
Novel p.G250A Mutation Associated with Chronic Pancreatitis Highlights Misfolding-Prone Region in Carboxypeptidase A1 (CPA1)

 , , , , and
, , , , and
Abstract
:1. Introduction
2. Results
2.1. A Novel CPA1 Mutation in an Early-Onset Case of CP
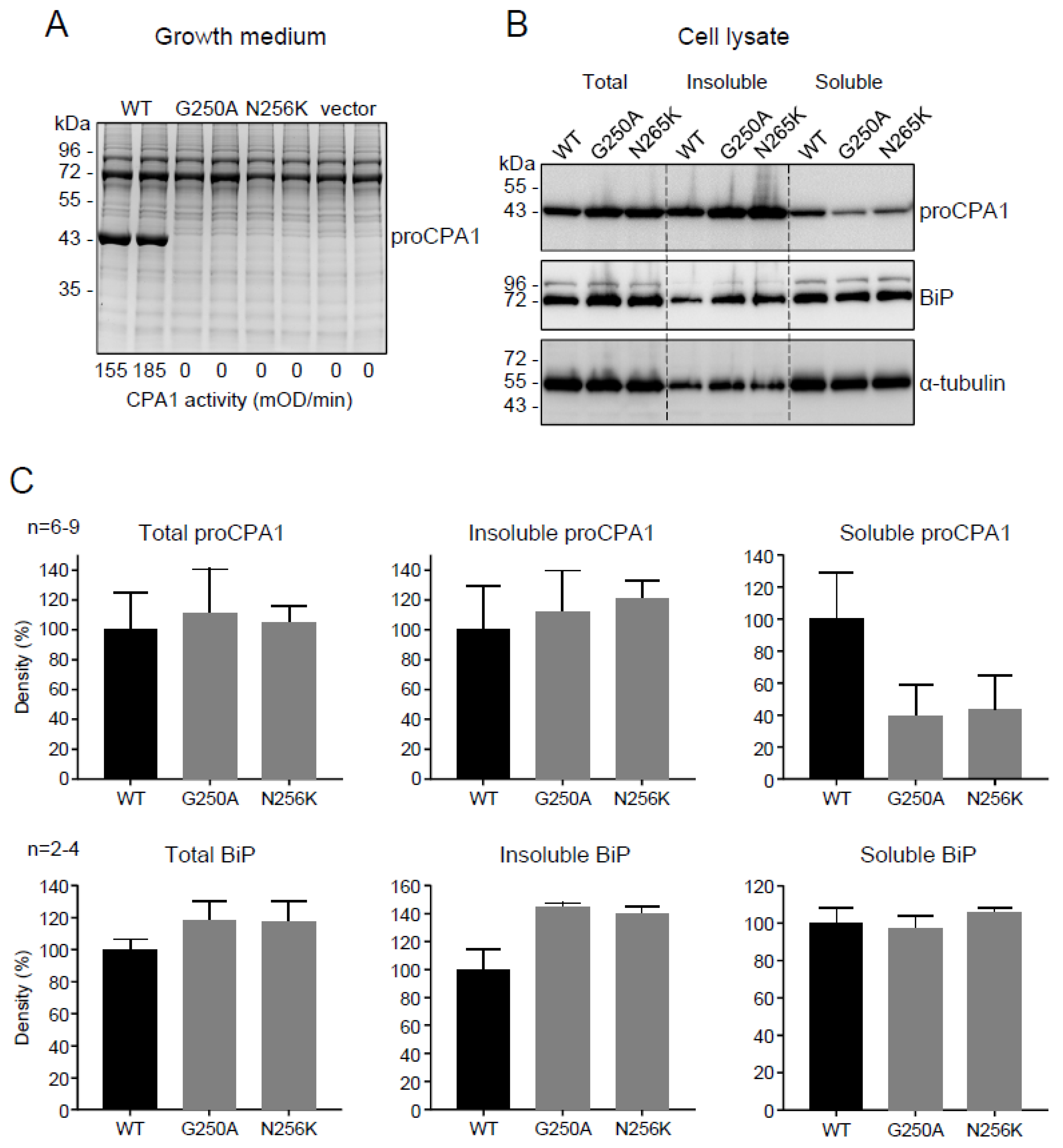
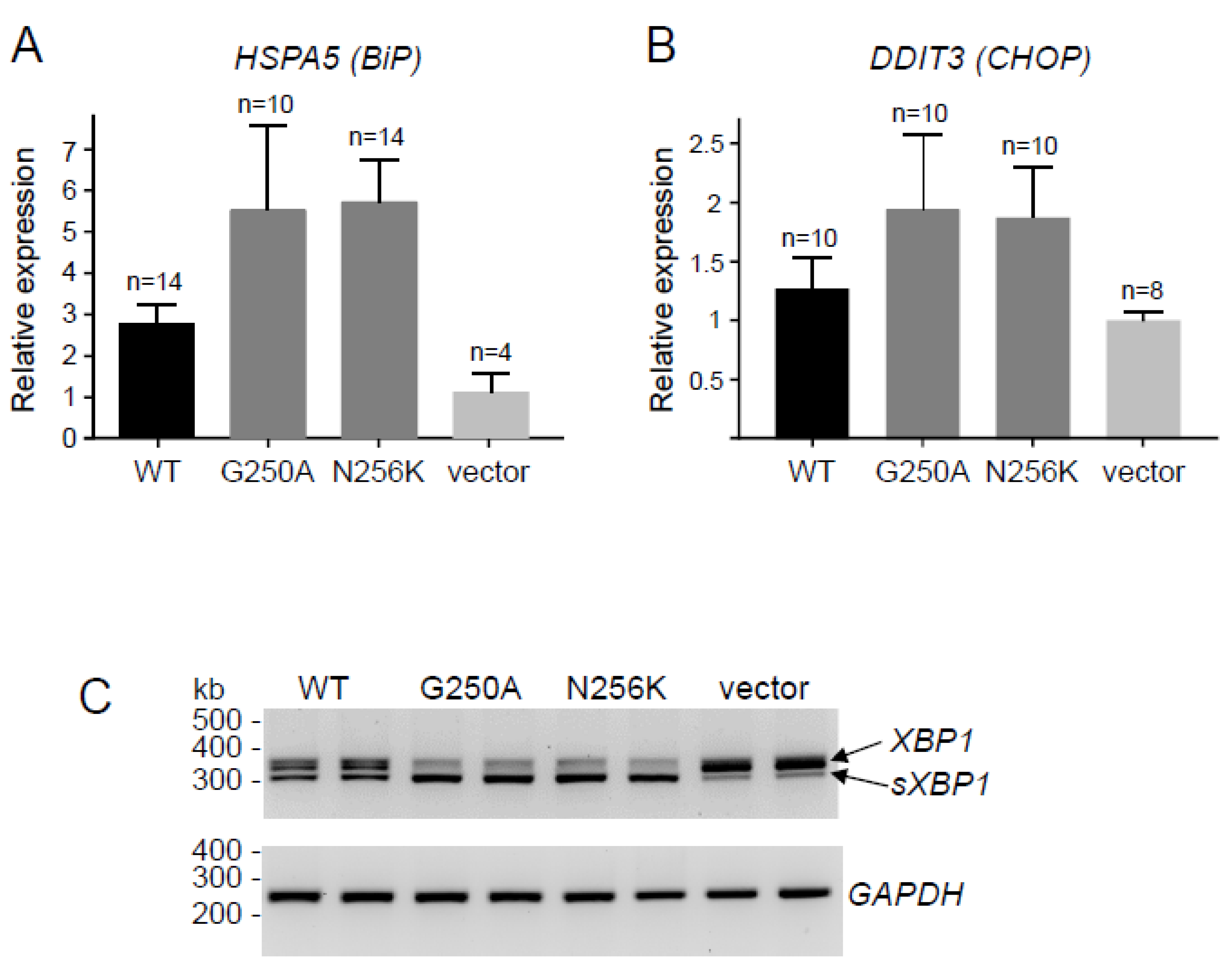
2.2. Functional Analysis of the p.G250A Mutation
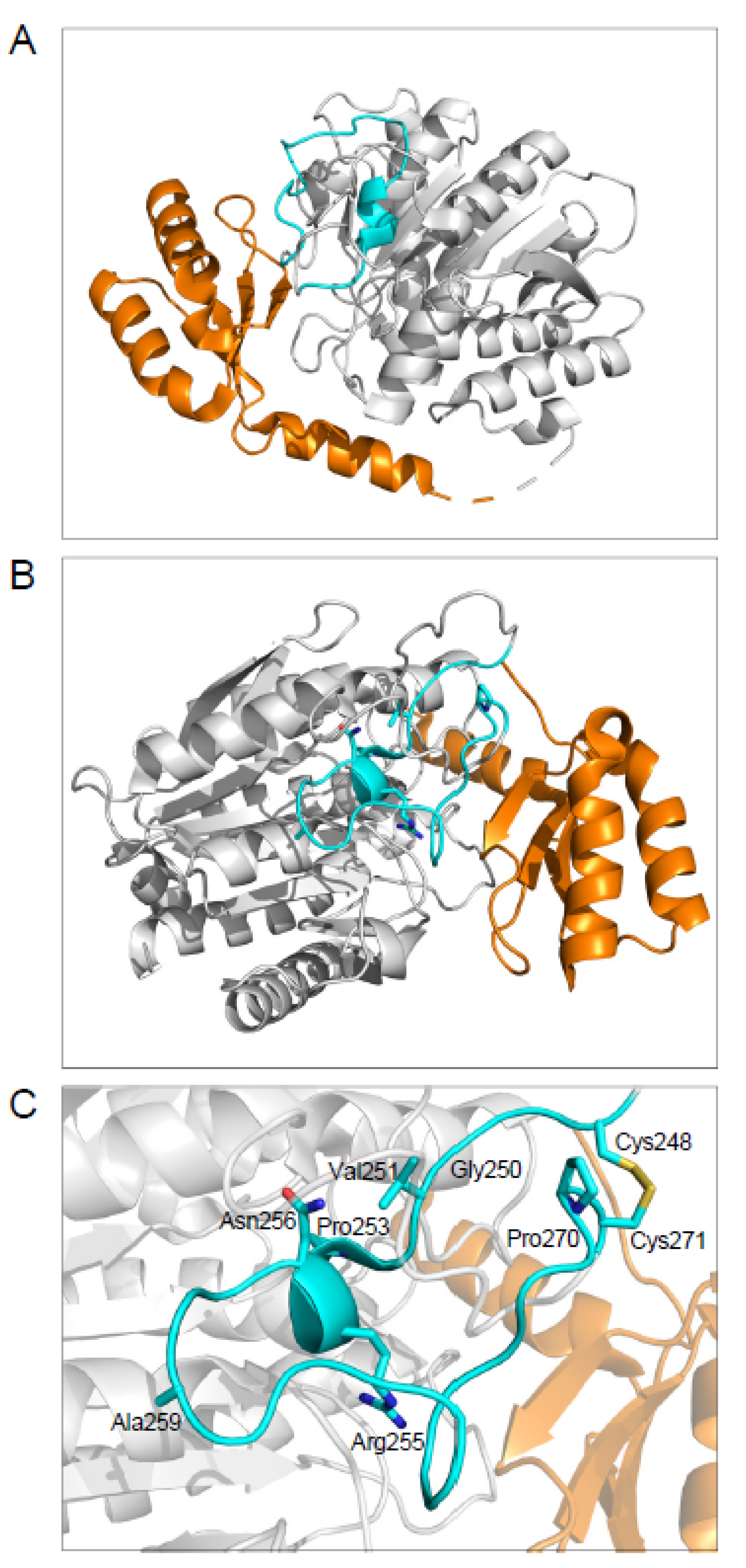
2.3. The Disulfide-Stabilized Loop in CPA1 Is Essential for Folding
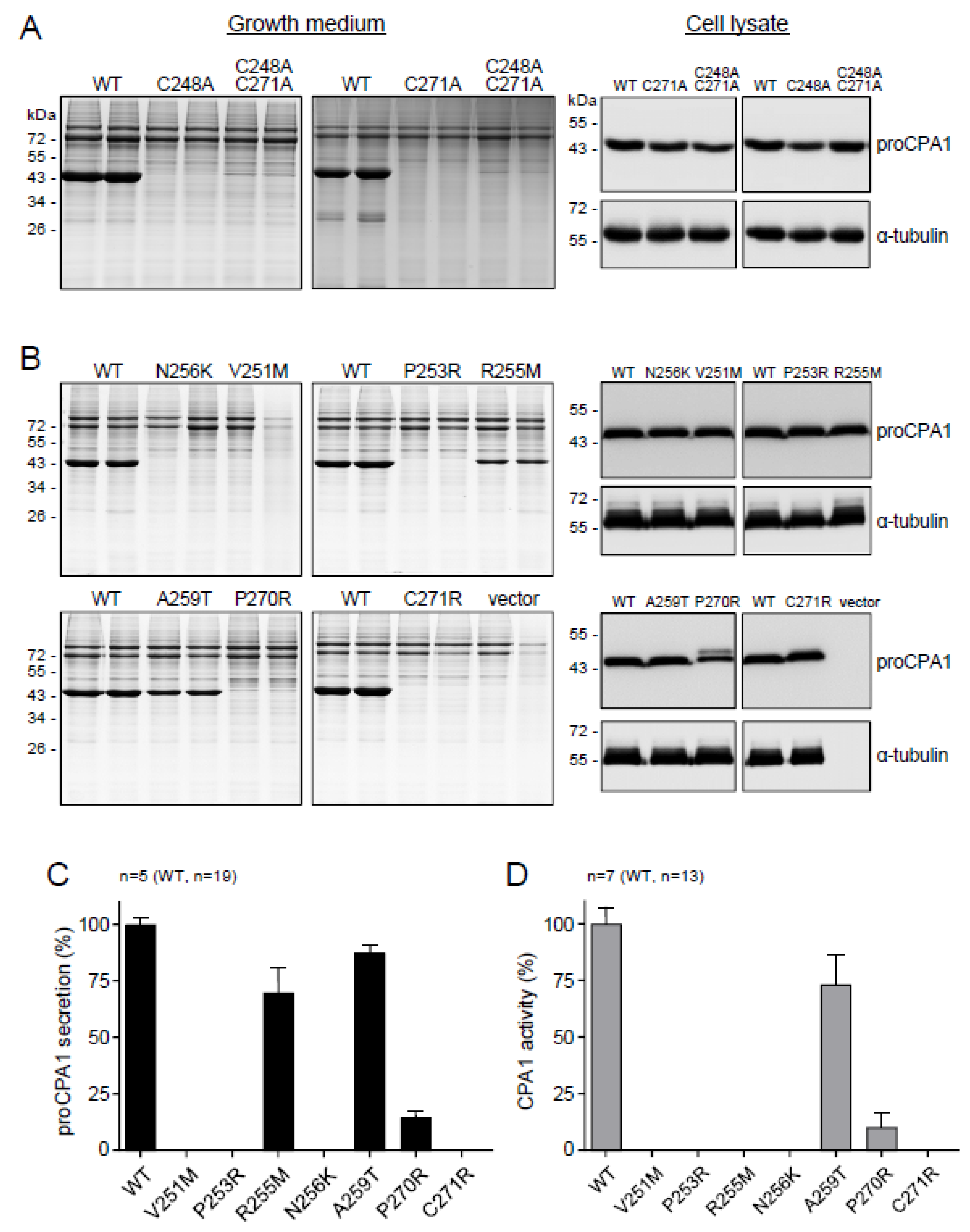
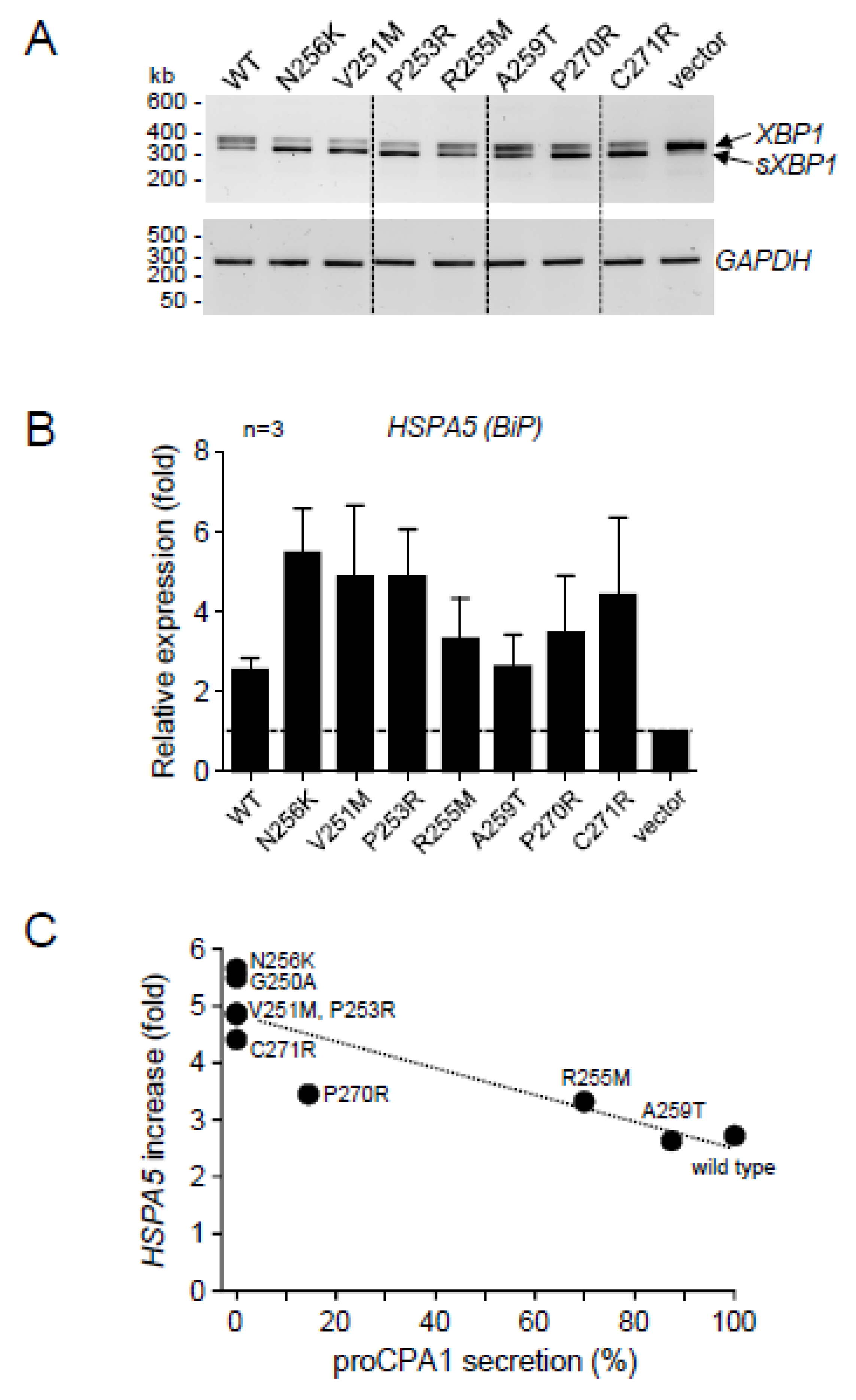
2.4. CPA1 Mutations within the Disulfide-Stabilized Loop
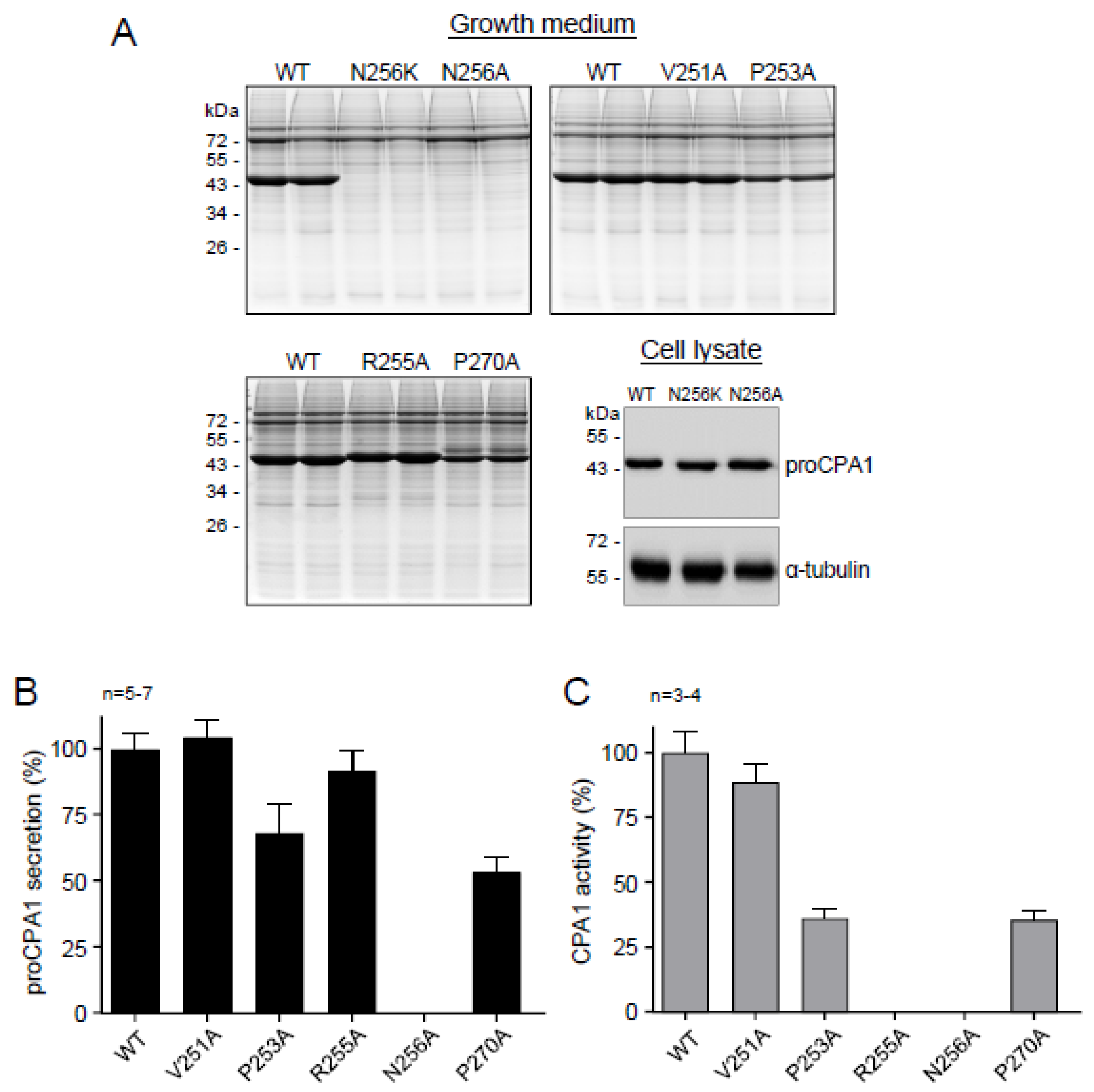
2.5. Ala Replacements Rescue Most CPA1 Mutants
3. Discussion
4. Methods
4.1. Accession Numbers and Nomenclature
4.2. Genetic Testing
4.3. Structural Modeling
4.4. Plasmid Construction and Mutagenesis
4.5. Cell Culture and Transfection
4.6. Measurement of Procarboxypeptidase Secretion
4.7. Measurement of Carboxypeptidase Activity
4.8. Preparation of Cell Lysates and Fractionation
4.9. Western Blotting
4.10. Reverse-Transcription (RT) Quantitative PCR (qPCR) to Measure HSPA5 (BiP) and DDIT3 (CHOP) mRNA
4.11. Measurement of X-Box Binding Protein 1 (XBP1) mRNA Splicing
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pascual, R.; Burgos, F.J.; Salva, M.; Soriano, F.; Mendez, E.; Aviles, F.X. Purification and properties of five different forms of human procarboxypeptidases. Eur. J. Biochem. 1989, 179, 609–616. [Google Scholar] [CrossRef] [PubMed]
- Vendrell, J.; Guasch, A.; Coll, M.; Villegas, V.; Billeter, M.; Wider, G.; Huber, R.; Wüthrich, K.; Avilés, F.X. Pancreatic procarboxypeptidases: Their activation processes related to the structural features of the zymogens and activation segments. Biol. Chem. Hoppe. Seyler. 1992, 373, 387–392. [Google Scholar] [CrossRef] [PubMed]
