
5′-Chalcogen-Substituted Nucleoside Pyrophosphate and Phosphate Monoester Analogues: Preparation and Hydrolysis Studies

 ,
,
Abstract
:1. Introduction
2. Results
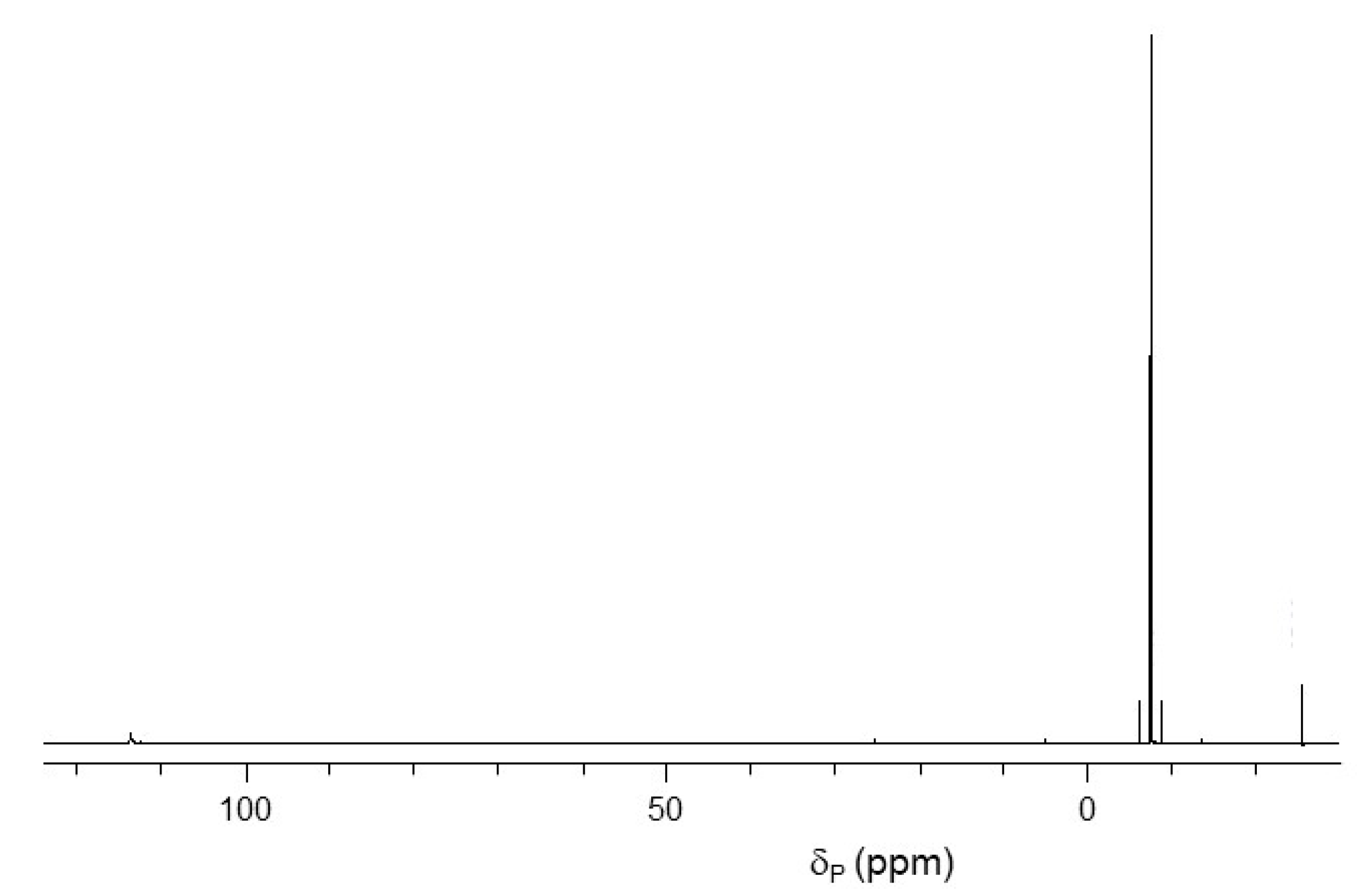
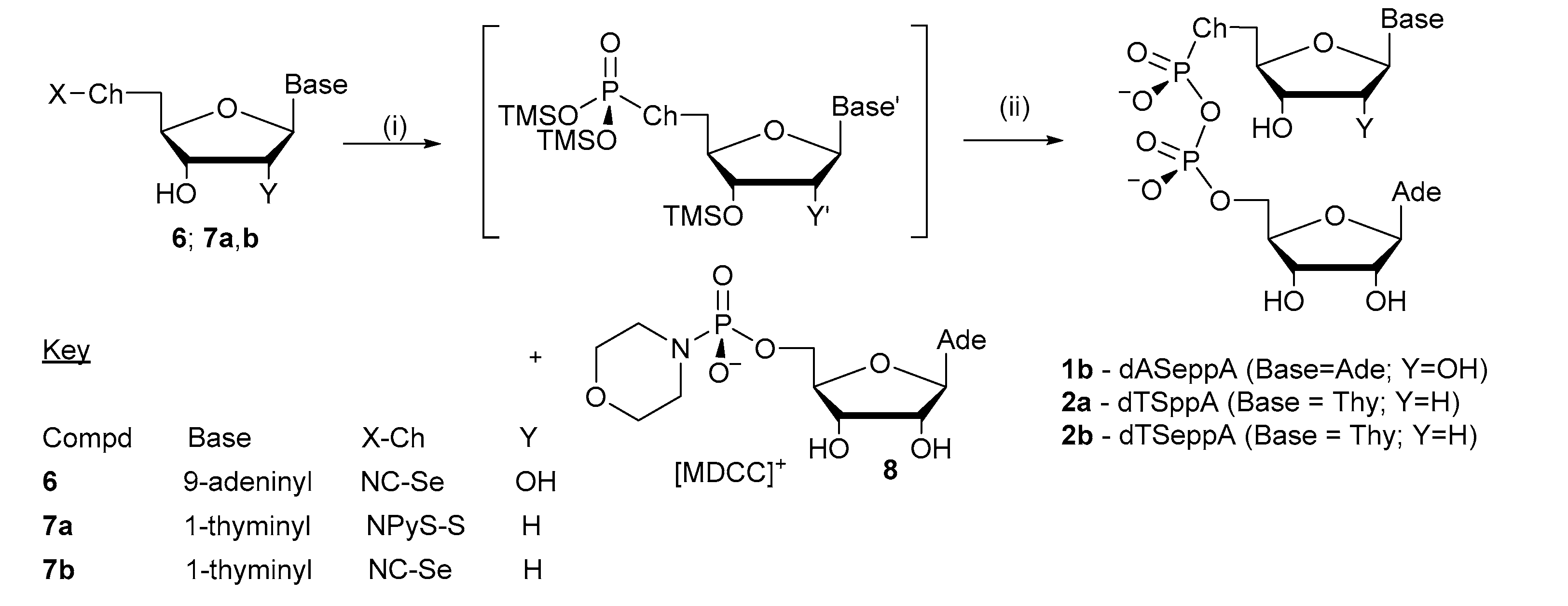
2.1. Synthesis of Pyrophosphorochalcogenolate-Linked Dinucleosides
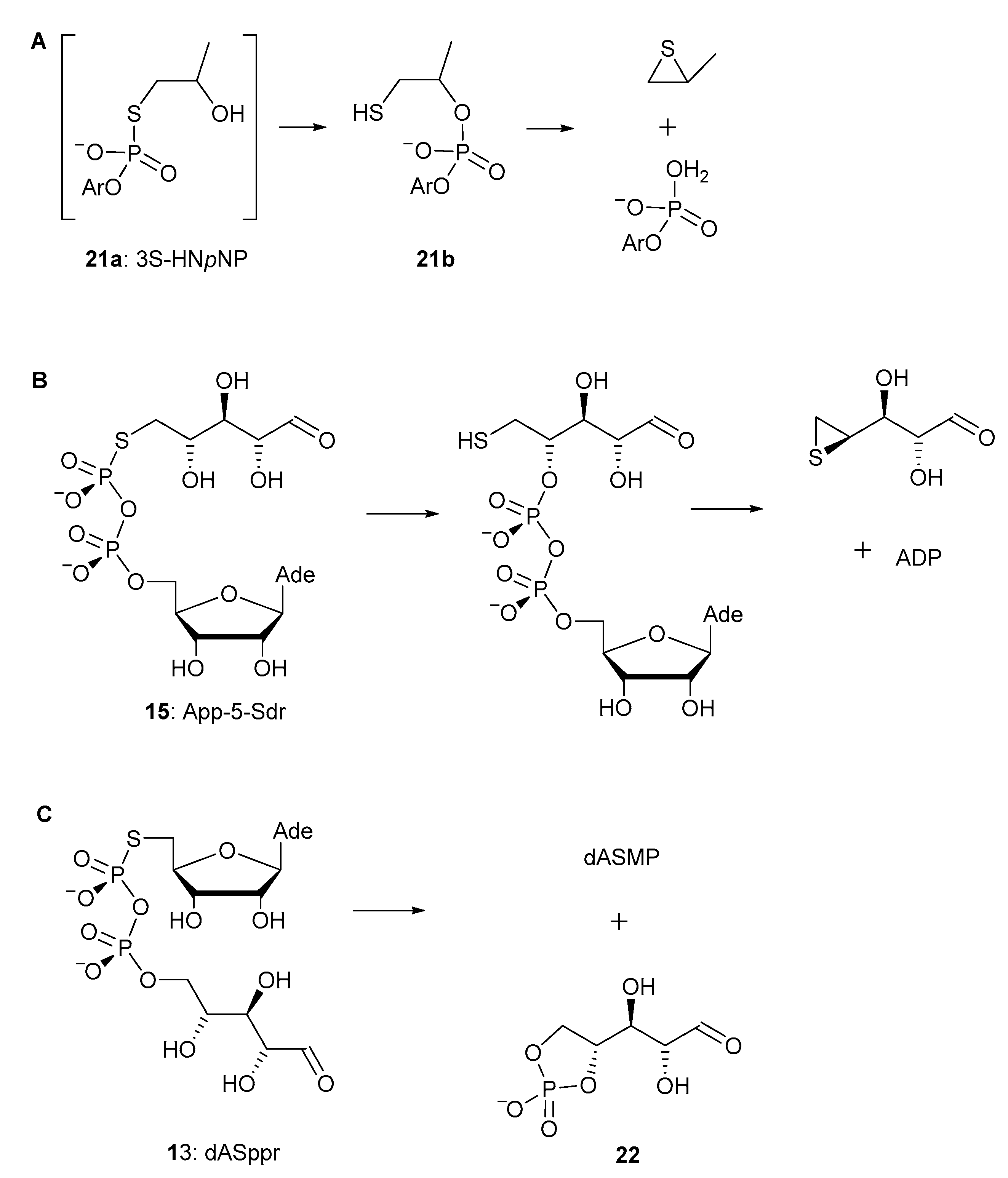
2.2. Stability of Pyrophorophorochalcogenate-Linked Dinucleosides
2.3. pKa Titrations and SVPDE Cleavage of Pyrophosphorochalcogenolate-Linked Dinucleosides
3. Discussion
4. Materials and Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Daniell, M.D.; Hill, J.S. A history of photodynamic therapy. ANZ J. Surg. 1991, 61, 340–348. [Google Scholar] [CrossRef] [PubMed]
- Drago, J.Z.; Modi, S.; Chandarlapaty, S. Unlocking the potential of antibody–drug conjugates for cancer therapy. Nat. Rev. Clin. Oncol. 2021, 18, 327–344. [Google Scholar] [CrossRef]
- Huang, Y.-F.; Shangguan, D.; Liu, H.; Phillips, J.A.; Zhang, X.; Chen, Y.; Tan, W. Molecular Assembly of an Aptamer–Drug Conjugate for Targeted Drug Delivery to Tumor Cells. ChemBioChem 2009, 10, 862–868. [Google Scholar] [CrossRef] [Green Version]
- Worm, D.J.; Els-Heindl, S.; Beck-Sickinger, A.G. Targeting of peptide-binding receptors on cancer cells with peptide-drug conjugates. Pept. Sci. 2020, 112, e24171. [Google Scholar] [CrossRef]
- Zolottsev, V.A.; Latysheva, A.S.; Pokrovsky, V.S.; Khan, I.I.; Misharin, A.Y. Promising applications of steroid conjugates for cancer research and treatment. Eur. J. Med. Chem. 2021, 210, 113089. [Google Scholar] [CrossRef]
- Low, P.S.; Kularatne, S.A. Folate-targeted therapeutic and imaging agents for cancer. Curr. Opin. Chem. Biol. 2009, 13, 256–262. [Google Scholar] [CrossRef] [PubMed]
