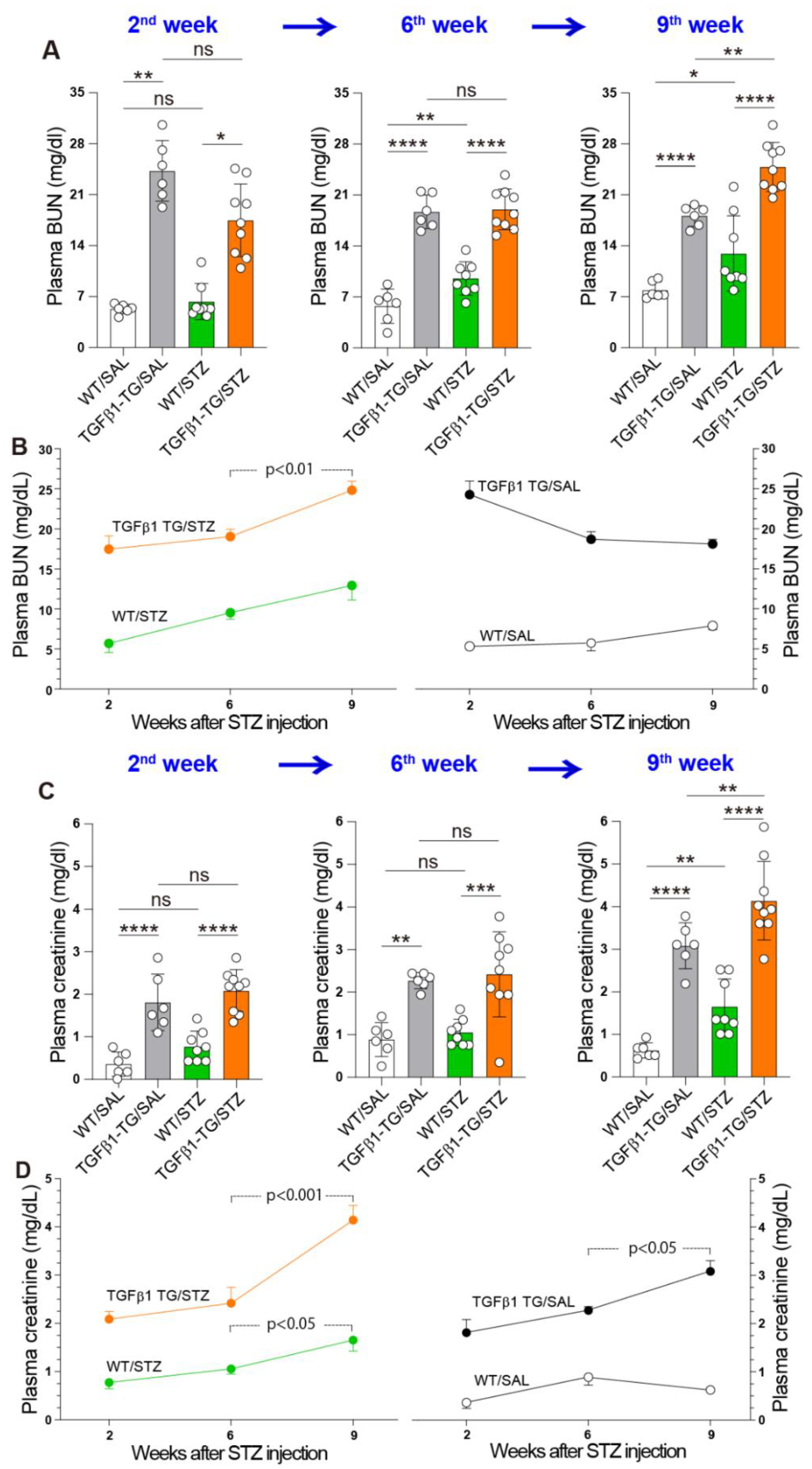
2.6. Progressive Renal Dysfunction in TGFβ1 TG Mice with DM
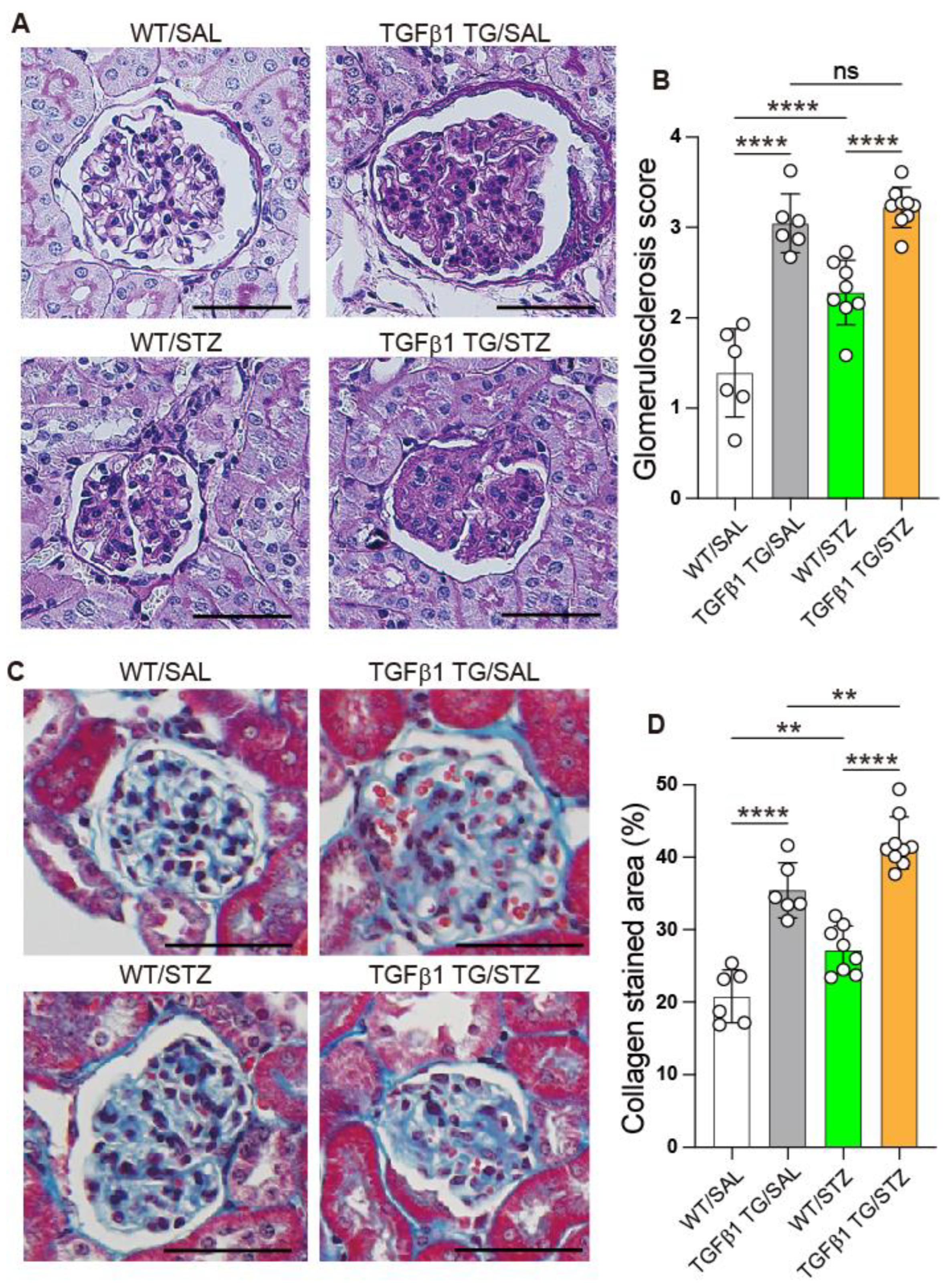
2.7. Progressive Glomerulosclerosis in Diabetic TGFβ1 TG Mice
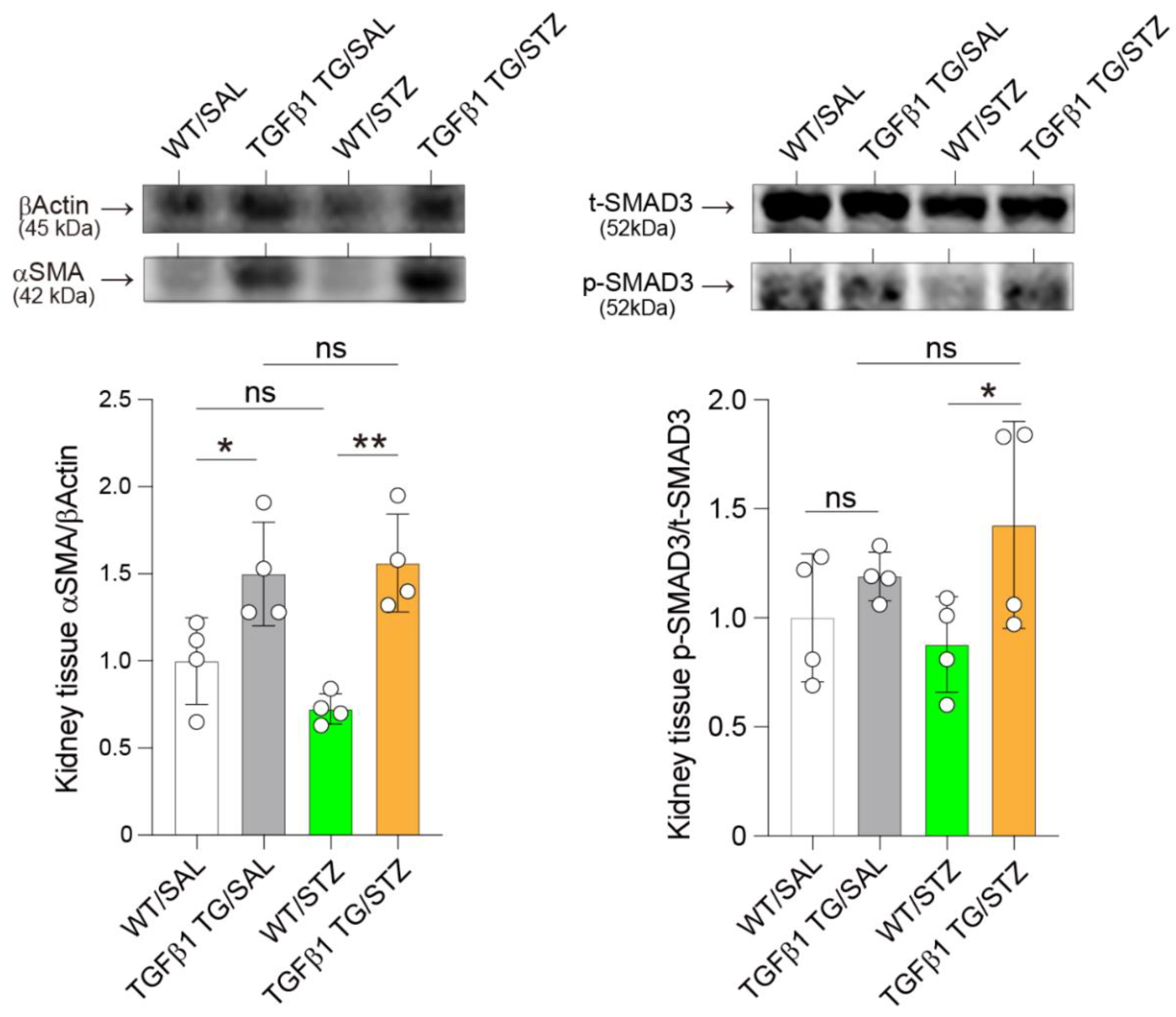
2.8. Elevated Renal Expression of Fibrosis Markers in Diabetic TGFβ1 TG Mice
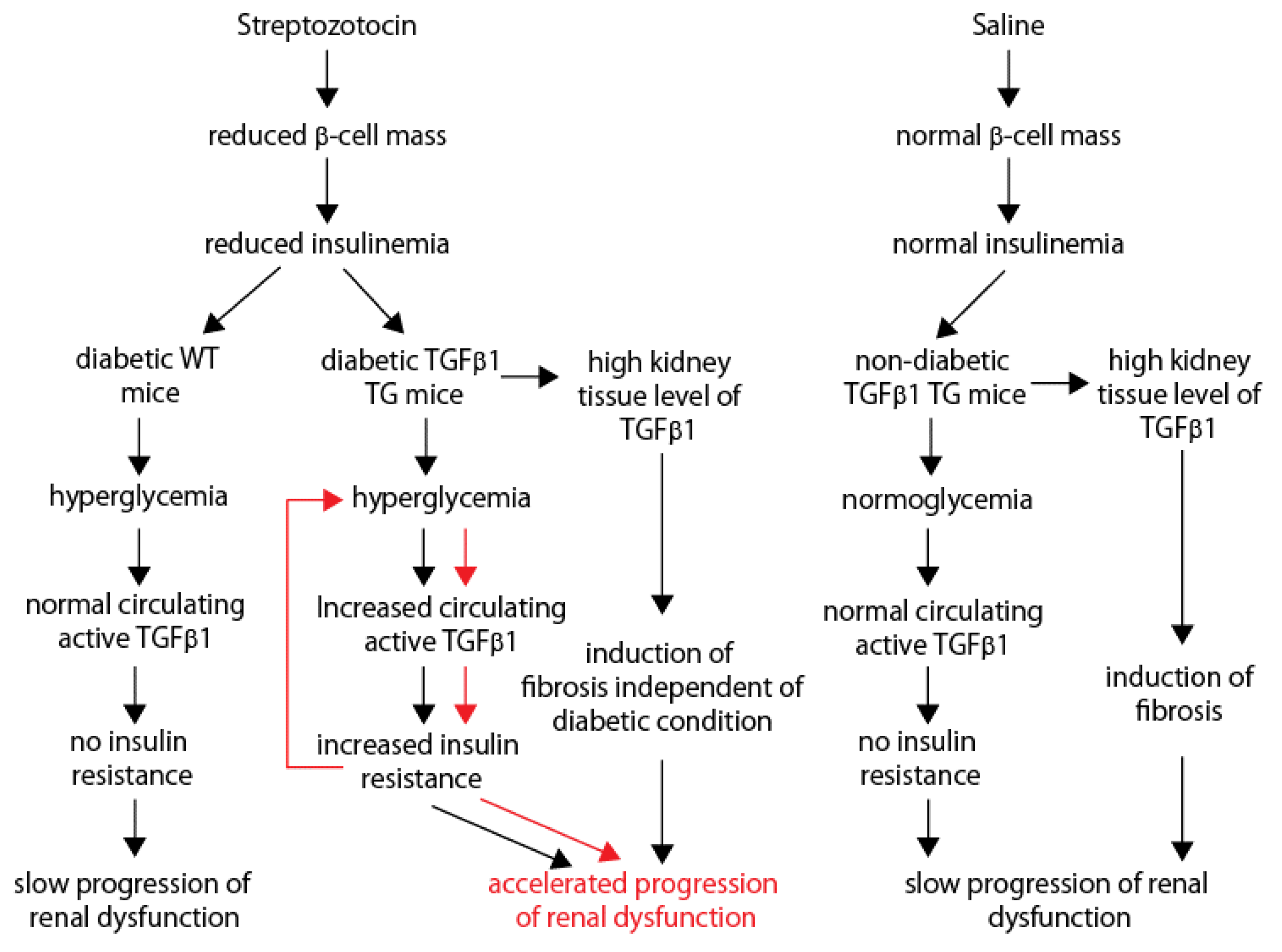
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Induction of Experimental Diabetes
4.3. Evaluation of Glucose Parameters
4.4. Histological Study
4.5. Cell Culture
4.6. Biochemical Analysis
4.7. Western Blotting
4.8. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- GBD 2013 Risk Factors Collaborators; Forouzanfar, M.H.; Alexander, L.; Anderson, H.R.; Bachman, V.F.; Biryukov, S.; Brauer, M.; Burnett, R.; Casey, D.; Coates, M.M.; et al. Global, regional, and national comparative risk assessment of 79 behavioural, environmental and occupational, and metabolic risks or clusters of risks, 1990–2015: A systematic analysis for the Global Burden of Disease Study 2015. Lancet 2016, 388, 1659–1724. [Google Scholar] [CrossRef] [Green Version]
- Saeedi, P.; Petersohn, I.; Salpea, P.; Malanda, B.; Karuranga, S.; Unwin, N.; Colagiuri, S.; Guariguata, L.; Motala, A.A.; Ogurtsova, K.; et al. Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: Results from the International Diabetes Federation Diabetes Atlas, 9(th) edition. Diabetes Res. Clin. Pract. 2019, 157, 107843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- World Health Organization. Diabetes [Internet]. Available online: https://www.who.int/news-room/fact-sheets/detail/diabetes (accessed on 1 September 2022).
