
DFT Simulation-Based Design of 1T-MoS2 Cathode Hosts for Li-S Batteries and Experimental Evaluation

 , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
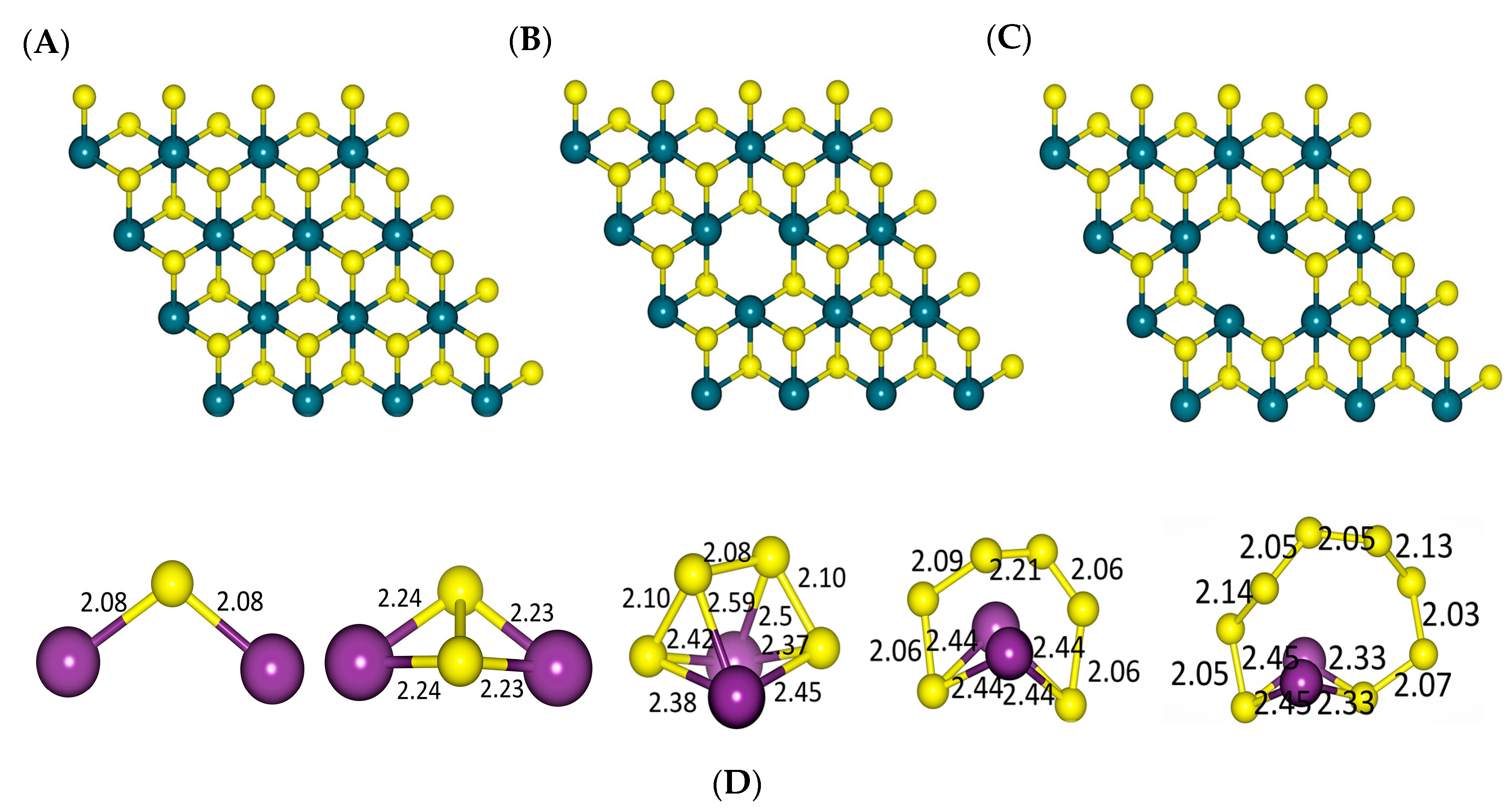
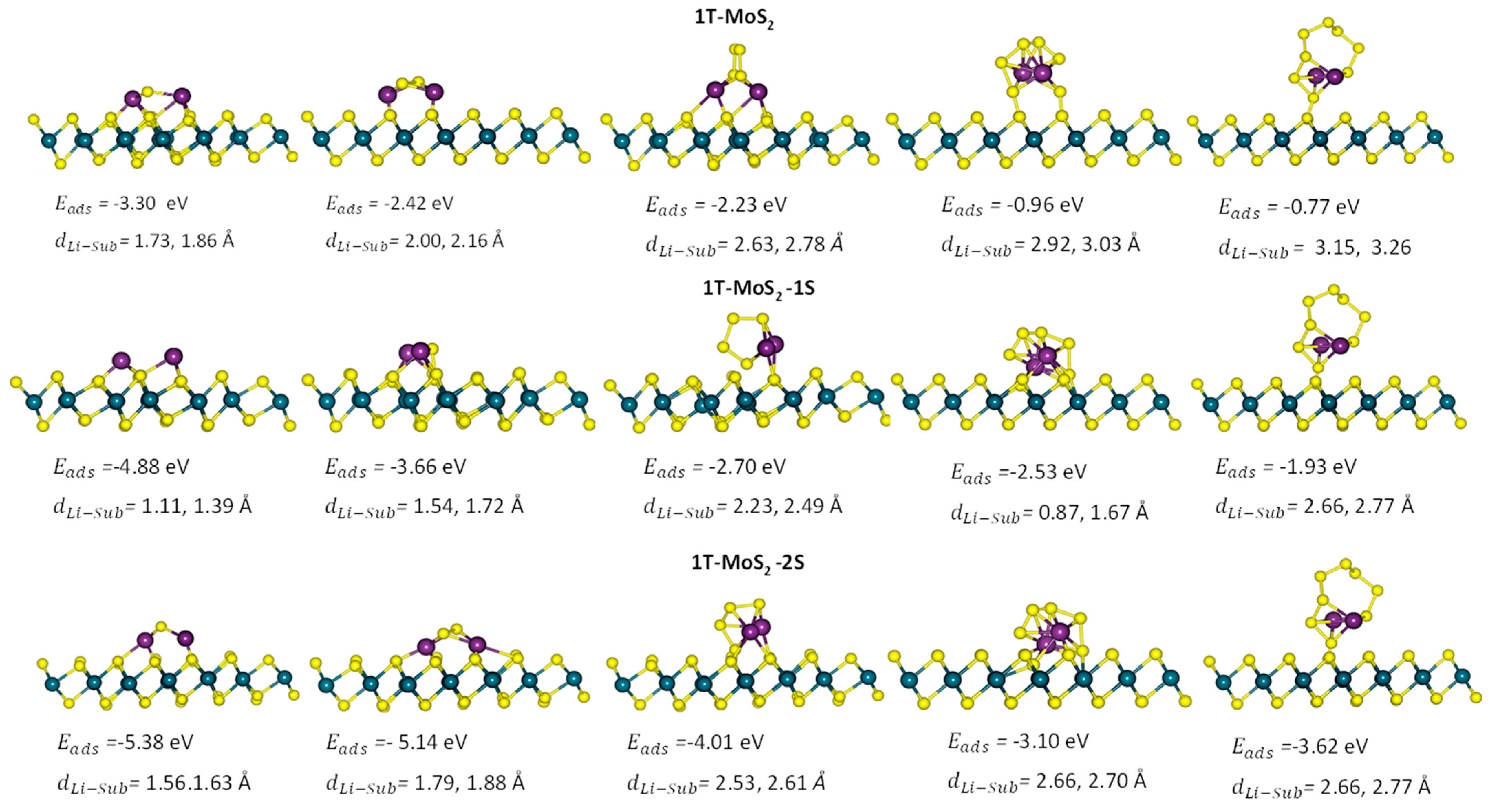
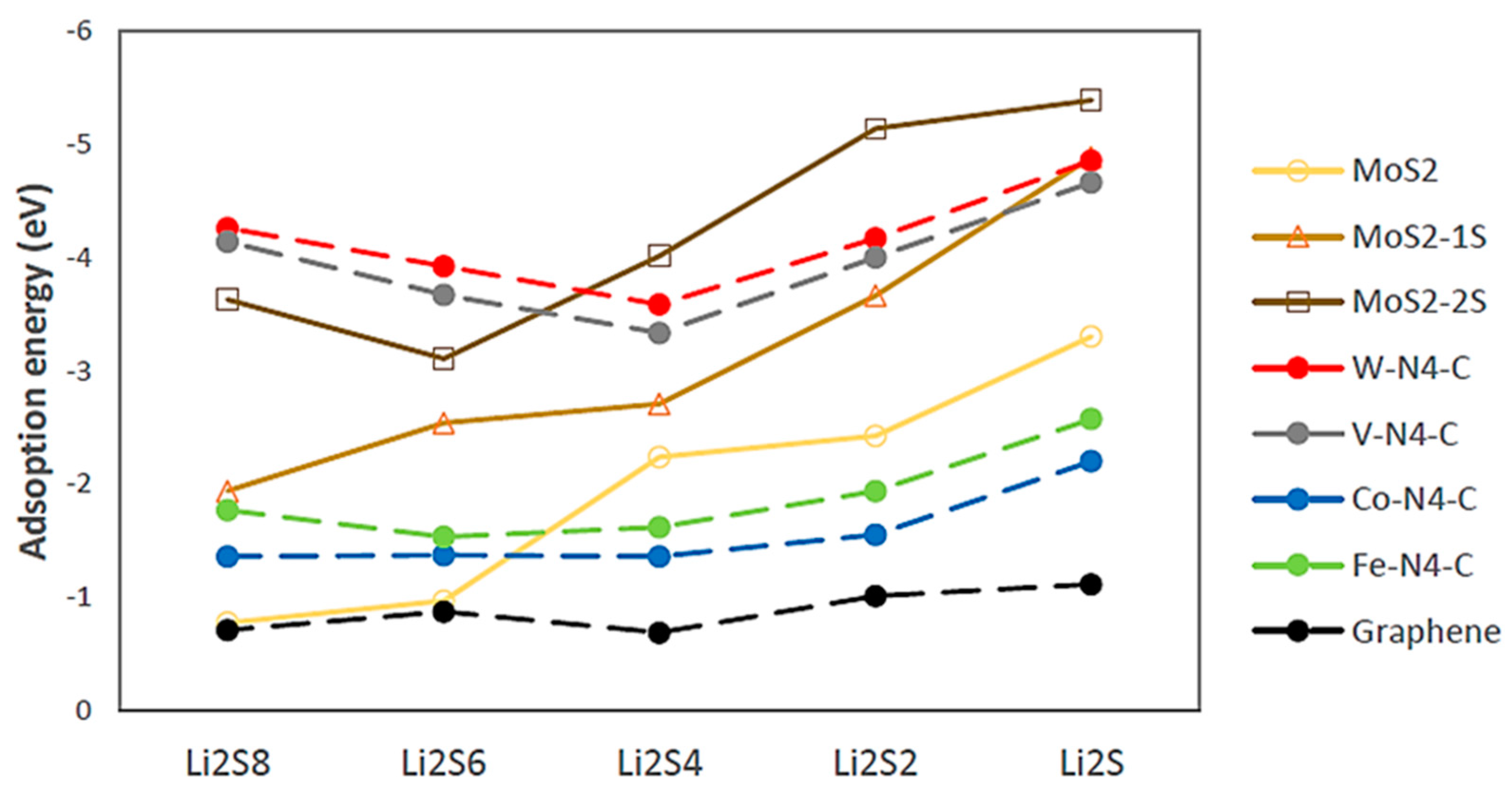
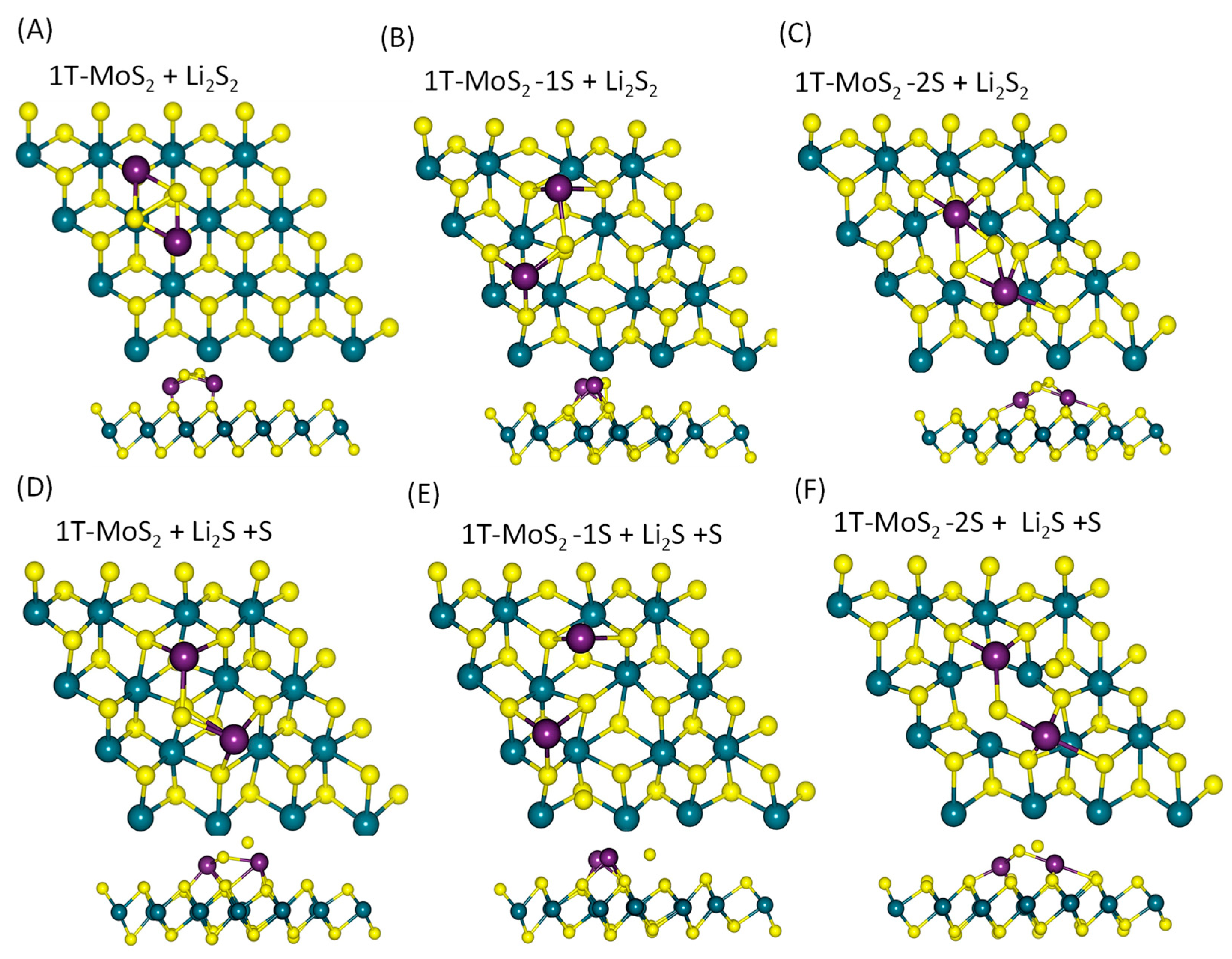
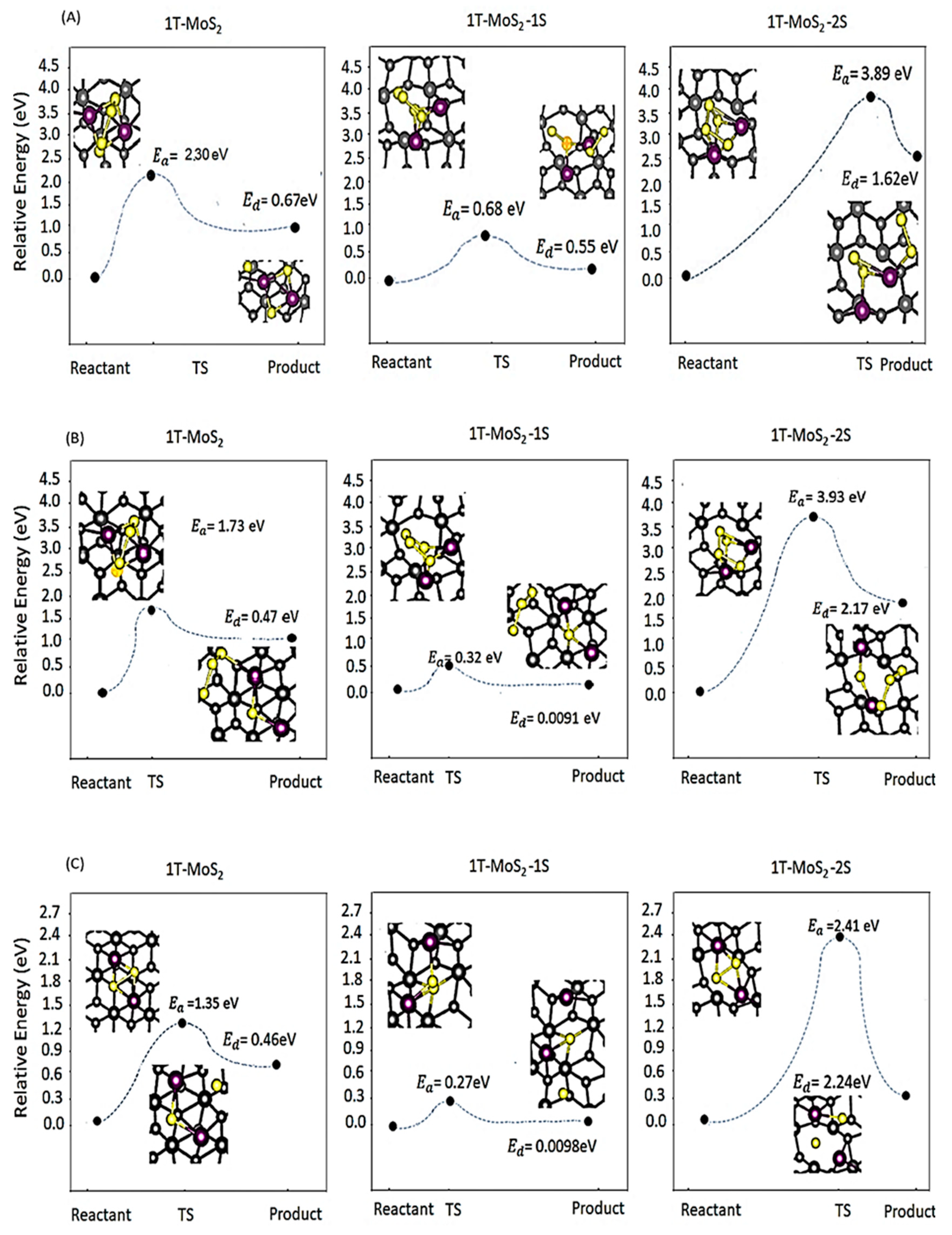
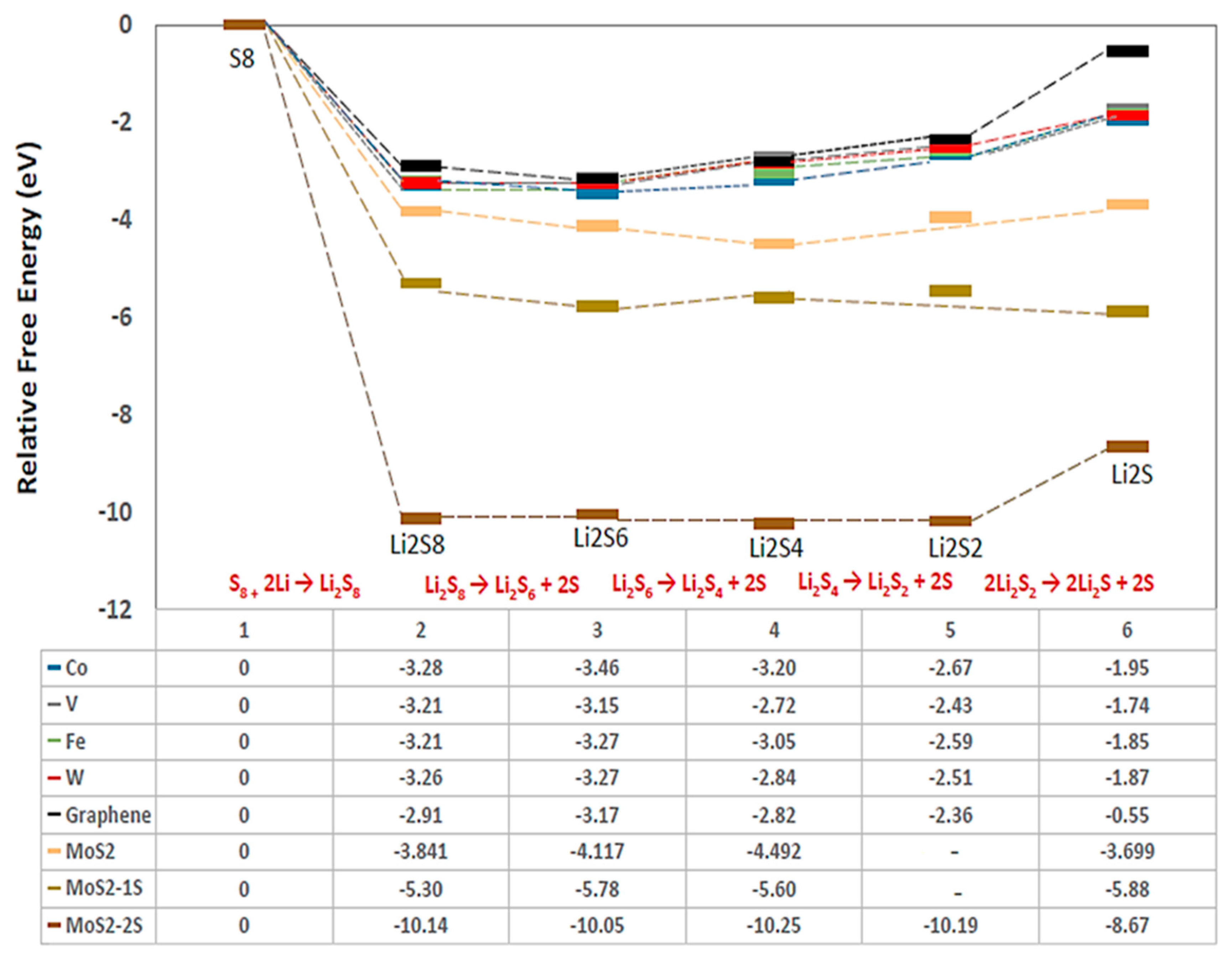
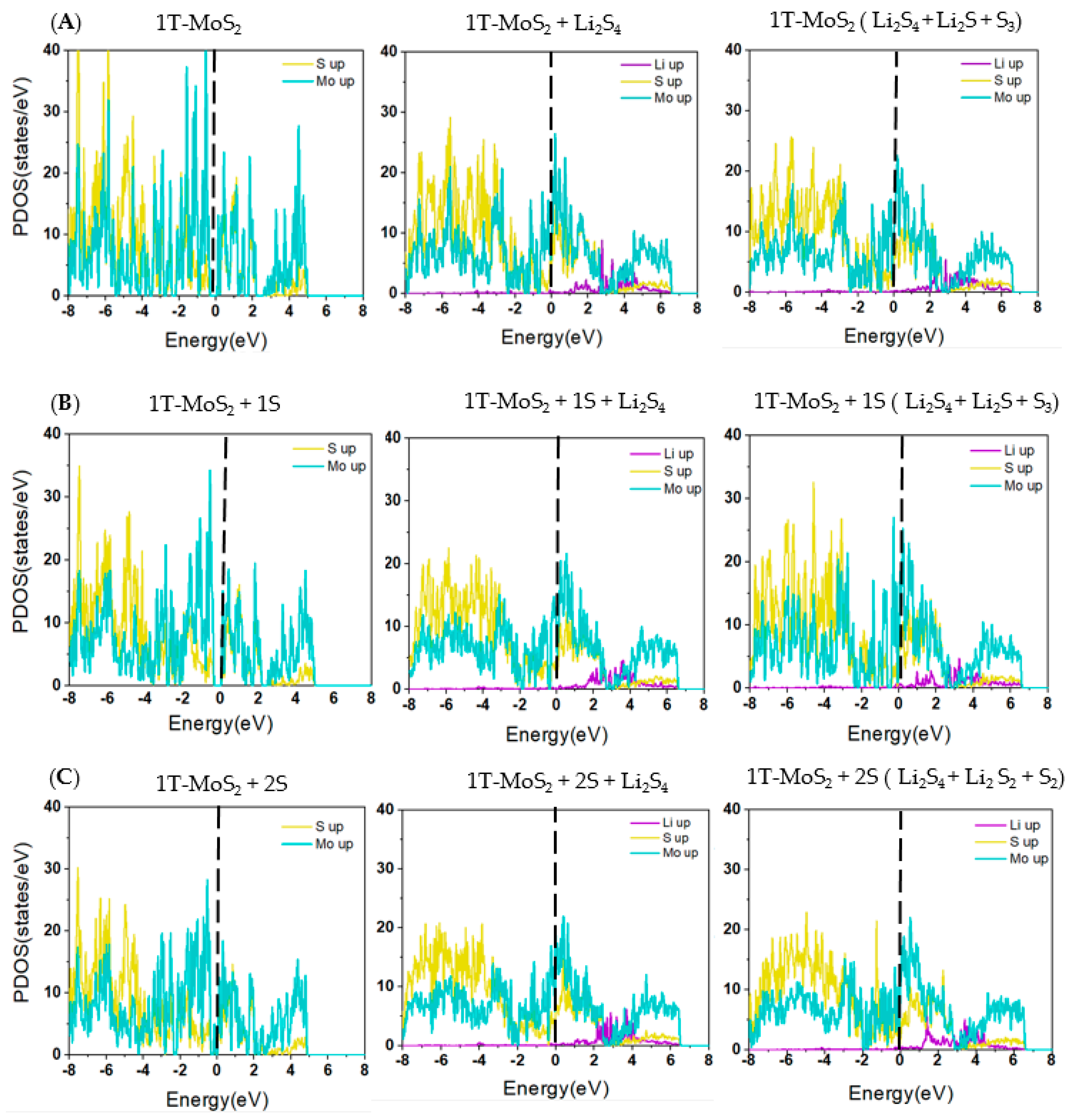
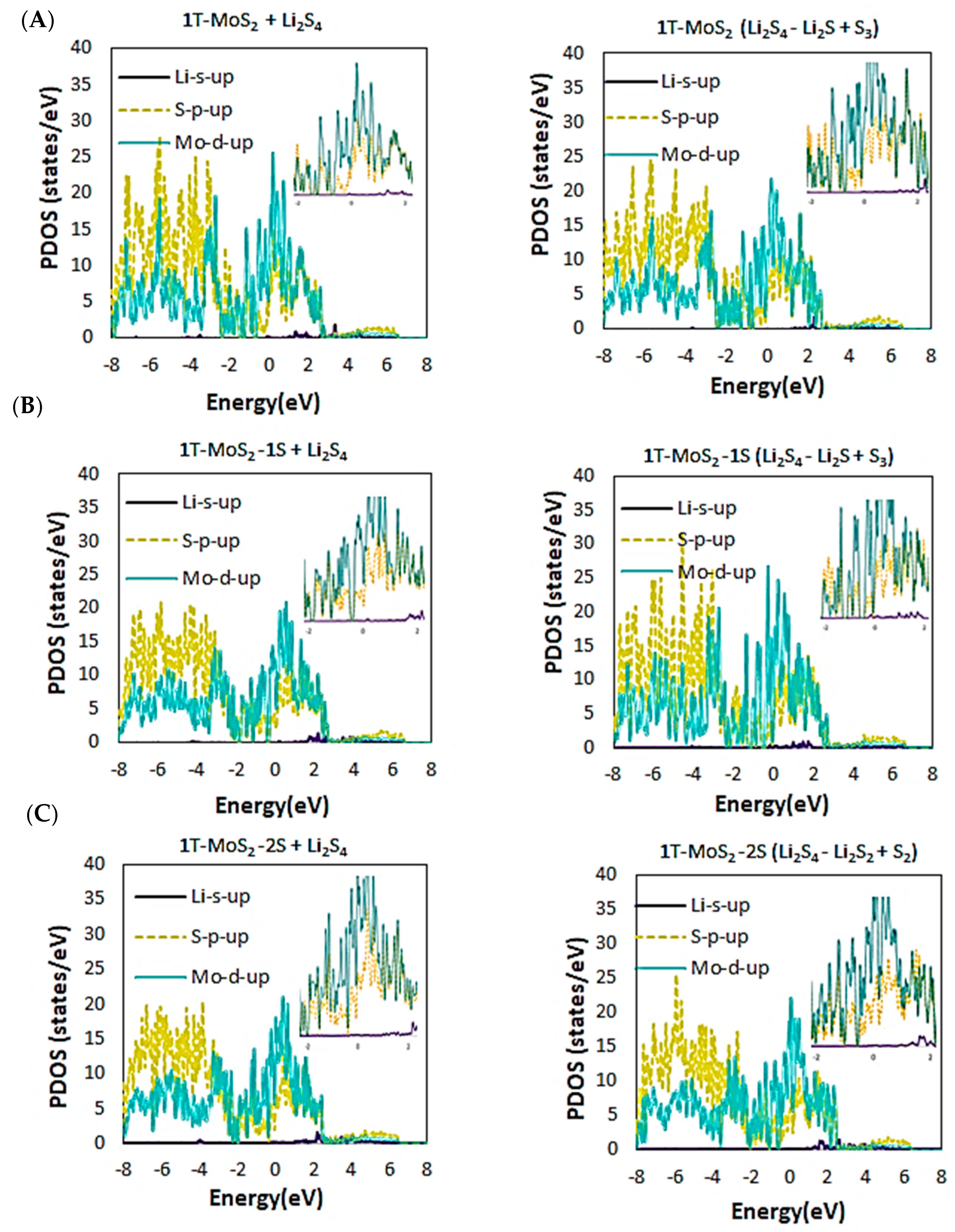
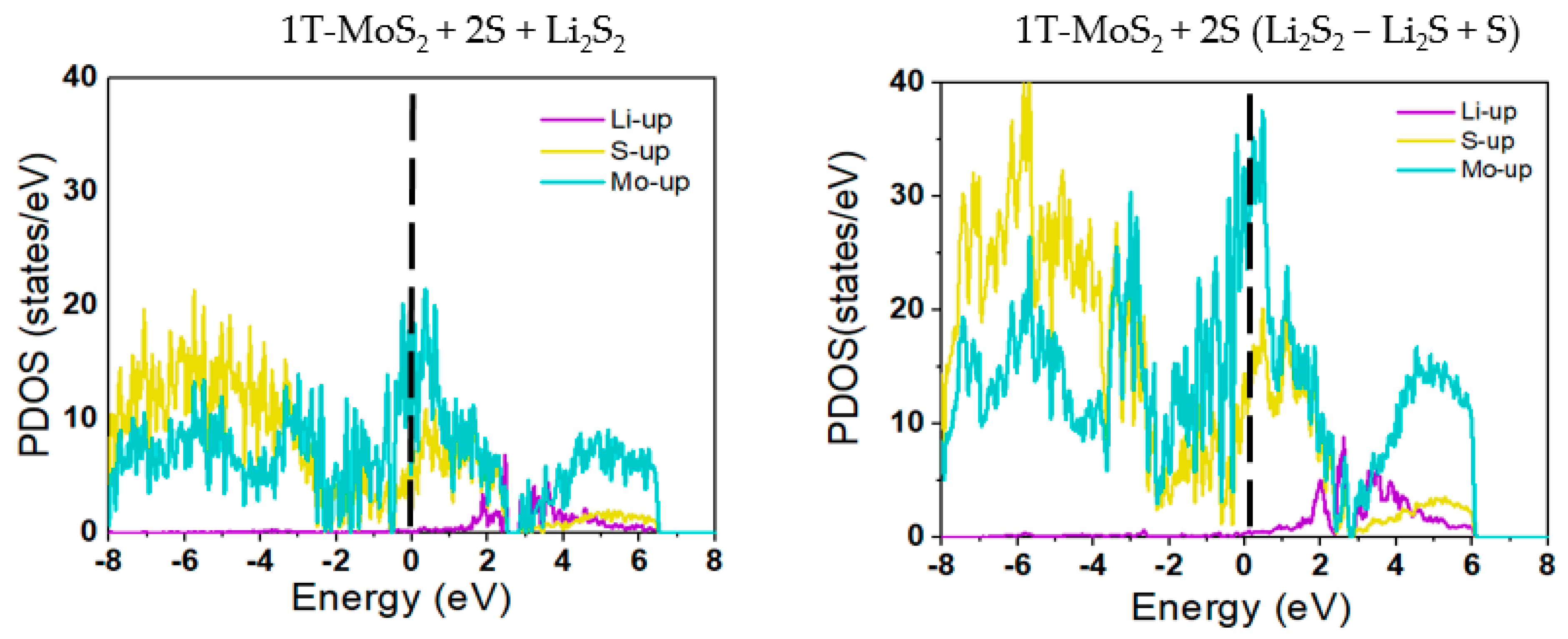
2.1. DFT Simulations
- (i)
- a two-step process: Li2S4 → Li2S2 + S2 followed by Li2S2 → Li2S + S;
- (ii)
- a one-step process: Li2S4 → Li2S + S3.
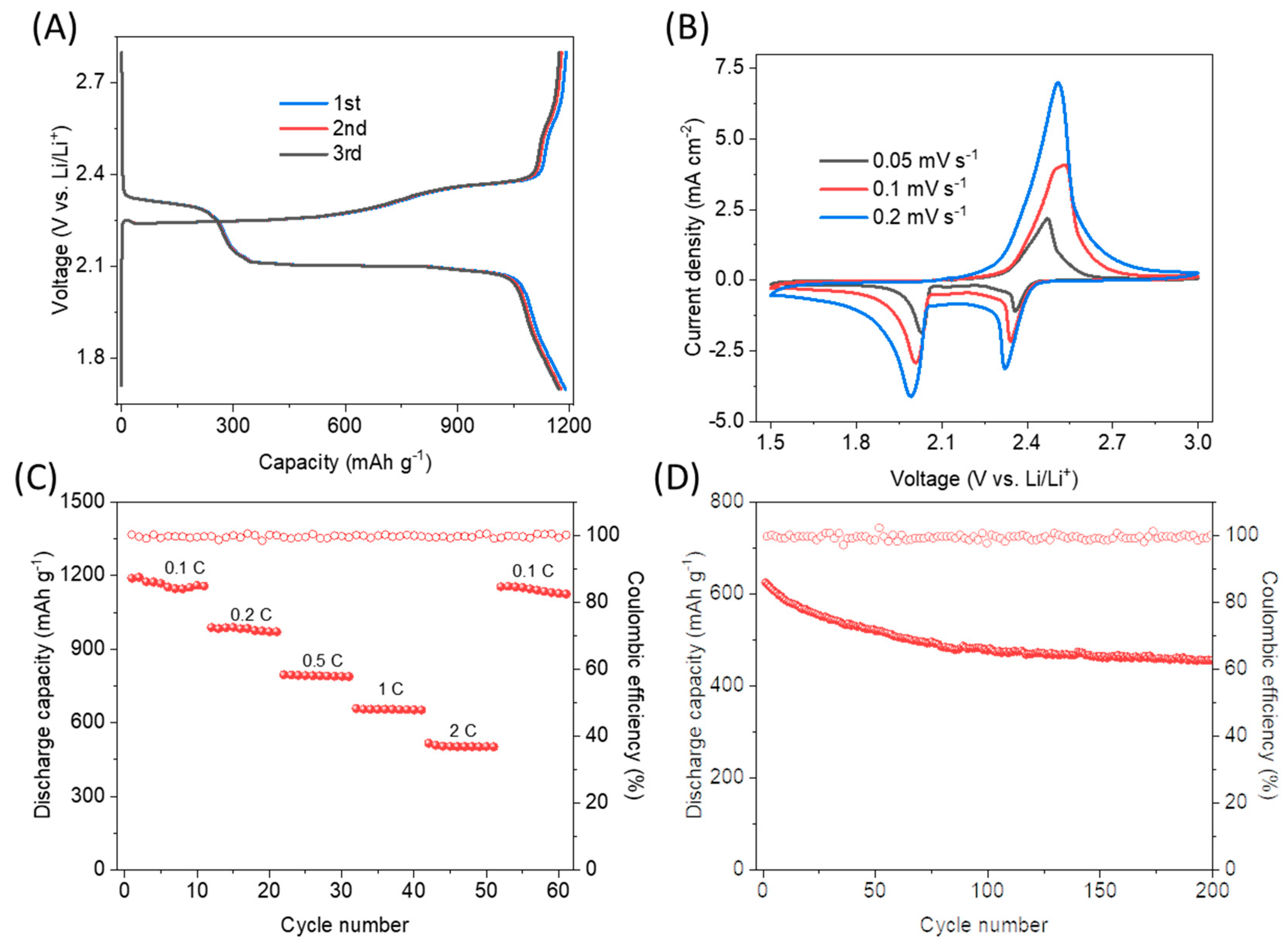
2.2. Experimental Evaluation
3. Methods and Materials
3.1. Computational Method
3.2. Computational Models
3.3. Materials and Experimental Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhu, K.; Wang, C.; Chi, Z.; Ke, F.; Yang, Y.; Wang, A.; Wang, W.; Miao, L. How far away are lithium-sulfur batteries from commercialization? Front. Energy Res. 2019, 7, 123. [Google Scholar] [CrossRef]
- Bruce, P.G.; Freunberger, S.A.; Hardwick, L.J.; Tarascon, J.-M. Li–O2 and Li–S batteries with high energy storage. Nat. Mater. 2012, 11, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Ding, B.; Wang, J.; Fan, Z.; Chen, S.; Lin, Q.; Lu, X.; Dou, H.; Nanjundan, A.K.; Yushin, G.; Zhang, X. Solid-state lithium–sulfur batteries: Advances, challenges and perspectives. Mater. Today 2020, 40, 114–131. [Google Scholar] [CrossRef]
- Reece, R.; Lekakou, C.; Smith, P.A. A structural supercapacitor based on activated carbon fabric and a solid electrolyte. Mater. Sci. Technol. 2019, 35, 368–375. [Google Scholar] [CrossRef]
- Choo, Y.; Halat, D.M.; Villaluenga, I.; Timachova, K.; Balsara, N.P. Diffusion and migration in polymer electrolytes. Prog. Polym. Sci. 2020, 103, 101220. [Google Scholar] [CrossRef] [Green Version]
- Reece, R.; Lekakou, C.; Smith, P.A. A high-performance structural supercapacitor. ACS Appl. Mater. Interfaces 2020, 12, 25683–25692. [Google Scholar] [CrossRef]