- Makovitzki, A.; Fink, A.; Shai, Y. Suppression of human solid tumor growth in mice by intratumor and systemic inoculation of histidine-rich and pH-dependent host defense-like lytic peptides. Cancer Res. 2009, 69, 3458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, M.; Zhang, F.; Wei, T.; Zuo, T.; Guan, Y.; Lin, G.; Shao, W. Smart linkers in polymer–drug conjugates for tumor-targeted delivery. J. Drug Target. 2016, 24, 475–491. [Google Scholar] [CrossRef] [PubMed]
- Kern, J.C.; Cancilla, M.; Dooney, D.; Kwasnjuk, K.; Zhang, R.; Beaumont, M.; Figueroa, I.; Hsieh, S.; Liang, L.; Tomazela, D.; et al. Discovery of Pyrophosphate Diesters as Tunable, Soluble, and Bioorthogonal Linkers for Site-Specific Antibody–Drug Conjugates. J. Am. Chem. Soc. 2016, 138, 1430–1445. [Google Scholar] [CrossRef]
- Kern, J.C.; Dooney, D.; Zhang, R.; Liang, L.; Brandish, P.E.; Cheng, M.; Feng, G.; Beck, A.; Bresson, D.; Firdos, J.; et al. Novel Phosphate Modified Cathepsin B Linkers: Improving Aqueous Solubility and Enhancing Payload Scope of ADCs. Bioconj. Chem. 2016, 27, 2081–2088. [Google Scholar] [CrossRef]
- Dragovich, P.S.; Adhikari, P.; Blake, R.A.; Blaquiere, N.; Chen, J.; Cheng, Y.-X.; den Besten, W.; Han, J.; Hartman, S.J.; He, J.T.; et al. Antibody-mediated delivery of chimeric protein degraders which target estrogen receptor alpha (ERα). Bioorg. Med. Chem. Lett. 2020, 30, 126907. [Google Scholar] [CrossRef] [PubMed]
- Kouvaris, J.R.; Kouloulias, V.E.; Vlahos, L.J. Amifostine: The first selective-target and broad-spectrum radioprotector. Oncologist 2007, 12, 738–747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kunwar, A.; Priyadarsini, K.I. Therapeutic applications of organoselenium compounds. Chapter 12 history and development of selenium-based radioprotectors: Distinctions between the inorganic and organic forms. In Organoselenium Compounds in Biology and Medicine: Synthesis, Biological and Therapeutic Treatments; The Royal Society of Chemistry: Cambridge, UK, 2018; pp. 317–341. [Google Scholar]
