
Modeling RTT Syndrome by iPSC-Derived Neurons from Male and Female Patients with Heterogeneously Severe Hot-Spot MECP2 Variants

 , , , , ,
, , , , ,  ,
,  ,
,
Abstract
:1. Introduction
2. Results
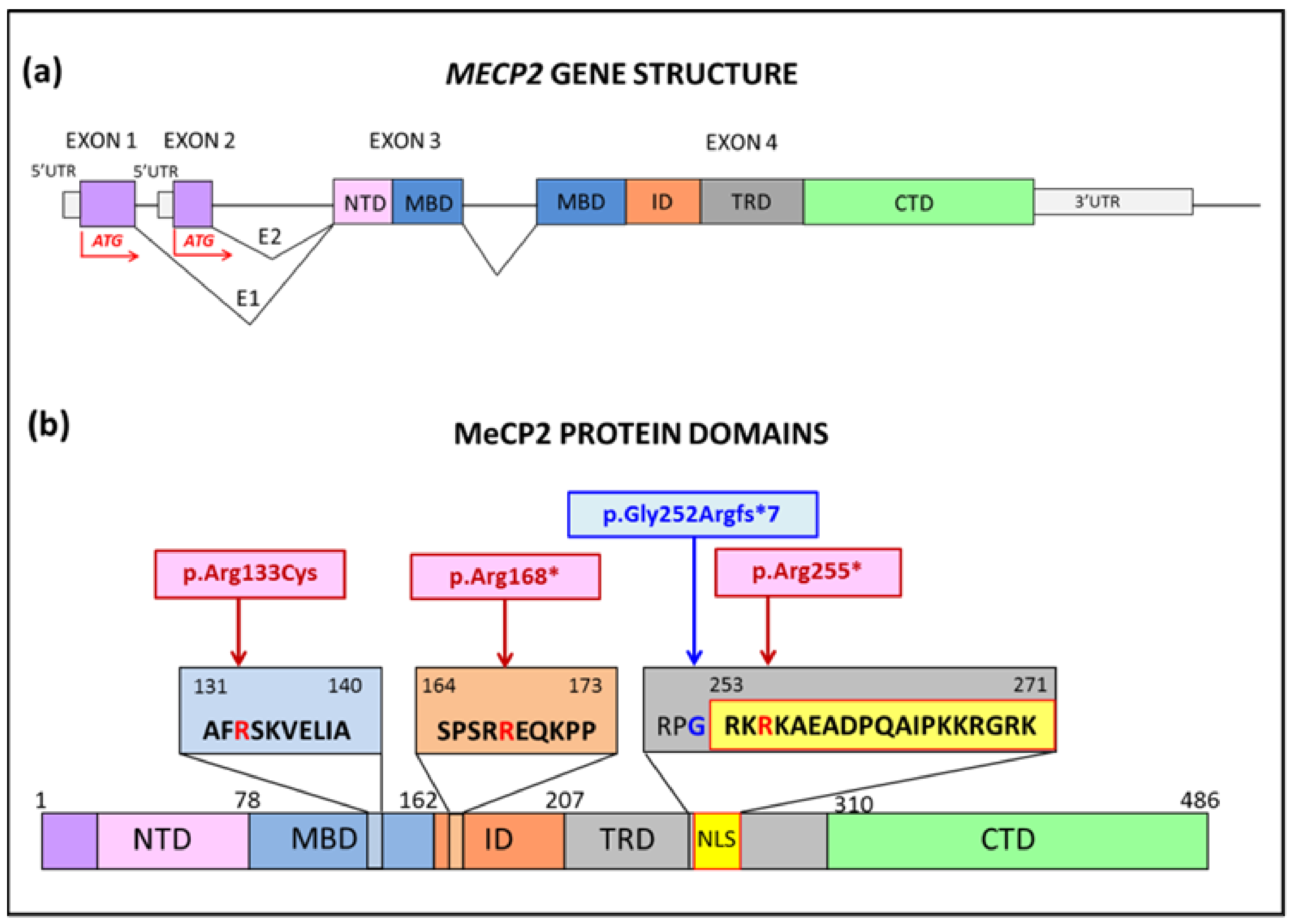
2.1. MECP2 Patients Selected for In Vitro Modeling
2.2. Patients Clinical Phenotype
2.3. From iPSCs to Neuron Differentiation
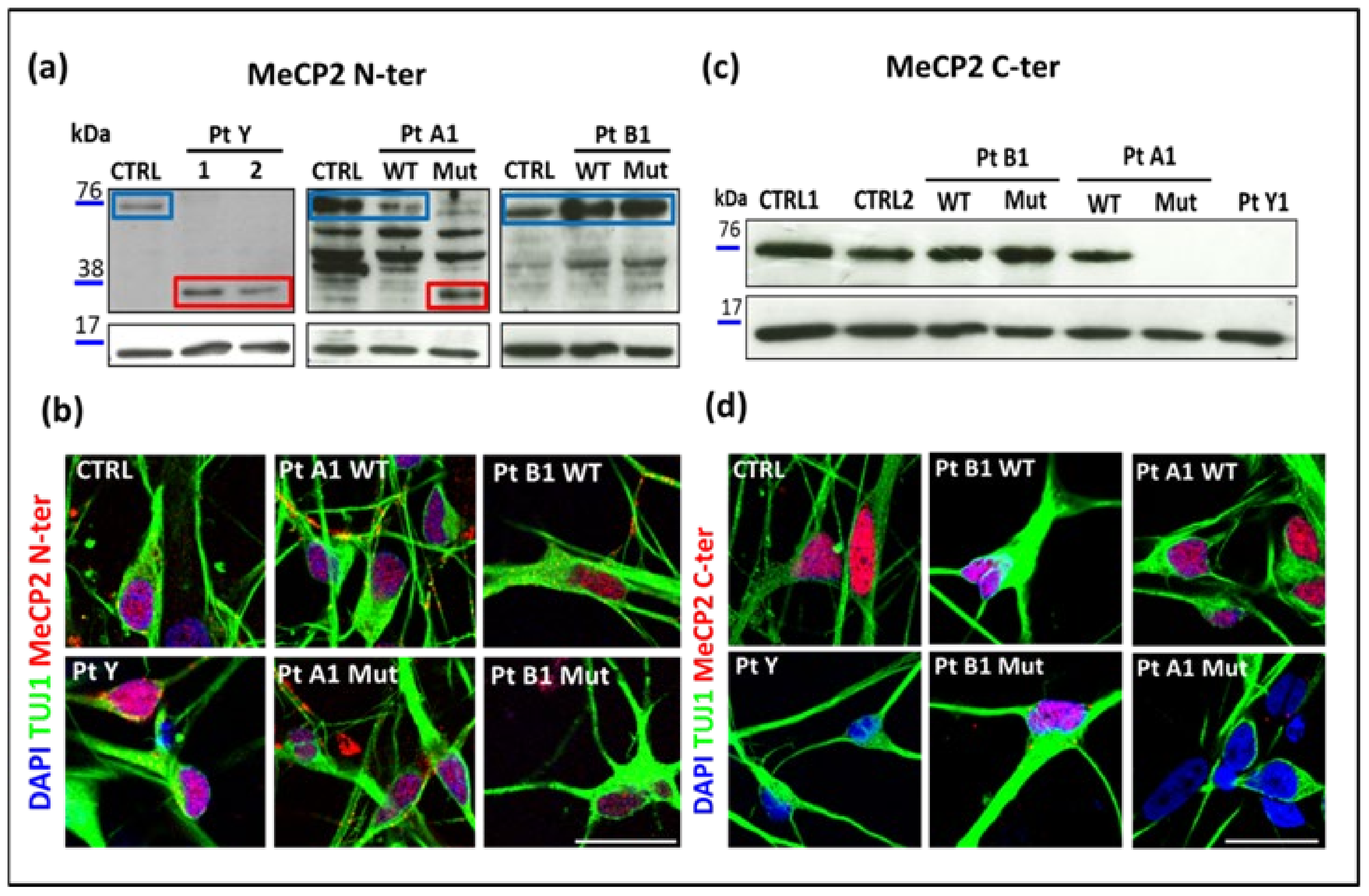
2.4. MeCP2 Protein in RTT Patients
2.5. Morphological Analyses of RTT Young Neurons
2.6. Electrophysiology of MECP2 Defective Cortical Neurons
3. Discussion
4. Materials and Methods
4.1. iPSC Generation and Characterization
4.1.1. SNP Array
4.1.2. Mutation Sequencing
- Forward 5′ AAGCAAAGGAAATCTGGCCG 3′
- Reverse 5′ GTCTCCTGCACAGATCGGAT 3′
4.1.3. The Human Androgen Receptor Assay
4.2. Cortical i-Neurons Generation
4.3. Protein Extraction and Western Blot
4.4. Immunofluorescence Staining
4.5. Morphological Analysis
4.6. Electrophysiological Recordings
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shahbazian, M.D. Insight into Rett Syndrome: MeCP2 Levels Display Tissue- and Cell-Specific Differences and Correlate with Neuronal Maturation. Hum. Mol. Genet. 2002, 11, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Kishi, N.; Macklis, J.D. MECP2 Is Progressively Expressed in Post-Migratory Neurons and Is Involved in Neuronal Maturation Rather than Cell Fate Decisions. Mol. Cell. Neurosci. 2004, 27, 306–321. [Google Scholar] [CrossRef] [PubMed]
- Ross, P.D.; Guy, J.; Selfridge, J.; Kamal, B.; Bahey, N.; Tanner, K.E.; Gillingwater, T.H.; Jones, R.A.; Loughrey, C.M.; McCarroll, C.S.; et al. Exclusive Expression of MeCP2 in the Nervous System Distinguishes between Brain and Peripheral Rett Syndrome-like Phenotypes. Hum. Mol. Genet. 2016, 25, ddw269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharifi, O.; Yasui, D.H. The Molecular Functions of MeCP2 in Rett Syndrome Pathology. Front. Genet. 2021, 12, 624290. [Google Scholar] [CrossRef]
- Chahrour, M.; Zoghbi, H.Y. The Story of Rett Syndrome: From Clinic to Neurobiology. Neuron 2007, 56, 422–437. [Google Scholar] [CrossRef] [Green Version]
- Belichenko, P.V.; Wright, E.E.; Belichenko, N.P.; Masliah, E.; Li, H.H.; Mobley, W.C.; Francke, U. Widespread Changes in Dendritic and Axonal Morphology in Mecp2 -Mutant Mouse Models of Rett Syndrome: Evidence for Disruption of Neuronal Networks. J. Comp. Neurol. 2009, 514, 240–258. [Google Scholar] [CrossRef]