- Harding, J.L.; Pavkov, M.E.; Magliano, D.J.; Shaw, J.E.; Gregg, E.W. Global trends in diabetes complications: A review of current evidence. Diabetologia 2019, 62, 3–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y.; Vanhoutte, P.M. Macro- and microvascular endothelial dysfunction in diabetes. J Diabetes 2017, 9, 434–449. [Google Scholar] [CrossRef] [Green Version]
- Strain, W.D.; Paldanius, P.M. Diabetes, cardiovascular disease and the microcirculation. Cardiovasc. Diabetol. 2018, 17, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hung, P.H.; Hsu, Y.C.; Chen, T.H.; Lin, C.L. Recent Advances in Diabetic Kidney Diseases: From Kidney Injury to Kidney Fibrosis. Int. J. Mol. Sci. 2021, 22, 11857. [Google Scholar] [CrossRef]
- Chen, Q.; Zhu, A.; Wang, J.; Huan, X. Comparative analysis of diabetic nephropathy and non-diabetic nephropathy disease. Saudi J. Biol. Sci. 2017, 24, 1815–1817. [Google Scholar] [CrossRef]
- Lei, Y.; Xiong, Y.; Zhang, L.; Yan, H.; Li, Z.; Cao, L.; Huang, J.; Gu, A.; Ni, Z.; Qian, J.; et al. Comparison of Long-Term Outcomes between Peritoneal Dialysis Patients with Diabetes as a Primary Renal Disease or as a Comorbid Condition. PLoS ONE 2015, 10, e0126549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Racki, S.; Zaputovic, L.; Vujicic, B.; Crncevic-Orlic, Z.; Dvornik, S.; Mavric, Z. Comparison of survival between diabetic and non-diabetic patients on maintenance hemodialysis: A single-centre experience. Diabetes Res. Clin. Pract. 2007, 75, 169–175. [Google Scholar] [CrossRef]
- Wang, L.; Wang, H.L.; Liu, T.T.; Lan, H.Y. TGF-Beta as a Master Regulator of Diabetic Nephropathy. Int. J. Mol. Sci. 2021, 22, 7881. [Google Scholar] [CrossRef]
- Sato, T.; Sakurai, H.; Okubo, K.; Kusuta, R.; Onogi, T.; Tsuboi, M. Current state of dialysis treatment and vascular access management in Japan. J. Vasc. Access 2019, 20, 10–14. [Google Scholar] [CrossRef] [PubMed]
- Bank, N. Mechanisms of diabetic hyperfiltration. Kidney Int. 1991, 40, 792–807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akhurst, R.J.; Hata, A. Targeting the TGFbeta signalling pathway in disease. Nat. Rev. Drug Discov. 2012, 11, 790–811. [Google Scholar] [CrossRef] [Green Version]
- D’Alessandro-Gabazza, C.N.; Kobayashi, T.; Boveda-Ruiz, D.; Takagi, T.; Toda, M.; Gil-Bernabe, P.; Miyake, Y.; Yasukawa, A.; Matsuda, Y.; Suzuki, N.; et al. Development and preclinical efficacy of novel transforming growth factor-beta1 short interfering RNAs for pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2012, 46, 397–406. [Google Scholar] [CrossRef] [PubMed]
- D’Alessandro-Gabazza, C.N.; Kobayashi, T.; Yasuma, T.; Toda, M.; Kim, H.; Fujimoto, H.; Hataji, O.; Takeshita, A.; Nishihama, K.; Okano, T.; et al. A Staphylococcus pro-apoptotic peptide induces acute exacerbation of pulmonary fibrosis. Nat. Commun. 2020, 11, 1539. [Google Scholar] [CrossRef] [Green Version]
- D’Alessandro-Gabazza, C.N.; Yasuma, T.; Kobayashi, T.; Toda, M.; Abdel-Hamid, A.M.; Fujimoto, H.; Hataji, O.; Nakahara, H.; Takeshita, A.; Nishihama, K.; et al. Inhibition of lung microbiota-derived proapoptotic peptides ameliorates acute exacerbation of pulmonary fibrosis. Nat. Commun. 2022, 13, 1558. [Google Scholar] [CrossRef]
- Takeshita, A.; Yasuma, T.; Nishihama, K.; D’Alessandro-Gabazza, C.N.; Toda, M.; Totoki, T.; Okano, Y.; Uchida, A.; Inoue, R.; Qin, L.; et al. Thrombomodulin ameliorates transforming growth factor-beta1-mediated chronic kidney disease via the G-protein coupled receptor 15/Akt signal pathway. Kidney Int. 2020, 98, 1179–1192. [Google Scholar] [CrossRef]
- Zhao, L.; Zou, Y.; Liu, F. Transforming Growth Factor-Beta1 in Diabetic Kidney Disease. Front. Cell Dev. Biol. 2020, 8, 187. [Google Scholar] [CrossRef] [Green Version]
- Jakus, V.; Sapak, M.; Kostolanska, J. Circulating TGF-beta1, glycation, and oxidation in children with diabetes mellitus type 1. Exp. Diabetes Res. 2012, 2012, 510902. [Google Scholar] [CrossRef] [Green Version]
- Qiao, Y.C.; Chen, Y.L.; Pan, Y.H.; Ling, W.; Tian, F.; Zhang, X.X.; Zhao, H.L. Changes of transforming growth factor beta 1 in patients with type 2 diabetes and diabetic nephropathy: A PRISMA-compliant systematic review and meta-analysis. Medicine 2017, 96, e6583. [Google Scholar] [CrossRef]