- Avilés, F.X.; Vendrell, J.; Guasch, A.; Coll, M.; Huber, R. Advances in metallo-procarboxypeptidases. Emerging details on the inhibition mechanism and on the activation process. Eur. J. Biochem. 1993, 211, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Ventura, S.; Gomis-Rüth, F.X.; Puigserver, A.; Avilés, F.X.; Vendrell, J. Pancreatic procarboxypeptidases: Oligomeric structures and activation processes revisited. Biol. Chem. 1997, 378, 161–165. [Google Scholar]
- Vendrell, J.; Querol, E.; Avilés, F.X. Metallocarboxypeptidases and their protein inhibitors. Structure, function and biomedical properties. Biochim. Biophys. Acta 2000, 1477, 284–298. [Google Scholar] [CrossRef]
- Scheele, G.; Bartelt, D.; Bieger, W. Characterization of human exocrine pancreatic proteins by two-dimensional isoelectric focusing/sodium dodecyl sulfate gel electrophoresis. Gastroenterology 1981, 80, 461–473. [Google Scholar] [CrossRef]
- Szmola, R.; Bence, M.; Carpentieri, A.; Szabó, A.; Costello, C.E.; Samuelson, J.; Sahin-Tóth, M. Chymotrypsin C is a co-activator of human pancreatic procarboxypeptidases A1 and A2. J. Biol. Chem. 2011, 286, 1819–1827. [Google Scholar] [CrossRef] [Green Version]
- Moulard, M.; Kerfelec, B.; Mallet, B.; Chapus, C. Identification of a procarboxypeptidase A-truncated protease E binary complex in human pancreatic juice. FEBS Lett. 1989, 250, 166–170. [Google Scholar] [CrossRef] [Green Version]
- Moulard, M.; Michon, T.; Kerfelec, B.; Chapus, C. Further studies on the human pancreatic binary complexes involving procarboxypeptidase A. FEBS Lett. 1990, 261, 179–183. [Google Scholar] [CrossRef] [Green Version]