- Pei, F.; Lin, L.; Ou, D.; Zheng, Z.; Mo, S.; Fang, X.; Zheng, N. Self-supporting sulfur cathodes enabled by two-dimensional carbon yolk-shell nanosheets for high-energy-density lithium-sulfur batteries. Nat. Commun. 2017, 8, 482. [Google Scholar] [CrossRef] [Green Version]
- Wu, P.; Chen, L.-H.; Xiao, S.-S.; Yu, S.; Wang, Z.; Li, Y.; Su, B.-L. Insight into the positive effect of porous hierarchy in S/C cathodes on the electrochemical performance of Li–S batteries. Nanoscale 2018, 10, 11861–11868. [Google Scholar] [CrossRef]
- Dent, M.; Jakubczyk, E.; Zhang, T.; Lekakou, C. Kinetics of sulphur dissolution in lithium–sulphur batteries. J. Phys. Energy 2022, 4, 24001. [Google Scholar] [CrossRef]
- Grabe, S.; Baboo, J.P.; Tennison, S.; Zhang, T.; Lekakou, C.; Andritsos, E.I.; Cai, Q.; Downes, S.; Hinder, S.; Watts, J.F. Sulfur infiltration and allotrope formation in porous cathode hosts for lithium-sulfur batteries. AIChE J. 2022, 68, e17638. [Google Scholar] [CrossRef]
- Lasetta, K.; Baboo, J.P.; Lekakou, C. Modeling and simulations of the sulfur infiltration in activated carbon fabrics during composite cathode fabrication for lithium-sulfur batteries. J. Compos. Sci. 2021, 5, 65. [Google Scholar] [CrossRef]
- Helen, M.; Diemant, T.; Schindler, S.; Behm, R.J.; Danzer, M.; Kaiser, U.; Fichtner, M.; Anji Reddy, M. Insight into sulfur confined in ultramicroporous carbon. ACS Omega 2018, 3, 11290–11299. [Google Scholar] [CrossRef] [PubMed]
- Chandula Wasalathilake, K.; Roknuzzaman, M.; Ostrikov, K.; Ayoko, G.A.; Yan, C. Interaction between functionalized graphene and sulfur compounds in a lithium-sulfur battery—A density functional theory investigation. RSC Adv. 2018, 8, 2271–2279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, L.-C.; Liang, J.; Zhou, G.-M.; Li, F.; Saito, R.; Cheng, H.-M. Understanding the interactions between lithium polysulfides and N-doped graphene using density functional theory calculations. Nano Energy 2016, 25, 203–210. [Google Scholar] [CrossRef]
- Hou, T.; Chen, X.; Peng, H.; Huang, J.; Li, B.; Zhang, Q.; Li, B. Design principles for heteroatom-doped nanocarbon to achieve strong anchoring of polysulfides for lithium-sulfur batteries. Small 2016, 12, 3283–3291. [Google Scholar] [CrossRef]