- Eguaogie, O.; Cooke, L.A.; Martin, P.M.L.; Ravalico, F.; Conway, L.P.; Hodgson, D.R.W.; Law, C.J.; Vyle, J.S. Synthesis of novel pyrophosphorothiolate-linked dinucleoside cap analogues in a ball mill. Org. Biomol. Chem. 2016, 14, 1201–1205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eguaogie, O.; Vyle, J.S. Vibration ball milling for the synthesis of 5′-Thioadenosine 5′-Pyrophosphate (P′→5′) adenosine (dASppA). In Current Protocols in Nucleic Acid Chemistry; John Wiley & Sons, Inc.: New York, NY, USA, 2017. [Google Scholar]
- Wojtczak, B.A.; Sikorski, P.J.; Fac-Dabrowska, K.; Nowicka, A.; Warminski, M.; Kubacka, D.; Nowak, E.; Nowotny, M.; Kowalska, J.; Jemielity, J. 5′-Phosphorothiolate Dinucleotide Cap Analogues: Reagents for Messenger RNA Modification and Potent Small-Molecular Inhibitors of Decapping Enzymes. J. Am. Chem. Soc. 2018, 140, 5987–5999. [Google Scholar] [CrossRef] [PubMed]
- Brear, P.; Freeman, G.R.; Shankey, M.C.; Trmčić, M.; Hodgson, D.R.W. Aqueous methods for the preparation of 5 ’-substituted guanosine derivatives. Chem. Commun. 2009, 4980–4981. [Google Scholar] [CrossRef] [PubMed]
- Wieland, T.; Lambert, R. Synthese und Eigenschaften des Barium-S-n-butylthiophosphates. Chem. Ber. 1956, 89, 2476–2482. [Google Scholar] [CrossRef]
- Lee, S.-Y.; Müller, C.E. Nucleotide pyrophosphatase/phosphodiesterase 1 (NPP1) and its inhibitors. MedChemComm 2017, 8, 823–840. [Google Scholar] [CrossRef] [PubMed]
- Kayrouz, C.M.; Huang, J.; Hauser, N.; Seyedsayamdost, M.R. Biosynthesis of selenium-containing small molecules in diverse microorganisms. Nature 2022, 610, 199–204. [Google Scholar] [CrossRef]
- Sierant, M.; Leszczynska, G.; Sadowska, K.; Komar, P.; Radzikowska-Cieciura, E.; Sochacka, E.; Nawrot, B. Escherichia coli tRNA 2-selenouridine synthase (SelU) converts S2U-RNA to Se2U-RNA via S-geranylated-intermediate. FEBS Lett. 2018, 592, 2248–2258. [Google Scholar] [CrossRef] [Green Version]
- Lipka, P.; Michalska, M. Efficient synthesis of S- and Se-(2,3,4,6-tetra-O-acetyl-β-d-glucosyl) thiophosphates and selenophosphates. Carbohydr. Res. 1983, 113, 317–320. [Google Scholar] [CrossRef]