- Carter, J.C.; Lanham, D.C.; Pham, D.; Bibat, G.; Naidu, S.; Kaufmann, W.E. Selective Cerebral Volume Reduction in Rett Syndrome: A Multiple-Approach MR Imaging Study. AJNR Am. J. Neuroradiol. 2008, 29, 436–441. [Google Scholar] [CrossRef]
- Pascual-Alonso, A.; Martínez-Monseny, A.F.; Xiol, C.; Armstrong, J. MECP2-Related Disorders in Males. Ijms 2021, 22, 9610. [Google Scholar] [CrossRef]
- Neul, J.L.; Fang, P.; Barrish, J.; Lane, J.; Caeg, E.B.; Smith, E.O.; Zoghbi, H.; Percy, A.; Glaze, D.G. Specific Mutations in Methyl-CpG-Binding Protein 2 Confer Different Severity in Rett Syndrome. Neurology 2008, 70, 1313–1321. [Google Scholar] [CrossRef] [Green Version]
- Cuddapah, V.A.; Pillai, R.B.; Shekar, K.V.; Lane, J.B.; Motil, K.J.; Skinner, S.A.; Tarquinio, D.C.; Glaze, D.G.; McGwin, G.; Kaufmann, W.E.; et al. Methyl-CpG-Binding Protein 2 ( MECP2 ) Mutation Type Is Associated with Disease Severity in Rett Syndrome. J. Med. Genet. 2014, 51, 152–158. [Google Scholar] [CrossRef]
- Ananiev, G.; Williams, E.C.; Li, H.; Chang, Q. Isogenic Pairs of Wild Type and Mutant Induced Pluripotent Stem Cell (IPSC) Lines from Rett Syndrome Patients as In Vitro Disease Model. PLoS ONE 2011, 6, e25255. [Google Scholar] [CrossRef] [PubMed]
- Colantuoni, C.; Jeon, O.-H.; Hyder, K.; Chenchik, A.; Khimani, A.H.; Narayanan, V.; Hoffman, E.P.; Kaufmann, W.E.; Naidu, S.; Pevsner, J. Gene Expression Profiling in Postmortem Rett Syndrome Brain: Differential Gene Expression and Patient Classification. Neurobiol. Dis. 2001, 8, 847–865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, R.Z.; Akbarian, S.; Tudor, M.; Jaenisch, R. Deficiency of Methyl-CpG Binding Protein-2 in CNS Neurons Results in a Rett-like Phenotype in Mice. Nat. Genet. 2001, 27, 327–331. [Google Scholar] [CrossRef] [PubMed]
- Guy, J.; Hendrich, B.; Holmes, M.; Martin, J.E.; Bird, A. A Mouse Mecp2-Null Mutation Causes Neurological Symptoms That Mimic Rett Syndrome. Nat. Genet. 2001, 27, 322–326. [Google Scholar] [CrossRef] [Green Version]
- Vashi, N.; Justice, M.J. Treating Rett Syndrome: From Mouse Models to Human Therapies. Mamm. Genome 2019, 30, 90–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eze, U.C.; Bhaduri, A.; Haeussler, M.; Nowakowski, T.J.; Kriegstein, A.R. Single-Cell Atlas of Early Human Brain Development Highlights Heterogeneity of Human Neuroepithelial Cells and Early Radial Glia. Nat. Neurosci 2021, 24, 584–594. [Google Scholar] [CrossRef]