- Szabó, A.; Pilsak, C.; Bence, M.; Witt, H.; Sahin-Tóth, M. Complex formation of human proelastases with procarboxypeptidases A1 and A2. J. Biol. Chem. 2016, 291, 17706–17716. [Google Scholar] [CrossRef] [Green Version]
- Párniczky, A.; Hegyi, E.; Tóth, A.Z.; Szücs, Á.; Szentesi, A.; Vincze, Á.; Izbéki, F.; Németh, B.C.; Hegyi, P.; Sahin-Tóth, M. Genetic analysis of human chymotrypsin-like elastases 3A and 3B (CELA3A and CELA3B) to assess the role of complex formation between proelastases and procarboxypeptidases in chronic pancreatitis. Int. J. Mol. Sci. 2016, 17, 2148. [Google Scholar] [CrossRef] [PubMed]
- Chapus, C.; Puigserver, A.; Kerfélec, B. The bovine pro-carboxypeptidase A-S6 ternary complex: A rare case of a secreted protein complex. Biochimie 1988, 70, 1143–1151. [Google Scholar] [CrossRef] [PubMed]
- Witt, H.; Beer, S.; Rosendahl, J.; Chen, J.M.; Chandak, G.R.; Masamune, A.; Bence, M.; Szmola, R.; Oracz, G.; Macek, M., Jr.; et al. Variants in CPA1 are strongly associated with early onset chronic pancreatitis. Nat. Genet. 2013, 45, 1216–1220. [Google Scholar] [CrossRef] [Green Version]
- Kujko, A.A.; Berki, D.M.; Oracz, G.; Wejnarska, K.; Antoniuk, J.; Wertheim-Tysarowska, K.; Kołodziejczyk, E.; Bal, J.; Sahin-Tóth, M.; Rygiel, A.M. A novel p.Ser282Pro CPA1 variant is associated with autosomal dominant hereditary pancreatitis. Gut 2017, 66, 1728–1730. [Google Scholar] [CrossRef]
- Németh, B.C.; Orekhova, A.; Zhang, W.; Nortman, S.A.; Thompson, T.; Hegyi, P.; Abu-El-Haija, M. Novel p.K374E variant of CPA1 causes misfolding-induced hereditary pancreatitis with autosomal dominant inheritance. Gut 2020, 69, 790–792. [Google Scholar] [CrossRef] [PubMed]