- Pervin, S.; Reddy, S.T.; Singh, R. Novel Roles of Follistatin/Myostatin in Transforming Growth Factor-beta Signaling and Adipose Browning: Potential for Therapeutic Intervention in Obesity Related Metabolic Disorders. Front. Endocrinol. 2021, 12, 653179. [Google Scholar] [CrossRef] [PubMed]
- Alessi, M.C.; Bastelica, D.; Morange, P.; Berthet, B.; Leduc, I.; Verdier, M.; Geel, O.; Juhan-Vague, I. Plasminogen activator inhibitor 1, transforming growth factor-beta1, and BMI are closely associated in human adipose tissue during morbid obesity. Diabetes 2000, 49, 1374–1380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fain, J.N.; Tichansky, D.S.; Madan, A.K. Transforming growth factor beta1 release by human adipose tissue is enhanced in obesity. Metabolism 2005, 54, 1546–1551. [Google Scholar] [CrossRef] [PubMed]
- Gordon, K.J.; Blobe, G.C. Role of transforming growth factor-beta superfamily signaling pathways in human disease. Biochim. Biophys. Acta 2008, 1782, 197–228. [Google Scholar] [CrossRef] [Green Version]
- Yadav, H.; Quijano, C.; Kamaraju, A.K.; Gavrilova, O.; Malek, R.; Chen, W.; Zerfas, P.; Zhigang, D.; Wright, E.C.; Stuelten, C.; et al. Protection from obesity and diabetes by blockade of TGF-beta/Smad3 signaling. Cell Metab. 2011, 14, 67–79. [Google Scholar] [CrossRef] [Green Version]
- Belosludtseva, N.V.; Starinets, V.S.; Pavlik, L.L.; Mikheeva, I.B.; Dubinin, M.V.; Belosludtsev, K.N. The Effect of S-15176 Difumarate Salt on Ultrastructure and Functions of Liver Mitochondria of C57BL/6 Mice with Streptozotocin/High-Fat Diet-Induced Type 2 Diabetes. Biology 2020, 9, 309. [Google Scholar] [CrossRef]
- Fernandez-Cabezudo, M.J.; George, J.A.; Bashir, G.; Mohamed, Y.A.; Al-Mansori, A.; Qureshi, M.M.; Lorke, D.E.; Petroianu, G.; Al-Ramadi, B.K. Involvement of Acetylcholine Receptors in Cholinergic Pathway-Mediated Protection Against Autoimmune Diabetes. Front. Immunol. 2019, 10, 1038. [Google Scholar] [CrossRef]
- Lopez-Soldado, I.; Fuentes-Romero, R.; Duran, J.; Guinovart, J.J. Effects of hepatic glycogen on food intake and glucose homeostasis are mediated by the vagus nerve in mice. Diabetologia 2017, 60, 1076–1083. [Google Scholar] [CrossRef] [Green Version]
- MacDonald, T.L.; Pattamaprapanont, P.; Cooney, E.M.; Nava, R.C.; Mitri, J.; Hafida, S.; Lessard, S.J. Canagliflozin Prevents Hyperglycemia-Associated Muscle Extracellular Matrix Accumulation and Improves the Adaptive Response to Aerobic Exercise. Diabetes 2022, 71, 881–893. [Google Scholar] [CrossRef]
- MacDonald, T.L.; Pattamaprapanont, P.; Pathak, P.; Fernandez, N.; Freitas, E.C.; Hafida, S.; Mitri, J.; Britton, S.L.; Koch, L.G.; Lessard, S.J. Hyperglycaemia is associated with impaired muscle signalling and aerobic adaptation to exercise. Nat. Metab. 2020, 2, 902–917. [Google Scholar] [CrossRef]
- Yasuma, T.; Yano, Y.; D’Alessandro-Gabazza, C.N.; Toda, M.; Gil-Bernabe, P.; Kobayashi, T.; Nishihama, K.; Hinneh, J.A.; Mifuji-Moroka, R.; Roeen, Z.; et al. Amelioration of Diabetes by Protein S. Diabetes 2016, 65, 1940–1951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leclercq, I.A.; Da Silva Morais, A.; Schroyen, B.; Van Hul, N.; Geerts, A. Insulin resistance in hepatocytes and sinusoidal liver cells: Mechanisms and consequences. J. Hepatol. 2007, 47, 142–156. [Google Scholar] [CrossRef]
- Chen, H.Y.; Ho, Y.J.; Chou, H.C.; Liao, E.C.; Tsai, Y.T.; Wei, Y.S.; Lin, L.H.; Lin, M.W.; Wang, Y.S.; Ko, M.L.; et al. The Role of Transforming Growth Factor-Beta in Retinal Ganglion Cells with Hyperglycemia and Oxidative Stress. Int. J. Mol. Sci. 2020, 21, 6482. [Google Scholar] [CrossRef] [PubMed]
- Esmatjes, E.; Flores, L.; Lario, S.; Claria, J.; Cases, A.; Inigo, P.; Campistol, J.M. Smoking increases serum levels of transforming growth factor-beta in diabetic patients. Diabetes Care 1999, 22, 1915–1916. [Google Scholar] [CrossRef] [PubMed]
- Huan, Y.; DeLoach, S.; Daskalakis, C.; Dunn, S.R.; Sharma, K.; Falkner, B. Regulation of transforming growth factor-beta1 by insulin in prediabetic African Americans. Kidney Int. 2010, 78, 318–324. [Google Scholar] [CrossRef] [Green Version]
- Pfeiffer, A.; Middelberg-Bisping, K.; Drewes, C.; Schatz, H. Elevated plasma levels of transforming growth factor-beta 1 in NIDDM. Diabetes Care 1996, 19, 1113–1117. [Google Scholar] [CrossRef] [PubMed]