- Zhang, L.; Liang, P.; Man, X.; Wang, D.; Huang, J.; Shu, H.; Liu, Z.; Wang, L. Fe, N co-doped graphene as a multi-functional anchor material for lithium-sulfur battery. J. Phys. Chem. Solids 2019, 126, 280–286. [Google Scholar] [CrossRef]
- Andritsos, E.I.; Lekakou, C.; Cai, Q. Single-atom catalysts as promising cathode materials for lithium-sulfur batteries. J. Phys. Chem. C 2021, 125, 18108–18118. [Google Scholar] [CrossRef]
- Zeng, Q.-W.; Hu, R.-M.; Chen, Z.-B.; Shang, J.-X. Single-atom Fe and N co-doped graphene for lithium-sulfur batteries: A density functional theory study. Mater. Res. Express 2019, 6, 95620. [Google Scholar] [CrossRef]
- Zhou, G.; Zhao, S.; Wang, T.; Yang, S.-Z.; Johannessen, B.; Chen, H.; Liu, C.; Ye, Y.; Wu, Y.; Peng, Y. Theoretical calculation guided design of single-atom catalysts toward fast kinetic and long-life Li–S batteries. Nano Lett. 2019, 20, 1252–1261. [Google Scholar] [CrossRef]
- Li, Y.; Wu, J.; Zhang, B.; Wang, W.; Zhang, G.; Seh, Z.W.; Zhang, N.; Sun, J.; Huang, L.; Jiang, J. Fast conversion and controlled deposition of lithium (poly) sulfides in lithium-sulfur batteries using high-loading cobalt single atoms. Energy Storage Mater. 2020, 30, 250–259. [Google Scholar] [CrossRef]
- Sun, M.; Wang, Z.; Li, X.; Li, H.; Jia, H.; Xue, X.; Jin, M.; Li, J.; Xie, Y.; Feng, M. Rational understanding of the catalytic mechanism of molybdenum carbide in polysulfide conversion in lithium–sulfur batteries. J. Mater. Chem. A 2020, 8, 11818–11823. [Google Scholar] [CrossRef]
- Zhang, J.; Duan, S.; You, C.; Wang, J.; Liu, H.; Guo, S.; Zhang, W.; Yang, R. In situ-grown tungsten carbide nanoparticles on nanocarbon as an electrocatalyst to promote the redox reaction kinetics of high-mass loading sulfur cathode for high volumetric performance. J. Mater. Chem. A 2020, 8, 22240–22250. [Google Scholar] [CrossRef]
- Liang, X.; Hart, C.; Pang, Q.; Garsuch, A.; Weiss, T.; Nazar, L.F. A highly efficient polysulfide mediator for lithium-sulfur batteries. Nat. Commun. 2015, 6, 5682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.-E.; Yin, K.; Qin, N.; Zhao, X.; Xia, F.-J.; Hu, Z.-Y.; Guo, G.; Cao, G.; Zhang, W. Oxygen-deficient titanium dioxide as a functional host for lithium-sulfur batteries. J. Mater. Chem. A 2019, 7, 10346–10353. [Google Scholar] [CrossRef]