- Nakano, E.; Geisz, A.; Masamune, A.; Niihori, T.; Hamada, S.; Kume, K.; Kakuta, Y.; Aoki, Y.; Matsubara, Y.; Ebert, K.; et al. Variants in pancreatic carboxypeptidase genes CPA2 and CPB1 are not associated with chronic pancreatitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 309, G688–G694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamura, K.; Yu, J.; Hata, T.; Suenaga, M.; Shindo, K.; Abe, T.; MacGregor-Das, A.; Borges, M.; Wolfgang, C.L.; Weiss, M.J.; et al. Mutations in the pancreatic secretory enzymes CPA1 and CPB1 are associated with pancreatic cancer. Proc. Natl. Acad. Sci. USA 2018, 115, 4767–4772. [Google Scholar] [CrossRef] [Green Version]
- Kawamoto, M.; Kohi, S.; Abe, T.; Dbouk, M.; Macgregor-Das, A.; Koi, C.; Song, K.; Borges, M.; Sugimine, R.; Laheru, D.; et al. Endoplasmic stress-inducing variants in CPB1 and CPA1 and risk of pancreatic cancer; a case-control study and meta-analysis. Int. J. Cancer 2022, 150, 1123–1133. [Google Scholar] [CrossRef]
- Hegyi, E.; Sahin-Tóth, M. Human CPA1 mutation causes digestive enzyme misfolding and chronic pancreatitis in mice. Gut 2019, 68, 301–312. [Google Scholar] [CrossRef]
- Wu, H.; Zhou, D.Z.; Berki, D.; Geisz, A.; Zou, W.B.; Sun, X.T.; Hu, L.H.; Zhao, Z.H.; Zhao, A.J.; He, L.; et al. No significant enrichment of rare functionally defective CPA1 variants in a large Chinese idiopathic chronic pancreatitis cohort. Hum. Mutat. 2017, 38, 959–963. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.-H.; Boulling, A.; Masson, E.; Cooper, D.N.; Li, Z.-S.; Férec, C.; Liao, Z.; Chen, J.-M. Most unambiguous loss-of-function CPA1 mutations are unlikely to predispose to chronic pancreatitis. Gut 2020, 69, 785–786. [Google Scholar] [CrossRef] [PubMed]