- Velasco, S.; Kedaigle, A.J.; Simmons, S.K.; Nash, A.; Rocha, M.; Quadrato, G.; Paulsen, B.; Nguyen, L.; Adiconis, X.; Regev, A.; et al. Individual Brain Organoids Reproducibly Form Cell Diversity of the Human Cerebral Cortex. Nature 2019, 570, 523–527. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [Green Version]
- Marchetto, M.C.N.; Carromeu, C.; Acab, A.; Yu, D.; Yeo, G.W.; Mu, Y.; Chen, G.; Gage, F.H.; Muotri, A.R. A Model for Neural Development and Treatment of Rett Syndrome Using Human Induced Pluripotent Stem Cells. Cell 2010, 143, 527–539. [Google Scholar] [CrossRef] [Green Version]
- Gomes, A.R.; Fernandes, T.G.; Cabral, J.M.S.; Diogo, M.M. Modeling Rett Syndrome with Human Pluripotent Stem Cells: Mechanistic Outcomes and Future Clinical Perspectives. Ijms 2021, 22, 3751. [Google Scholar] [CrossRef]
- Haase, F.D.; Coorey, B.; Riley, L.; Cantrill, L.C.; Tam, P.P.L.; Gold, W.A. Pre-Clinical Investigation of Rett Syndrome Using Human Stem Cell-Based Disease Models. Front. Neurosci. 2021, 15, 698812. [Google Scholar] [CrossRef] [PubMed]
- Lancaster, M.A.; Renner, M.; Martin, C.-A.; Wenzel, D.; Bicknell, L.S.; Hurles, M.E.; Homfray, T.; Penninger, J.M.; Jackson, A.P.; Knoblich, J.A. Cerebral Organoids Model Human Brain Development and Microcephaly. Nature 2013, 501, 373–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marton, R.M.; Pașca, S.P. Organoid and Assembloid Technologies for Investigating Cellular Crosstalk in Human Brain Development and Disease. Trends Cell Biol. 2020, 30, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Kim, J.; Zhou, L.; Wengert, E.; Zhang, L.; Wu, Z.; Carromeu, C.; Muotri, A.R.; Marchetto, M.C.N.; Gage, F.H.; et al. KCC2 Rescues Functional Deficits in Human Neurons Derived from Patients with Rett Syndrome. Proc. Natl. Acad. Sci. USA 2016, 113, 751–756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.J.; Savas, J.N.; Miller, M.T.; Hu, X.; Carromeu, C.; Lavallée-Adam, M.; Freitas, B.C.G.; Muotri, A.R.; Yates, J.R.; Ghosh, A. Proteomic Analyses Reveal Misregulation of LIN28 Expression and Delayed Timing of Glial Differentiation in Human IPS Cells with MECP2 Loss-of-Function. PLoS ONE 2019, 14, e0212553. [Google Scholar] [CrossRef]