- Borecka, B.; Chojnowski, J.; Cypryk, M.; Michalski, J.; Zielinska, J. Synthetic and mechanistic aspects of the reaction of trialkylsilyl halides with thio and seleno esters of phosphorus. J. Organomet. Chem. 1979, 171, 17–34. [Google Scholar] [CrossRef]
- Glass, R.S.; Singh, W.P.; Jung, W.; Veres, Z.; Scholz, T.D.; Stadtman, T. Monoselenophosphate: Synthesis, characterization, and identity with the prokaryotic biological selenium donor, compound SePX. Biochemistry 1993, 32, 12555–12559. [Google Scholar] [CrossRef]
- Ora, M.; Hanski, A. Stepwise mechanism of hydroxide ion catalyzed cyclization of uridine 3’-thiophosphates. Helv. Chim. Acta 2011, 94, 1563–1574. [Google Scholar] [CrossRef]
- Ora, M.; Järvi, J.; Oivanen, M.; Lönnberg, H. Hydrolytic reactions of the phosphorodithioate analogue of uridylyl(3’,5’)uridine: Kinetics and mechanisms for the cleavage, desulfurization, and isomerization of the internucleosidic linkage. J. Org. Chem. 2000, 65, 2651–2657. [Google Scholar] [CrossRef]
- Liu, X.; Reese, C.B. Uridylyl-(3′→5′)-(5′-thiouridine). An exceptionally base-labile di-ribonucleoside phosphate analogue. Tetrahedron Lett. 1995, 36, 3413–3416. [Google Scholar] [CrossRef]
- Thomson, J.B.; Patel, B.K.; Jimenez, V.; Eckart, K.; Eckstein, F. Synthesis and properties of diuridine phosphate analogues containing thio and amino modifications. J. Org. Chem. 1996, 61, 6273–6281. [Google Scholar] [CrossRef] [PubMed]
- Liu, X. Reese, CB 3’-thiouridyl-(3’,5’)-uridine. Tetrahedron Lett. 1996, 37, 925–928. [Google Scholar] [CrossRef]
- Weinstein, L.B.; Earnshaw, D.J.; Cosstick, R.; Cech, T.R. Synthesis and characterization of an RNA dinucleotide containing a 3’-S-Phosphorothiolate linkage. J. Am. Chem. Soc. 1996, 118, 10341–10350. [Google Scholar] [CrossRef]