- Abe, T.; Blackford, A.L.; Tamura, K.; Ford, M.; McCormick, P.; Chuidian, M.; Almario, J.A.; Borges, M.; Lennon, A.M.; Shin, E.J.; et al. Deleterious Germline Mutations Are a Risk Factor for Neoplastic Progression Among High-Risk Individuals Undergoing Pancreatic Surveillance. J. Clin. Oncol. 2019, 37, 1070–1080. [Google Scholar] [CrossRef] [PubMed]
- Sahin-Tóth, M. Genetic risk in chronic pancreatitis: The misfolding-dependent pathway. Curr. Opin. Gastroenterol. 2017, 33, 390–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szmola, R.; Sahin-Tóth, M. Pancreatitis-associated chymotrypsinogen C (CTRC) mutant elicits endoplasmic reticulum stress in pancreatic acinar cells. Gut 2010, 59, 365–372. [Google Scholar] [CrossRef]
- Beer, S.; Zhou, J.; Szabó, A.; Keiles, S.; Chandak, G.R.; Witt, H.; Sahin-Tóth, M. Comprehensive functional analysis of chymotrypsin C (CTRC) variants reveals distinct loss-of-function mechanisms associated with pancreatitis risk. Gut 2013, 62, 1616–1624. [Google Scholar] [CrossRef] [Green Version]
- Kereszturi, E.; Szmola, R.; Kukor, Z.; Simon, P.; Weiss, F.U.; Lerch, M.M.; Sahin-Tóth, M. Hereditary pancreatitis caused by mutation-induced misfolding of human cationic trypsinogen: A novel disease mechanism. Hum. Mutat. 2009, 30, 575–582. [Google Scholar] [CrossRef] [Green Version]
- Balázs, A.; Hegyi, P.; Sahin-Tóth, M. Pathogenic cellular role of the p.L104P human cationic trypsinogen variant in chronic pancreatitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2016, 310, G477–G486. [Google Scholar] [CrossRef] [Green Version]