- Fernandes, T.G.; Duarte, S.T.; Ghazvini, M.; Gaspar, C.; Santos, D.C.; Porteira, A.R.; Rodrigues, G.M.C.; Haupt, S.; Rombo, D.M.; Armstrong, J.; et al. Neural Commitment of Human Pluripotent Stem Cells under Defined Conditions Recapitulates Neural Development and Generates Patient-Specific Neural Cells. Biotechnol. J. 2015, 10, 1578–1588. [Google Scholar] [CrossRef]
- Ohashi, M.; Korsakova, E.; Allen, D.; Lee, P.; Fu, K.; Vargas, B.S.; Cinkornpumin, J.; Salas, C.; Park, J.C.; Germanguz, I.; et al. Loss of MECP2 Leads to Activation of P53 and Neuronal Senescence. Stem Cell Rep. 2018, 10, 1453–1463. [Google Scholar] [CrossRef]
- Xiang, Y.; Tanaka, Y.; Patterson, B.; Hwang, S.-M.; Hysolli, E.; Cakir, B.; Kim, K.-Y.; Wang, W.; Kang, Y.-J.; Clement, E.M.; et al. Dysregulation of BRD4 Function Underlies the Functional Abnormalities of MeCP2 Mutant Neurons. Mol. Cell 2020, 79, 84–98. [Google Scholar] [CrossRef]
- Kim, K.-Y.; Hysolli, E.; Park, I.-H. Neuronal Maturation Defect in Induced Pluripotent Stem Cells from Patients with Rett Syndrome. Proc. Natl. Acad. Sci. USA 2011, 108, 14169–14174. [Google Scholar] [CrossRef] [Green Version]
- Yoo, M.; Carromeu, C.; Kwon, O.; Muotri, A.; Schachner, M. The L1 Adhesion Molecule Normalizes Neuritogenesis in Rett Syndrome-Derived Neural Precursor Cells. Biochem. Biophys. Res. Commun. 2017, 494, 504–510. [Google Scholar] [CrossRef]
- Cacciatori, E.; Lelii, M.; Russo, S.; Alari, V.; Masciadri, M.; Guez, S.; Patria, M.F.; Marchisio, P.; Milani, D. Sleep Disordered Breathing and Daytime Hypoventilation in a Male with MECP2 Mutation. Am. J. Med. Genet. 2020, 182, 2982–2987. [Google Scholar] [CrossRef] [PubMed]
- Bu, Q.; Wang, A.; Hamzah, H.; Waldman, A.; Jiang, K.; Dong, Q.; Li, R.; Kim, J.; Turner, D.; Chang, Q. CREB Signaling Is Involved in Rett Syndrome Pathogenesis. J. Neurosci. 2017, 37, 3671–3685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.; Miller, E.C.; Pozzo-Miller, L. Dendritic Spine Dysgenesis in Rett Syndrome. Front. Neuroanat. 2014, 8, 97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Han, X.; Blanchi, B.; Guan, W.; Ge, W.; Yu, Y.-C.; Sun, Y.E. Graded and Pan-Neural Disease Phenotypes of Rett Syndrome Linked with Dosage of Functional MeCP2. Protein Cell 2021, 12, 639–652. [Google Scholar] [CrossRef] [PubMed]