- Elzagheid, M.I.; Oivanen, M.; Klika, K.D.; Jones, B.C.N.M.; Cosstick, R.; Lönnberg, H. Hydrolytic reactions of 3 ’-deoxy-3 ’-thioinosylyl-(3’->5’)uridine; An RNA dinucleotide containing a 3’-S-phosphorothiolate linkage. Nucleosides Nucleotides Nucleic Acids 1999, 18, 2093–2108. [Google Scholar] [CrossRef]
- Eguaogie, O.; Conlon, P.F.; Ravalico, F.; Sweet, J.S.T.; Elder, T.B.; Conway, L.P.; Lennon, M.E.; Hodgson, D.R.W.; Vyle, J.S. Nucleophilic displacement reactions of 5′-derivatised nucleosides in a vibration ball mill. Beilstein J. Org. Chem. 2017, 13, 87–92. [Google Scholar] [CrossRef]
- Mikkola, S. Hydrolytic reactions of diadenosine 5’,5’-triphosphate. Org. Biomol. Chem. 2004, 2, 770–776. [Google Scholar] [CrossRef]
- Kirby, A.J.; Varvoglis, A.G. The Reactivity of Phosphate Esters. Monoester Hydrolysis. J. Am. Chem. Soc. 1967, 89, 415–423. [Google Scholar] [CrossRef]
- Duarte, F.; Barrozo, A.; Åqvist, J.; Williams, N.H.; Kamerlin, S.C.L. The Competing Mechanisms of Phosphate Monoester Dianion Hydrolysis. J. Am. Chem. Soc. 2016, 138, 10664–10673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iyer, S.; Hengge, A.C. The effects of sulfur substitution for the nucleophile and bridging oxygen atoms in reactions of hydroxyalkyl phosphate esters. J. Org. Chem. 2008, 73, 4819–4829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, T.M.; Mallette, M.F.; Mumma, R.O. Isolation and Characterization of 5’-S-methyl-5’-thioadenosine. Biochemistry 1968, 7, 1399–1406. [Google Scholar] [CrossRef] [PubMed]
- Huhta, E.; Parjanen, A.; Mikkola, S. A kinetic study on the chemical cleavage of nucleoside diphosphate sugars. Carbohydr. Res. 2010, 345, 696–703. [Google Scholar] [CrossRef]
- Jaakkola, J.; Nieminen, A.; Kivelä, H.; Korhonen, H.; Tähtinen, P.; Mikkola, S. Kinetic and NMR spectroscopic study on the chemical stability and reactions pathways of sugar nucleotides. Nucleoides Nucleotides Nucleic Acids 2021, 40, 178–193. [Google Scholar] [CrossRef]
- Miller, D.L.; Westheimer, F.H. Hydrolysis of γ-phenylpropyl di- and triphosphates. J. Am. Chem. Soc. 1966, 88, 1507–1511. [Google Scholar] [CrossRef]