- Thiel, F.; Reiser, M.; Weiss, F.U. A rare PRSS1 p.S127C mutation is associated with chronic pancreatitis and causes misfolding-induced ER-stress. Pancreatology 2022. Epub ahead of print. [Google Scholar] [CrossRef]
- Szabó, A.; Xiao, X.; Haughney, M.; Spector, A.; Sahin-Tóth, M.; Lowe, M.E. A novel mutation in PNLIP causes pancreatic triglyceride lipase deficiency through protein misfolding. Biochim. Biophys. Acta 2015, 1852, 1372–1379. [Google Scholar] [CrossRef] [Green Version]
- Toldi, V.; Kassay, N.; Szabó, A. Missense PNLIP mutations impeding pancreatic lipase secretion cause protein misfolding and endoplasmic reticulum stress. Pancreatology 2021, 21, 1317–1325. [Google Scholar] [CrossRef]
- Xiao, X.; Jones, G.; Sevilla, W.A.; Stolz, D.B.; Magee, K.E.; Haughney, M.; Mukherjee, A.; Wang, Y.; Lowe, M.E. A Carboxyl Ester Lipase (CEL) Mutant Causes Chronic Pancreatitis by Forming Intracellular Aggregates That Activate Apoptosis. J. Biol. Chem. 2016, 291, 23224–23236. [Google Scholar] [CrossRef] [PubMed]
- Cassidy, B.M.; Zino, S.; Fjeld, K.; Molven, A.; Lowe, M.E.; Xiao, X. Single nucleotide polymorphisms in CEL-HYB1 increase risk for chronic pancreatitis through proteotoxic misfolding. Hum. Mutat. 2020, 41, 1967–1978. [Google Scholar] [CrossRef] [PubMed]
- Tjora, E.; Gravdal, A.; Engjom, T.; Cnop, M.; Johansson, B.B.; Dimcevski, G.G.; Molven, A.; Fjeld, K. Protein misfolding in combination with other risk factors in CEL-HYB1-mediated chronic pancreatitis. Eur. J. Gastroenterol. Hepatol. 2021, 33, 839–843. [Google Scholar] [CrossRef] [PubMed]