- Zhou, G.; Tian, H.; Jin, Y.; Tao, X.; Liu, B.; Zhang, R.; Seh, Z.W.; Zhuo, D.; Liu, Y.; Sun, J.; et al. Catalytic oxidation of Li2S on the surface of metal sulfides for Li-S batteries. Proc. Natl. Acad. Sci. USA 2017, 114, 840–845. [Google Scholar] [CrossRef] [Green Version]
- Dong, S.; Sun, X.; Wang, Z. Trapping polysulfide on two-dimensional molybdenum disulfide for Li–S batteries through phase selection with optimized binding. Beilstein J. Nanotechnol. 2019, 10, 774–780. [Google Scholar] [CrossRef]
- Fan, C.; Zheng, Y.; Zhang, X.; Shi, Y.; Liu, S.; Wang, H.; Wu, X.; Sun, H.; Zhang, J. High-performance and low-temperature lithium–sulfur batteries: Synergism of thermodynamic and kinetic regulation. Adv. Energy Mater. 2018, 8, 1703638. [Google Scholar] [CrossRef]
- Hao, Z.; Yuan, L.; Chen, C.; Xiang, J.; Li, Y.; Huang, Z.; Hu, P.; Huang, Y. TiN as a simple and efficient polysulfide immobilizer for lithium–sulfur batteries. J. Mater. Chem. A 2016, 4, 17711–17717. [Google Scholar] [CrossRef]
- Li, X.; Guo, G.; Qin, N.; Deng, Z.; Lu, Z.; Shen, D.; Zhao, X.; Li, Y.; Su, B.-L.; Wang, H.-E. SnS2/TiO2 nanohybrids chemically bonded on nitrogen-doped graphene for lithium–sulfur batteries: Synergy of vacancy defects and heterostructures. Nanoscale 2018, 10, 15505–15512. [Google Scholar] [CrossRef]
- Hu, L.; Dai, C.; Liu, H.; Li, Y.; Shen, B.; Chen, Y.; Bao, S.; Xu, M. Double-shelled NiO-NiCo2O4 heterostructure@ carbon hollow nanocages as an efficient sulfur host for advanced lithium-sulfur batteries. Adv. Energy Mater. 2018, 8, 1800709. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, Q.; Sun, Y.K. Recent progress of advanced binders for Li-S batteries. J. Power Sources 2018, 396, 19–32. [Google Scholar] [CrossRef]
- Wang, H.-E.; Li, X.; Qin, N.; Zhao, X.; Cheng, H.; Cao, G.; Zhang, W. Sulfur-deficient MoS2 grown inside hollow mesoporous carbon as a functional polysulfide mediator. J. Mater. Chem. A 2019, 7, 12068–12074. [Google Scholar] [CrossRef]