- Hodge, R.D.; Bakken, T.E.; Miller, J.A.; Smith, K.A.; Barkan, E.R.; Graybuck, L.T.; Close, J.L.; Long, B.; Johansen, N.; Penn, O.; et al. Conserved Cell Types with Divergent Features in Human versus Mouse Cortex. Nature 2019, 573, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Yazdani, M.; Deogracias, R.; Guy, J.; Poot, R.A.; Bird, A.; Barde, Y.-A. Disease Modeling Using Embryonic Stem Cells: MeCP2 Regulates Nuclear Size and RNA Synthesis in Neurons. Stem Cells 2012, 30, 2128–2139. [Google Scholar] [CrossRef] [PubMed]
- Quach, T.T.; Stratton, H.J.; Khanna, R.; Kolattukudy, P.E.; Honnorat, J.; Meyer, K.; Duchemin, A.-M. Intellectual Disability: Dendritic Anomalies and Emerging Genetic Perspectives. Acta Neuropathol. 2021, 141, 139–158. [Google Scholar] [CrossRef] [PubMed]
- Alari, V.; Russo, S.; Rovina, D.; Garzo, M.; Crippa, M.; Calzari, L.; Scalera, C.; Concolino, D.; Castiglioni, E.; Giardino, D.; et al. Generation of Three IPSC Lines (IAIi002, IAIi004, IAIi003) from Rubinstein-Taybi Syndrome 1 Patients Carrying CREBBP Non Sense c.4435G>T, p.(Gly1479*) and c.3474G>A, p.(Trp1158*) and Missense c.4627G>T, p.(Asp1543Tyr) Mutations. Stem Cell Res. 2019, 40, 101553. [Google Scholar] [CrossRef]
- Alari, V.; Russo, S.; Terragni, B.; Ajmone, P.F.; Sironi, A.; Catusi, I.; Calzari, L.; Concolino, D.; Marotta, R.; Milani, D.; et al. iPSC-Derived Neurons of CREBBP- and EP300 -Mutated Rubinstein-Taybi Syndrome Patients Show Morphological Alterations and Hypoexcitability. Stem Cell Res. 2018, 30, 130–140. [Google Scholar] [CrossRef]
- Alari, V.; Scalmani, P.; Ajmone, P.F.; Perego, S.; Avignone, S.; Catusi, I.; Lonati, P.A.; Borghi, M.O.; Finelli, P.; Terragni, B.; et al. Histone Deacetylase Inhibitors Ameliorate Morphological Defects and Hypoexcitability of IPSC-Neurons from Rubinstein-Taybi Patients. Int. J. Mol. Sci. 2021, 22, 5777. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pt | Age | Age at Diagnosis | Clinical Signs at Onset | Epilepsy Onset | D.R. | Motor Function | Speech | Communication | Behavioural Problems | S | GI Problems | GF | M | CSS |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A1 | 7 years | 3 years | Regression, hand stereotypies (24 months) | 5 years | Yes | Independent walking | Absent | Maintained through eye contact | Absent | No | No | Yes | Yes | 20 |
| A2 | 10 years | 1 year | Developmental delay, hand stereotypies (12 months) | 24 months | Yes | Never acquired deambulation | Absent | Absent | Yes | Yes | Mild | No | Yes | 33 |