- Testero, S.A.; Granados, C.; Fernández, D.; Gallego, P.; Covaleda, G.; Reverter, D.; Vendrell, J.; Avilés, F.X.; Pallarès, I.; Mobashery, S. Discovery of Mechanism-Based Inactivators for Human Pancreatic Carboxypeptidase A from a Focused Synthetic Library. ACS Med. Chem. Lett. 2017, 8, 1122–1127. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.-T.; Zou, W.-B.; Cao, Y.; Wang, Y.-C.; Deng, S.-J.; Cooper, D.N.; Férec, C.; Li, Z.-S.; Chen, J.-M.; Liao, Z. The CEL-HYB1 Hybrid Allele Promotes Digestive Enzyme Misfolding and Pancreatitis in Mice. Cell. Mol. Gastroenterol. Hepatol. 2022, 14, 55–74. [Google Scholar] [CrossRef]
- Orekhova, A.; Geisz, A.; Sahin-Tóth, M. Ethanol feeding accelerates pancreatitis progression in CPA1 N256K mutant mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2020, 318, G694–G704. [Google Scholar] [CrossRef]
- Németh, B.C.; Demcsák, A.; Geisz, A.; Sahin-Tóth, M. Misfolding-induced chronic pancreatitis in CPA1 N256K mutant mice is unaffected by global deletion of Ddit3/Chop. Sci. Rep. 2022, 12, 6357. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Exon | Nucleotide Change | Amino-Acid Change | Number of CP Carriers | Number of Non-CP Carriers | Gnomad Allele Frequency |
|---|---|---|---|---|---|
| exon 7 | c.749G>C | p.G250A | 1 | 0 | absent |
| exon 7 | c.751G>A | p.V251M | 7 | 0 | 6/251,446 |
| exon 7 | c.758C>G | p.P253R | 1 | 0 | absent |
| exon 7 | c.764G>T | p.R255M | 1 | 0 | absent |
| exon 7 | c.768C>G | p.N256K | 7 | 0 | absent |
| exon 7 | c.775G>A | p.A259T | 0 | 1 | 1/31,398 |
| exon 7 | c.776C>T | p.A259V | 0 | 1 | 3/282,782 |
| exon 8 | c.809C>G | p.P270R | 1 | 0 | absent |
| exon 8 | c.811T>C | p.C271R | 2 | 0 | absent |
| Mutation | Secretion (%) This Study | Secretion (%) Published | Activity (%) This Study | Activity (%) Published |
|---|---|---|---|---|
| p.G250A | 0 | -- | 0 | -- |
| p.V251M | 0 | 0 | 0 | 0 |