- Ora, M.; Murtola, M. Hydrolytic reactions of 3’-N-phosphoramidate and 3’-N-thiophosphoramidate analogs of thymidylyl-3’,5’-thymidine. Org. Biomol. Chem. 2004, 2, 593–600. [Google Scholar] [CrossRef]
- Smith, M.; Drummond, G.I.; Khorana, H.G. Cyclic phosphates. IV Ribonucleoside 3’,5’-cyclic phosphates. A general method of synthesis and some properties. J. Am. Chem. Soc. 1961, 83, 698–706. [Google Scholar] [CrossRef]
- Lehikoinen, P.; Mattinen, J.; Lönnberg, H. Mechanism for the solvolytic decompositions of nucleoside analogs. B. Reactions of adenine nucleosides with aqueous alkalis—Kinetics and mechanisms. J. Org. Chem. 1986, 20, 3819–3823. [Google Scholar] [CrossRef]
- Reich, H.J.; Hondal, R.J. Why nature chose selenium. ACS Chem. Biol. 2016, 11, 821–841. [Google Scholar] [CrossRef]
- Rizvi, M.A.; Zaki, M.; Afzal, M.; Mane, M.; Kumar, M.; Shah, B.A.; Srivastav, S.; Srikrishna, S.; Peerzada, G.M.; Tabassum, S. Nuclear blebbing of biologically active organoselenium compound towards human cervical cancer cell (HeLa): In vitro DNA/HSA binding, cleavage and cell imaging studies. Eur. J. Med. Chem. 2015, 90, 876–888. [Google Scholar] [CrossRef]
- Hodgson, D.R.W. Chapter five-physicochemical aspects of aqueous and nonaqueous approaches to the preparation of nucleosides, nucleotides and phosphate ester mimics. In Advances in Physical Organic Chemistry; Williams, I.H., Williams, N.H., Eds.; Academic Press: Cambridge, MA, USA, 2017; Volume 51, pp. 187–219. [Google Scholar]
- Stern, N.; Major, D.T.; Gottlieb, H.E.; Weizman, D.; Fischer, B. What is the conformation of physiologically-active dinucleoside polyphosphates in solution? Conformational analysis of free dinucleoside polyphosphates by NMR and molecular dynamics simulations. Org. Biomol. Chem. 2010, 8, 4637–4652. [Google Scholar] [CrossRef]