- Manthiram, A.; Fu, Y.; Chung, S.-H.; Zu, C.; Su, Y.-S. Rechargeable lithium–sulfur batteries. Chem. Rev. 2014, 114, 11751–11787. [Google Scholar] [CrossRef] [PubMed]
- Al Salem, H.; Babu, G.; Rao, C.V.; Arava, L.M.R. Electrocatalytic polysulfide traps for controlling redox shuttle process of Li−S batteries. Am. Chem. Soc. 2015, 137, 11542–11545. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Fan, L.; Tian, D.; Wu, X.; Qiu, Y.; Zhao, C.; Guan, B.; Wang, Y.; Zhang, N.; Sun, K. Rational design of hierarchical SnO2/1T-MoS2 nanoarray electrode for ultralong-life Li–S batteries. ACS Energy Lett. 2018, 3, 1627–1633. [Google Scholar] [CrossRef]
- Liu, Y.; Cui, C.; Liu, Y.; Liu, W.; Wei, J. Application of MoS2 in the cathode of lithium sulfur batteries. RSC Adv. 2020, 10, 7384–7395. [Google Scholar] [CrossRef] [Green Version]
- Jayabal, S.; Wu, J.; Chen, J.; Geng, D.; Meng, X. Metallic 1T-MoS2 nanosheets and their composite materials: Preparation, properties and emerging applications. Mater. Today Energy 2018, 10, 264–279. [Google Scholar] [CrossRef]
- Sun, D.; Huang, D.; Wang, H.; Xu, G.-L.; Zhang, X.; Zhang, R.; Tang, Y.; Abd EI-Hady, D.; Alshitari, W.; Saad AL-Bogami, A.; et al. 1T MoS2 nanosheets with extraordinary sodium storage properties via thermal-driven ion intercalation assisted exfoliation of bulky MoS2. Nano Energy 2019, 61, 361–369. [Google Scholar] [CrossRef]
- Gupta, D.; Chauhan, V.; Kumar, R. A comprehensive review on synthesis and applications of molybdenum disulfide (MoS2) material: Past and recent developments. Inorg. Chem. Commun. 2020, 121, 108200. [Google Scholar] [CrossRef]
- You, Y.; Ye, Y.; Wei, M.; Sun, W.; Tang, Q.; Zhang, J.; Chen, X.; Li, H.; Xu, J. Three-dimensional MoS2/rGO foams as efficient sulfur hosts for high-performance lithium-sulfur batteries. Chem. Eng. J. 2019, 355, 671–678. [Google Scholar] [CrossRef]
- Yu, B.; Chen, Y.; Wang, Z.; Chen, D.; Wang, X.; Zhang, W.; He, J.; He, W. 1T-MoS2 nanotubes wrapped with N-doped graphene as highly-efficient absorbent and electrocatalyst for Li–S batteries. J. Power Sources 2020, 447, 227364. [Google Scholar] [CrossRef]