| p.P253R | 0 | 0 | 0 | 0 |
| p.R255M | 69.8 ± 10.9 | 86 | 0 | 0 |
| p.N256K | 0 | 0 | 0 | 0 |
| p.A259T | 87.4 ± 3.4 | 82 | 73.2 ± 13.3 | 85 |
| p.P270R | 14.4 ± 2.6 | 14 | 10.0 ± 6.3 | 9 |
| p.C271R | 0 | 0 | 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sándor, M.; Thiel, F.G.; Schmid, M.; Demcsák, A.; Morales Granda, N.C.; Németh, B.C.; Vajda, S.; Hoerning, A.; Sahin-Tóth, M. Novel p.G250A Mutation Associated with Chronic Pancreatitis Highlights Misfolding-Prone Region in Carboxypeptidase A1 (CPA1). Int. J. Mol. Sci. 2022, 23, 15463. https://doi.org/10.3390/ijms232415463
Sándor M, Thiel FG, Schmid M, Demcsák A, Morales Granda NC, Németh BC, Vajda S, Hoerning A, Sahin-Tóth M. Novel p.G250A Mutation Associated with Chronic Pancreatitis Highlights Misfolding-Prone Region in Carboxypeptidase A1 (CPA1). International Journal of Molecular Sciences. 2022; 23(24):15463. https://doi.org/10.3390/ijms232415463
Chicago/Turabian StyleSándor, Máté, Franziska G. Thiel, Margit Schmid, Alexandra Demcsák, Nataly C. Morales Granda, Balázs Csaba Németh, Sandor Vajda, André Hoerning, and Miklós Sahin-Tóth. 2022. "Novel p.G250A Mutation Associated with Chronic Pancreatitis Highlights Misfolding-Prone Region in Carboxypeptidase A1 (CPA1)" International Journal of Molecular Sciences 23, no. 24: 15463. https://doi.org/10.3390/ijms232415463
APA StyleSándor, M., Thiel, F. G., Schmid, M., Demcsák, A., Morales Granda, N. C., Németh, B. C., Vajda, S., Hoerning, A., & Sahin-Tóth, M. (2022). Novel p.G250A Mutation Associated with Chronic Pancreatitis Highlights Misfolding-Prone Region in Carboxypeptidase A1 (CPA1). International Journal of Molecular Sciences, 23(24), 15463. https://doi.org/10.3390/ijms232415463