- Baranowski, M.R.; Nowicka, A.; Jemielity, J.; Kowalska, J. A fluorescent HTS assay for phosphohydrolases based on nucleoside 5′-fluorophosphates: Its application in screening for inhibitors of mRNA decapping scavenger and PDE-I. Org. Biomol. Chem. 2016, 14, 4595–4604. [Google Scholar] [CrossRef]
- Ullah, A.; Ullah, K.; Ali, H.; Betzel, C.; Ur Rehman, S. The Sequence and a Three-Dimensional Structural Analysis Reveal Substrate Specificity Among Snake Venom Phosphodiesterases. Toxins 2019, 11, 625. [Google Scholar] [CrossRef] [Green Version]
- Halkides, C.J.; Frey, P.A. The mechanism of the hydrolysis of μ-monothiopyrophosphate. J. Am. Chem. Soc. 1991, 113, 9843–9848. [Google Scholar] [CrossRef]
- Knight, W.B.; Sem, D.S.; Smith, K.; Miziorko, H.M.; Rendina, A.R.; Cleland, W.W. Phosphorylated thiosugars—Synthesis, properties, and reactivity in enzymatic reactions. Biochemistry 1991, 30, 4970–4977. [Google Scholar] [CrossRef] [PubMed]
- Oivanen, M.; Lönnberg, H. Kinetics and mechanisms for reactions of adenosine 2’- and 3’-monophosphates in aqueous acid: Competition between phosphate migration, dephosphorylation, and depurination. J. Org. Chem. 1989, 54, 2556–2560. [Google Scholar] [CrossRef]
- Lightcap, E.S.; Frey, P.A. Discrete monomeric metaphosphate anion as an intermediate in the hydrolysis of μ-monothiopyrophosphate. J. Am. Chem. Soc. 1992, 114, 9750–9755. [Google Scholar] [CrossRef]
- Lightcap, E.S.; Frey, P.A. Evidence for monomeric metaphosphate as an intermediate in the hydrolysis of μ-monothiopyrophosphate. J. Am. Chem. Soc. 1991, 113, 9415–9416. [Google Scholar] [CrossRef]
- Ma, B.; Meredith, C.; Schaefer, H.F. The quest for a metaphosphate intermediate. The mechanism for hydrolysis of pyrophosphates with and without catalysis. J. Phys. Chem. 1995, 99, 3815–3822. [Google Scholar] [CrossRef]