- Romano, M.; Guagnano, M.T.; Pacini, G.; Vigneri, S.; Falco, A.; Marinopiccoli, M.; Manigrasso, M.R.; Basili, S.; Davi, G. Association of inflammation markers with impaired insulin sensitivity and coagulative activation in obese healthy women. J. Clin. Endocrinol. Metab. 2003, 88, 5321–5326. [Google Scholar] [CrossRef] [PubMed]
- Shankland, S.J.; Scholey, J.W.; Ly, H.; Thai, K. Expression of transforming growth factor-beta 1 during diabetic renal hypertrophy. Kidney Int. 1994, 46, 430–442. [Google Scholar] [CrossRef] [Green Version]
- Iwano, M.; Kubo, A.; Nishino, T.; Sato, H.; Nishioka, H.; Akai, Y.; Kurioka, H.; Fujii, Y.; Kanauchi, M.; Shiiki, H.; et al. Quantification of glomerular TGF-beta 1 mRNA in patients with diabetes mellitus. Kidney Int. 1996, 49, 1120–1126. [Google Scholar] [CrossRef] [Green Version]
- Sharma, K.; Ziyadeh, F.N. Hyperglycemia and diabetic kidney disease. The case for transforming growth factor-beta as a key mediator. Diabetes 1995, 44, 1139–1146. [Google Scholar] [CrossRef]
- Yamamoto, T.; Nakamura, T.; Noble, N.A.; Ruoslahti, E.; Border, W.A. Expression of transforming growth factor beta is elevated in human and experimental diabetic nephropathy. Proc. Natl. Acad. Sci. USA 1993, 90, 1814–1818. [Google Scholar] [CrossRef] [Green Version]
- Ziyadeh, F.N.; Sharma, K.; Ericksen, M.; Wolf, G. Stimulation of collagen gene expression and protein synthesis in murine mesangial cells by high glucose is mediated by autocrine activation of transforming growth factor-beta. J. Clin. Investig. 1994, 93, 536–542. [Google Scholar] [CrossRef] [PubMed]
- Han, D.C.; Isono, M.; Hoffman, B.B.; Ziyadeh, F.N. High glucose stimulates proliferation and collagen type I synthesis in renal cortical fibroblasts: Mediation by autocrine activation of TGF-beta. J. Am. Soc. Nephrol. 1999, 10, 1891–1899. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, B.B.; Sharma, K.; Zhu, Y.; Ziyadeh, F.N. Transcriptional activation of transforming growth factor-beta1 in mesangial cell culture by high glucose concentration. Kidney Int. 1998, 54, 1107–1116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rocco, M.V.; Chen, Y.; Goldfarb, S.; Ziyadeh, F.N. Elevated glucose stimulates TGF-beta gene expression and bioactivity in proximal tubule. Kidney Int. 1992, 41, 107–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weigert, C.; Sauer, U.; Brodbeck, K.; Pfeiffer, A.; Haring, H.U.; Schleicher, E.D. AP-1 proteins mediate hyperglycemia-induced activation of the human TGF-beta1 promoter in mesangial cells. J. Am. Soc. Nephrol. 2000, 11, 2007–2016. [Google Scholar] [CrossRef]
- Wolf, G.; Sharma, K.; Chen, Y.; Ericksen, M.; Ziyadeh, F.N. High glucose-induced proliferation in mesangial cells is reversed by autocrine TGF-beta. Kidney Int. 1992, 42, 647–656. [Google Scholar] [CrossRef] [Green Version]
- McGowan, T.A.; Dunn, S.R.; Falkner, B.; Sharma, K. Stimulation of urinary TGF-beta and isoprostanes in response to hyperglycemia in humans. Clin. J. Am. Soc. Nephrol. 2006, 1, 263–268. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Qi, X.; Shao, Y.; Li, Y.; Fu, X.; Feng, S.; Wu, Y. High glucose induced-macrophage activation through TGF-beta-activated kinase 1 signaling pathway. Inflamm. Res. 2016, 65, 655–664. [Google Scholar] [CrossRef]
- Sharma, K.; Ziyadeh, F.N.; Alzahabi, B.; McGowan, T.A.; Kapoor, S.; Kurnik, B.R.; Kurnik, P.B.; Weisberg, L.S. Increased renal production of transforming growth factor-beta1 in patients with type II diabetes. Diabetes 1997, 46, 854–859. [Google Scholar] [CrossRef]
- Munger, J.S.; Harpel, J.G.; Gleizes, P.E.; Mazzieri, R.; Nunes, I.; Rifkin, D.B. Latent transforming growth factor-beta: Structural features and mechanisms of activation. Kidney Int. 1997, 51, 1376–1382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersen, H.; Friis, U.G.; Hansen, P.B.; Svenningsen, P.; Henriksen, J.E.; Jensen, B.L. Diabetic nephropathy is associated with increased urine excretion of proteases plasmin, prostasin and urokinase and activation of amiloride-sensitive current in collecting duct cells. Nephrol. Dial. Transplant. 2015, 30, 781–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellasi, A.; Di Micco, L.; Santoro, D.; Marzocco, S.; De Simone, E.; Cozzolino, M.; Di Lullo, L.; Guastaferro, P.; Di Iorio, B.; UBI study investigators. Correction of metabolic acidosis improves insulin resistance in chronic kidney disease. BMC Nephrol. 2016, 17, 158. [Google Scholar] [CrossRef] [PubMed]