| B1 | 19 years | 5 years | Regression, absent speech, hand stereotypies (30 months) | 24 months | No | Independent walking | Single words | Maintained through eye contact | Absent | Mild | Mild | Yes | Yes | 17 |
| B2 | 45 years | 30 years | Regression, autistic features, hand stereotypies (18 months) | 42 months | No | Independent walking | Absent | Maintained through eye contact | Absent | Mild | Mild | No | NA | 18 |
| B3 | 22 years | 3 years | Regression, hand stereotypies (18 months) | 18 months | No | Independent walking | Absent | Maintained through eye contact | Absent | Mild | No | Yes | Yes | 17 |
| Y | dead | 11 months | Decreased fetal movements and fetal growth restriction, oculogyric crisis and generalized hypotonia (prenatal-neonatal) | - | No | NA | NA | NA | NA | NA | Yes (constipation) | Yes | Yes |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Perego, S.; Alari, V.; Pietra, G.; Lamperti, A.; Vimercati, A.; Camporeale, N.; Garzo, M.; Cogliati, F.; Milani, D.; Vignoli, A.; et al. Modeling RTT Syndrome by iPSC-Derived Neurons from Male and Female Patients with Heterogeneously Severe Hot-Spot MECP2 Variants. Int. J. Mol. Sci. 2022, 23, 14491. https://doi.org/10.3390/ijms232214491
Perego S, Alari V, Pietra G, Lamperti A, Vimercati A, Camporeale N, Garzo M, Cogliati F, Milani D, Vignoli A, et al. Modeling RTT Syndrome by iPSC-Derived Neurons from Male and Female Patients with Heterogeneously Severe Hot-Spot MECP2 Variants. International Journal of Molecular Sciences. 2022; 23(22):14491. https://doi.org/10.3390/ijms232214491
Chicago/Turabian StylePerego, Sara, Valentina Alari, Gianluca Pietra, Andrea Lamperti, Alessandro Vimercati, Nicole Camporeale, Maria Garzo, Francesca Cogliati, Donatella Milani, Aglaia Vignoli, and et al. 2022. "Modeling RTT Syndrome by iPSC-Derived Neurons from Male and Female Patients with Heterogeneously Severe Hot-Spot MECP2 Variants" International Journal of Molecular Sciences 23, no. 22: 14491. https://doi.org/10.3390/ijms232214491
APA StylePerego, S., Alari, V., Pietra, G., Lamperti, A., Vimercati, A., Camporeale, N., Garzo, M., Cogliati, F., Milani, D., Vignoli, A., Peron, A., Larizza, L., Pizzorusso, T., & Russo, S. (2022). Modeling RTT Syndrome by iPSC-Derived Neurons from Male and Female Patients with Heterogeneously Severe Hot-Spot MECP2 Variants. International Journal of Molecular Sciences, 23(22), 14491. https://doi.org/10.3390/ijms232214491