- Dirlam, P.T.; Park, J.; Simmonds, A.G.; Domanik, K.; Arrington, C.B.; Schaefer, J.L.; Oleshko, V.P.; Kleine, T.S.; Char, K.; Glass, R.S.; et al. lemental Sulfur and molybdenum disulfide composites for Li–S batteries with long cycle life and high-rate capability. ACS Appl. Mater. Interfaces 2016, 8, 13437–13448. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Dai, C.; Lim, J.-M.; Chen, Y.; Lian, X.; Wang, M.; Li, Y.; Xiao, P.; Henkelman, G.; Xu, M. A highly efficient double-hierarchical sulfur host for advanced lithium-sulfur batteries. Chem. Sci. 2018, 9, 666–675. [Google Scholar] [CrossRef] [Green Version]
- Abraham, A.M.; Kammampata, S.P.; Ponnurangam, S.; Thangadurai, V. Efficient Synthesis and characterization of robust MoS2 and S cathode for advanced Li–S battery: Combined experimental and theoretical studies. ACS Appl. Mater. Interfaces 2019, 11, 35729–35737. [Google Scholar] [CrossRef]
- Xu, Z.-L.; Onofrio, N.; Wang, J. Boosting the anchoring and catalytic capability of MoS2 for high-loading lithium sulfur batteries. J. Mater. Chem. A 2020, 8, 17646–17656. [Google Scholar] [CrossRef]
- Zhang, Y.; Mu, Z.; Yang, C.; Xu, Z.; Zhang, S.; Zhang, X.; Li, Y.; Lai, J.; Sun, Z.; Yang, Y.; et al. Rational design of MXene/1T-2H MoS2-C Nanohybrids for high-performance lithium–sulfur batteries. Adv. Funct. Mat. 2018, 28, 1707578. [Google Scholar] [CrossRef]
- He, J.; Hartmann, G.; Lee, M.; Hwang, G.S.; Chen, Y.; Manthiram, A. Freestanding 1T MoS2/graphene heterostructures as a highly efficient electrocatalyst for lithium polysulfides in Li–S batteries. Energy Environ. Sci. 2019, 12, 344–350. [Google Scholar]
- Lin, H.; Yang, L.; Jiang, X.; Li, G.; Zhang, T.; Yao, Q.; Zheng, G.W.; Lee, J.Y. Electrocatalysis of polysulfide conversion by sulfur-deficient MoS2 nanoflakes for lithium–sulfur batteries. Energy Environ. Sci. 2017, 10, 1476–1486. [Google Scholar] [CrossRef] [Green Version]
- Kamphaus, E.P.; Balbuena, P.B. Long-Chain polysulfide retention at the cathode of Li–S batteries. J. Phys. Chem. C 2016, 120, 4296–4305. [Google Scholar] [CrossRef]
- Zhang, Q.; Wang, Y.; Seh, Z.W.; Fu, Z.; Zhang, R.; Cui, Y. Understanding the anchoring effect of two-dimensional layered materials for lithium–sulfur batteries. Nano Lett. 2015, 15, 3780–3786. [Google Scholar] [CrossRef]