- Crawford, S.E.; Stellmach, V.; Murphy-Ullrich, J.E.; Ribeiro, S.M.; Lawler, J.; Hynes, R.O.; Boivin, G.P.; Bouck, N. Thrombospondin-1 is a major activator of TGF-beta1 in vivo. Cell 1998, 93, 1159–1170. [Google Scholar] [CrossRef] [Green Version]
- Fakhruddin, S.; Alanazi, W.; Jackson, K.E. Diabetes-Induced Reactive Oxygen Species: Mechanism of Their Generation and Role in Renal Injury. J. Diabetes Res. 2017, 2017, 8379327. [Google Scholar] [CrossRef] [Green Version]
- Khalil, N. TGF-beta: From latent to active. Microbes Infect. 1999, 1, 1255–1263. [Google Scholar] [CrossRef]
- Sawada, K.; Toyoda, M.; Kaneyama, N.; Shiraiwa, S.; Moriya, H.; Miyatake, H.; Tanaka, E.; Yamamoto, N.; Miyauchi, M.; Kimura, M.; et al. Upregulation of alpha3beta1-Integrin in Podocytes in Early-Stage Diabetic Nephropathy. J. Diabetes Res. 2016, 2016, 9265074. [Google Scholar] [CrossRef] [Green Version]
- McIlvenna, L.C.; Patten, R.K.; McAinch, A.J.; Rodgers, R.J.; Stepto, N.K.; Moreno-Asso, A. Transforming Growth Factor Beta 1 Alters Glucose Uptake but Not Insulin Signalling in Human Primary Myotubes From Women With and Without Polycystic Ovary Syndrome. Front. Endocrinol. 2021, 12, 732338. [Google Scholar] [CrossRef]
- Stepto, N.K.; Hiam, D.; Gibson-Helm, M.; Cassar, S.; Harrison, C.L.; Hutchison, S.K.; Joham, A.E.; Canny, B.J.; Moreno-Asso, A.; Strauss, B.J.; et al. Exercise and insulin resistance in PCOS: Muscle insulin signalling and fibrosis. Endocr. Connect. 2020, 9, 346–359. [Google Scholar] [CrossRef] [Green Version]
- Stepto, N.K.; Moreno-Asso, A.; McIlvenna, L.C.; Walters, K.A.; Rodgers, R.J. Molecular Mechanisms of Insulin Resistance in Polycystic Ovary Syndrome: Unraveling the Conundrum in Skeletal Muscle? J. Clin. Endocrinol. Metab. 2019, 104, 5372–5381. [Google Scholar] [CrossRef]
- Herder, C.; Zierer, A.; Koenig, W.; Roden, M.; Meisinger, C.; Thorand, B. Transforming growth factor-beta1 and incident type 2 diabetes: Results from the MONICA/KORA case-cohort study, 1984–2002. Diabetes Care 2009, 32, 1921–1923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seong, H.A.; Manoharan, R.; Ha, H. Smad proteins differentially regulate obesity-induced glucose and lipid abnormalities and inflammation via class-specific control of AMPK-related kinase MPK38/MELK activity. Cell Death Dis. 2018, 9, 471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, C.K.; Leuenberger, N.; Tan, M.J.; Yan, Y.W.; Chen, Y.; Kambadur, R.; Wahli, W.; Tan, N.S. Smad3 deficiency in mice protects against insulin resistance and obesity induced by a high-fat diet. Diabetes 2011, 60, 464–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dantas, W.S.; Roschel, H.; Murai, I.H.; Gil, S.; Davuluri, G.; Axelrod, C.L.; Ghosh, S.; Newman, S.S.; Zhang, H.; Shinjo, S.K.; et al. Exercise-Induced Increases in Insulin Sensitivity After Bariatric Surgery Are Mediated By Muscle Extracellular Matrix Remodeling. Diabetes 2020, 69, 1675–1691. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Wei, X.; Hu, H.; Yang, D.; Zhang, B.; Fan, X.; Liu, J.; He, H.; Oh, Y.; Wu, Q.; et al. The saturated fatty acid palmitate induces insulin resistance through Smad3-mediated down-regulation of FNDC5 in myotubes. Biochem. Biophys. Res. Commun. 2019, 520, 619–626. [Google Scholar] [CrossRef]
- Alshehade, S.; Alshawsh, M.A.; Murugaiyah, V.; Asif, M.; Alshehade, O.; Almoustafa, H.; Al Zarzour, R.H. The role of protein kinases as key drivers of metabolic dysfunction-associated fatty liver disease progression: New insights and future directions. Life Sci. 2022, 305, 120732. [Google Scholar] [CrossRef]
- Demir, S.; Wolff, G.; Wieder, A.; Maida, A.; Buhler, L.; Brune, M.; Hautzinger, O.; Feuchtinger, A.; Poth, T.; Szendroedi, J.; et al. TSC22D4 interacts with Akt1 to regulate glucose metabolism. Sci. Adv. 2022, 8, eabo5555. [Google Scholar] [CrossRef]
- Wang, L.; Ma, R.; Flavell, R.A.; Choi, M.E. Requirement of mitogen-activated protein kinase kinase 3 (MKK3) for activation of p38alpha and p38delta MAPK isoforms by TGF-beta 1 in murine mesangial cells. J. Biol. Chem. 2002, 277, 47257–47262. [Google Scholar] [CrossRef] [Green Version]