- De Cordoba, B.R.F.; Moreno, H.; Valencia, K.; Perurena, N.; Ruedas, P.; Walle, T.; Pezonaga-Torres, A.; Hinojosa, J.; Guruceaga, E.; Pineda-Lucena, A.; et al. Tumor ENPP1 (CD203a)/Haptoglobin Axis Exploits Myeloid-Derived Suppressor Cells to Promote Post-Radiotherapy Local Recurrence in Breast Cancer. Cancer Discov. 2022, 12, 1356–1377. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Substrate | Conditions | k/10−6 s−1 |
|---|---|---|
| 5′-dTSMP (4a) | pH 3, 298 K | 152 |
| pH 7, 298 K | 5.19 | |
| pH 7, 308 K | 17.1 | |
| pH 7, 318 K | 58.4 | |
| pH 11, 298 K | 0.34 | |
| 5′-dTSeMP (4b) | pH 3, 298 K | 53.1 |
| pH 7, 298 K | 16.8 | |
| pH 7, 303 K | 75.1 | |
| pH 7, 313 K | 297 | |
| pH 11, 298 K | 14.4 | |
| 5′-TMP (4c) | pH 3, 363 K | 2.60 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mikkola, S.; Eguaogie, O.; Nieminen, A.; Conlon, P.F.; Jakeman, D.L.; Moore, K.; Lane, I.C.; Vyle, J.S. 5′-Chalcogen-Substituted Nucleoside Pyrophosphate and Phosphate Monoester Analogues: Preparation and Hydrolysis Studies. Int. J. Mol. Sci. 2022, 23, 15582. https://doi.org/10.3390/ijms232415582
Mikkola S, Eguaogie O, Nieminen A, Conlon PF, Jakeman DL, Moore K, Lane IC, Vyle JS. 5′-Chalcogen-Substituted Nucleoside Pyrophosphate and Phosphate Monoester Analogues: Preparation and Hydrolysis Studies. International Journal of Molecular Sciences. 2022; 23(24):15582. https://doi.org/10.3390/ijms232415582
Chicago/Turabian StyleMikkola, Satu, Olga Eguaogie, Anu Nieminen, Patrick F. Conlon, David L. Jakeman, Keith Moore, Ian C. Lane, and Joseph S. Vyle. 2022. "5′-Chalcogen-Substituted Nucleoside Pyrophosphate and Phosphate Monoester Analogues: Preparation and Hydrolysis Studies" International Journal of Molecular Sciences 23, no. 24: 15582. https://doi.org/10.3390/ijms232415582
APA StyleMikkola, S., Eguaogie, O., Nieminen, A., Conlon, P. F., Jakeman, D. L., Moore, K., Lane, I. C., & Vyle, J. S. (2022). 5′-Chalcogen-Substituted Nucleoside Pyrophosphate and Phosphate Monoester Analogues: Preparation and Hydrolysis Studies. International Journal of Molecular Sciences, 23(24), 15582. https://doi.org/10.3390/ijms232415582