- Zhang, Q.; Zhang, X.; Li, M.; Liu, J.; Wu, Y. Sulfur-deficient MoS2−x promoted lithium polysulfides conversion in lithium-sulfur battery: A first-principles study. Appl. Surf. Sci. 2019, 487, 452–463. [Google Scholar] [CrossRef]
- Assary, R.S.; Curtiss, L.A.; Moore, J.S. Toward a Molecular Understanding of energetics in Li–S batteries using nonaqueous electrolytes: A high-level quantum chemical study. J. Phys. Chem. C 2014, 118, 11545–11558. [Google Scholar] [CrossRef]
- Jayabal, S.; Saranya, G.; Liu, Y.; Geng, D.; Meng, X. Unravelling the synergy effects of defect-rich 1T-MoS2/carbon nanotubes for the hydrogen evolution reaction by experimental and calculational studies. Sustain. Energy Fuels 2019, 3, 2100–2110. [Google Scholar] [CrossRef]
- Yang, J.; Wang, Y.; Lagos, M.J.; Manichev, V.; Fullon, R.; Song, X.; Voiry, D.; Chakraborty, S.; Zhang, W.; Batson, P.E. Single atomic vacancy catalysis. ACS Nano 2019, 13, 9958–9964. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Li, Q.; Qin, X.; Liang, G.; Han, W.; Zhou, D.; He, Y.-B.; Li, B.; Kang, F. Suppressing self-discharge and shuttle effect of lithium–sulfur batteries with V2O5-decoratChed carbon nanofiber interlayer. Small 2017, 13, 1602539. [Google Scholar] [CrossRef]
- Shi, H.; Dong, Y.; Zhou, F.; Chen, J.; Wu, Z.-S. 2D hybrid interlayer of electrochemically exfoliated graphene and Co(OH)2 nanosheet as a bi-functionalized polysulfide barrier for high-performance lithium–sulfur batteries. J. Phys. Energy 2019, 1, 015002. [Google Scholar] [CrossRef]
- Baboo, J.P.; Yatoo, M.A.; Dent, M.; Hojaji Najafabadi, E.; Lekakou, C.; Slade, R.; Hinder, S.J.; Watts, J.F. Exploring different binders for a LiFePO4 battery, battery testing, modeling and simulations. Energies 2022, 15, 2332. [Google Scholar] [CrossRef]
- Cho, J.; Ryu, S.; Gong, Y.J.; Pyo, S.; Yun, H.; Kim, H.; Lee, J.; Yoo, J.; Kim, Y.S. Nitrogen-doped MoS2 as a catalytic sulfur host for lithium-sulfur batteries. Chem. Eng. J. 2022, 439, 135568. [Google Scholar] [CrossRef]