- Berria, R.; Wang, L.; Richardson, D.K.; Finlayson, J.; Belfort, R.; Pratipanawatr, T.; De Filippis, E.A.; Kashyap, S.; Mandarino, L.J. Increased collagen content in insulin-resistant skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2006, 290, E560–E565. [Google Scholar] [CrossRef]
- Richardson, D.K.; Kashyap, S.; Bajaj, M.; Cusi, K.; Mandarino, S.J.; Finlayson, J.; DeFronzo, R.A.; Jenkinson, C.P.; Mandarino, L.J. Lipid infusion decreases the expression of nuclear encoded mitochondrial genes and increases the expression of extracellular matrix genes in human skeletal muscle. J. Biol. Chem. 2005, 280, 10290–10297. [Google Scholar] [CrossRef]
- Tam, C.S.; Chaudhuri, R.; Hutchison, A.T.; Samocha-Bonet, D.; Heilbronn, L.K. Skeletal muscle extracellular matrix remodeling after short-term overfeeding in healthy humans. Metabolism 2017, 67, 26–30. [Google Scholar] [CrossRef] [PubMed]
- Watts, R.; McAinch, A.J.; Dixon, J.B.; O’Brien, P.E.; Cameron-Smith, D. Increased Smad signaling and reduced MRF expression in skeletal muscle from obese subjects. Obesity 2013, 21, 525–528. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.S.; Kang, L.; Wasserman, D.H. The extracellular matrix and insulin resistance. Trends Endocrinol. Metab. 2015, 26, 357–366. [Google Scholar] [CrossRef] [Green Version]
- Bonnevie-Nielsen, V.; Steffes, M.W.; Lernmark, A. A major loss in islet mass and B-cell function precedes hyperglycemia in mice given multiple low doses of streptozotocin. Diabetes 1981, 30, 424–429. [Google Scholar] [CrossRef] [PubMed]
- King, A.J. The use of animal models in diabetes research. Br. J. Pharmacol. 2012, 166, 877–894. [Google Scholar] [CrossRef] [Green Version]
- Like, A.A.; Rossini, A.A. Streptozotocin-induced pancreatic insulitis: New model of diabetes mellitus. Science 1976, 193, 415–417. [Google Scholar] [CrossRef]
- Okano, Y.; Takeshita, A.; Yasuma, T.; Toda, M.; Nishihama, K.; Fridman D’Alessandro, V.; Inoue, C.; D’Alessandro-Gabazza, C.N.; Kobayashi, T.; Yano, Y.; et al. Protective Role of Recombinant Human Thrombomodulin in Diabetes Mellitus. Cells 2021, 10, 2237. [Google Scholar] [CrossRef]
- Nishihama, K.; Yasuma, T.; Yano, Y.; CN, D.A.-G.; Toda, M.; Hinneh, J.A.; Baffour Tonto, P.; Takeshita, A.; Totoki, T.; Mifuji-Moroka, R.; et al. Anti-apoptotic activity of human matrix metalloproteinase-2 attenuates diabetes mellitus. Metabolism 2018, 82, 88–99. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
D’Alessandro, V.F.; Takeshita, A.; Yasuma, T.; Toda, M.; D’Alessandro-Gabazza, C.N.; Okano, Y.; Tharavecharak, S.; Inoue, C.; Nishihama, K.; Fujimoto, H.; et al. Transforming Growth Factorβ1 Overexpression Is Associated with Insulin Resistance and Rapidly Progressive Kidney Fibrosis under Diabetic Conditions. Int. J. Mol. Sci. 2022, 23, 14265. https://doi.org/10.3390/ijms232214265
D’Alessandro VF, Takeshita A, Yasuma T, Toda M, D’Alessandro-Gabazza CN, Okano Y, Tharavecharak S, Inoue C, Nishihama K, Fujimoto H, et al. Transforming Growth Factorβ1 Overexpression Is Associated with Insulin Resistance and Rapidly Progressive Kidney Fibrosis under Diabetic Conditions. International Journal of Molecular Sciences. 2022; 23(22):14265. https://doi.org/10.3390/ijms232214265
Chicago/Turabian StyleD’Alessandro, Valeria Fridman, Atsuro Takeshita, Taro Yasuma, Masaaki Toda, Corina N. D’Alessandro-Gabazza, Yuko Okano, Suphachai Tharavecharak, Chisa Inoue, Kota Nishihama, Hajime Fujimoto, and et al. 2022. "Transforming Growth Factorβ1 Overexpression Is Associated with Insulin Resistance and Rapidly Progressive Kidney Fibrosis under Diabetic Conditions" International Journal of Molecular Sciences 23, no. 22: 14265. https://doi.org/10.3390/ijms232214265
APA StyleD’Alessandro, V. F., Takeshita, A., Yasuma, T., Toda, M., D’Alessandro-Gabazza, C. N., Okano, Y., Tharavecharak, S., Inoue, C., Nishihama, K., Fujimoto, H., Kobayashi, T., Yano, Y., & Gabazza, E. C. (2022). Transforming Growth Factorβ1 Overexpression Is Associated with Insulin Resistance and Rapidly Progressive Kidney Fibrosis under Diabetic Conditions. International Journal of Molecular Sciences, 23(22), 14265. https://doi.org/10.3390/ijms232214265