- Zhai, S.; Abraham, A.M.; Chen, B.; Fan, Z.; Hu, J.; Cai, Z.; Thangadurai, V. Abundant Canadian pine with polysulfide redox mediating ZnS/CuS nanocomposite to attain high-capacity lithium sulfur battery. Carbon 2022, 195, 253–262. [Google Scholar] [CrossRef]
- Clark, S.J.; Segall, M.D.; Pickard, C.J.; Hasnip, P.J.; Probert, M.I.J.; Refson, K.; Payne, M.C. First principles methods using CASTEP. Zeitschrift Krist. Cryst. Mater. 2005, 220, 567–570. [Google Scholar] [CrossRef] [Green Version]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef] [PubMed]
- Babar, S.; Lekakou, C. Molecular modeling of electrolyte and polysulfide ions for lithium-sulfur batteries. Ionics 2021, 27, 635–642. [Google Scholar] [CrossRef]
- Lu, Y.-C.; He, Q.; Gasteiger, H.A. Probing the lithium−sulfur redox reactions: A rotating-ring disk electrode study. J. Phys. Chem. C 2014, 118, 5733–5741. [Google Scholar] [CrossRef]
- Acerce, M.; Voiry, D.; Chhowalla, M. Metallic 1T phase MoS2 nanosheets as supercapacitor electrode materials. Nat. Nanotechnol. 2015, 10, 313–318. [Google Scholar] [CrossRef] [PubMed]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hojaji, E.; Andritsos, E.I.; Li, Z.; Chhowalla, M.; Lekakou, C.; Cai, Q. DFT Simulation-Based Design of 1T-MoS2 Cathode Hosts for Li-S Batteries and Experimental Evaluation. Int. J. Mol. Sci. 2022, 23, 15608. https://doi.org/10.3390/ijms232415608
Hojaji E, Andritsos EI, Li Z, Chhowalla M, Lekakou C, Cai Q. DFT Simulation-Based Design of 1T-MoS2 Cathode Hosts for Li-S Batteries and Experimental Evaluation. International Journal of Molecular Sciences. 2022; 23(24):15608. https://doi.org/10.3390/ijms232415608
Chicago/Turabian StyleHojaji, Elaheh, Eleftherios I. Andritsos, Zhuangnan Li, Manish Chhowalla, Constantina Lekakou, and Qiong Cai. 2022. "DFT Simulation-Based Design of 1T-MoS2 Cathode Hosts for Li-S Batteries and Experimental Evaluation" International Journal of Molecular Sciences 23, no. 24: 15608. https://doi.org/10.3390/ijms232415608
APA StyleHojaji, E., Andritsos, E. I., Li, Z., Chhowalla, M., Lekakou, C., & Cai, Q. (2022). DFT Simulation-Based Design of 1T-MoS2 Cathode Hosts for Li-S Batteries and Experimental Evaluation. International Journal of Molecular Sciences, 23(24), 15608. https://doi.org/10.3390/ijms232415608