
The Chromatin Structure at the MECP2 Gene and In Silico Prediction of Potential Coding and Non-Coding MECP2 Splice Variants



Abstract
:
1. Introduction
2. Results
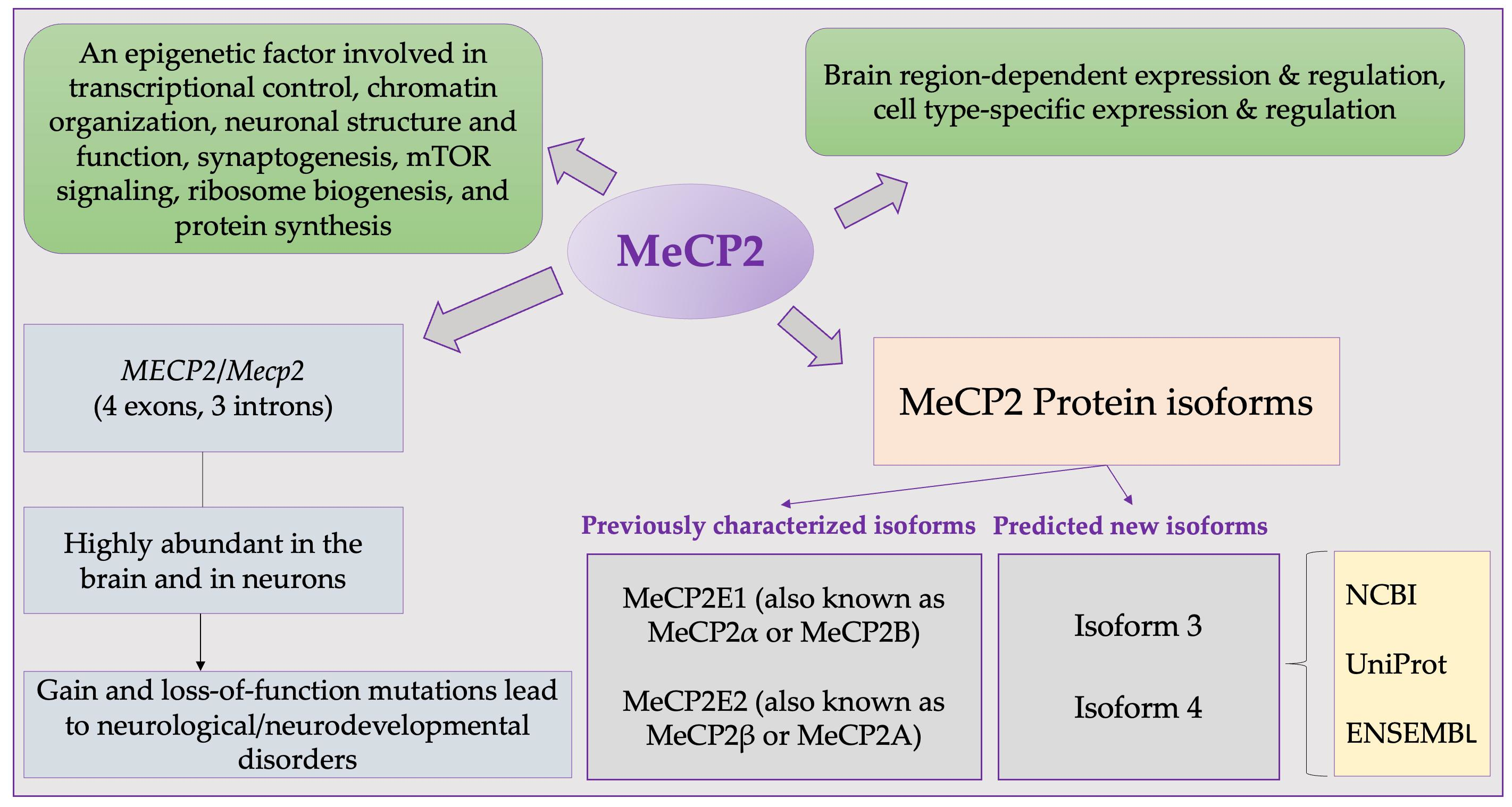
2.1. MECP2 Transcripts and Encoded Protein Isoforms
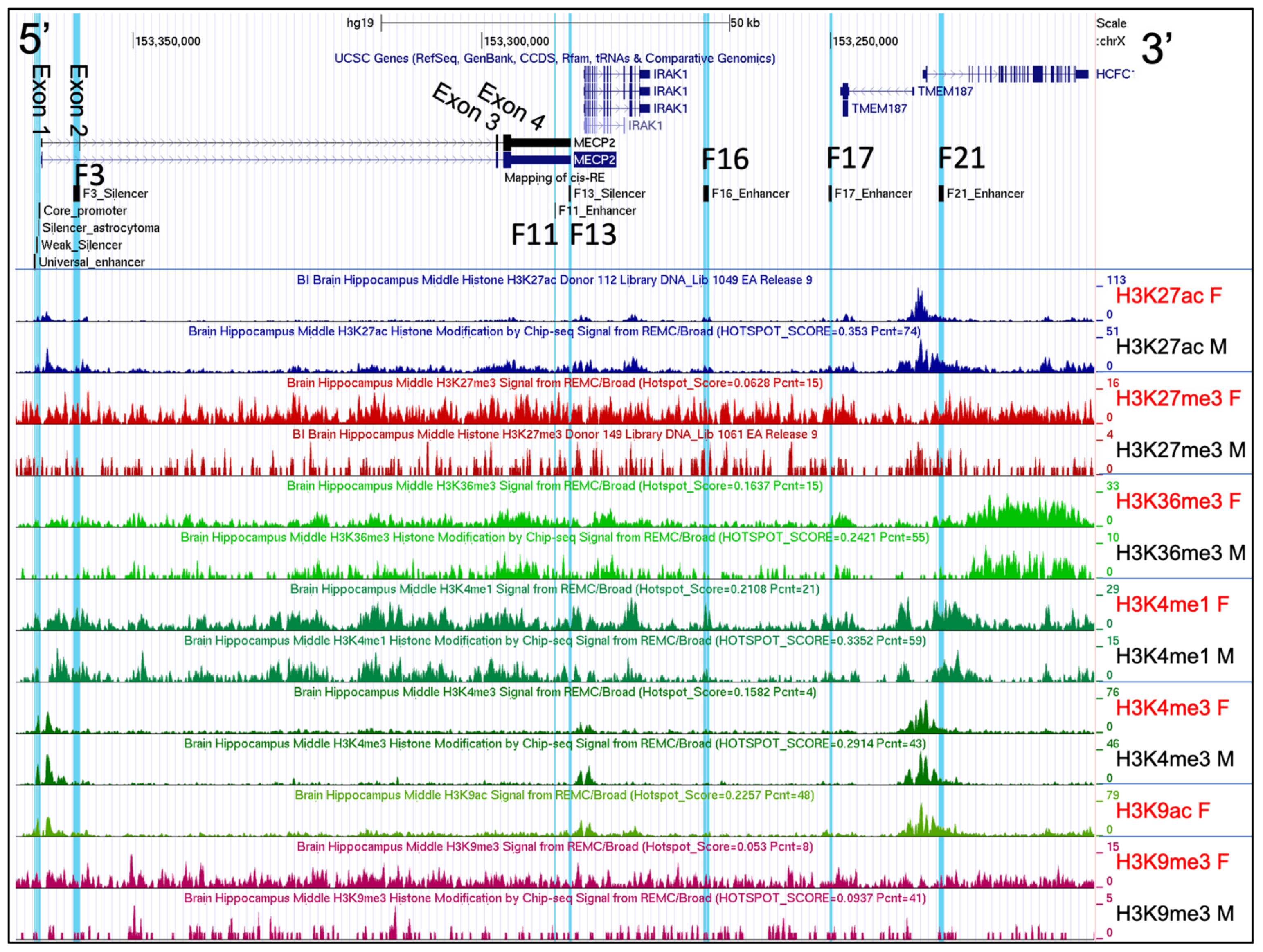
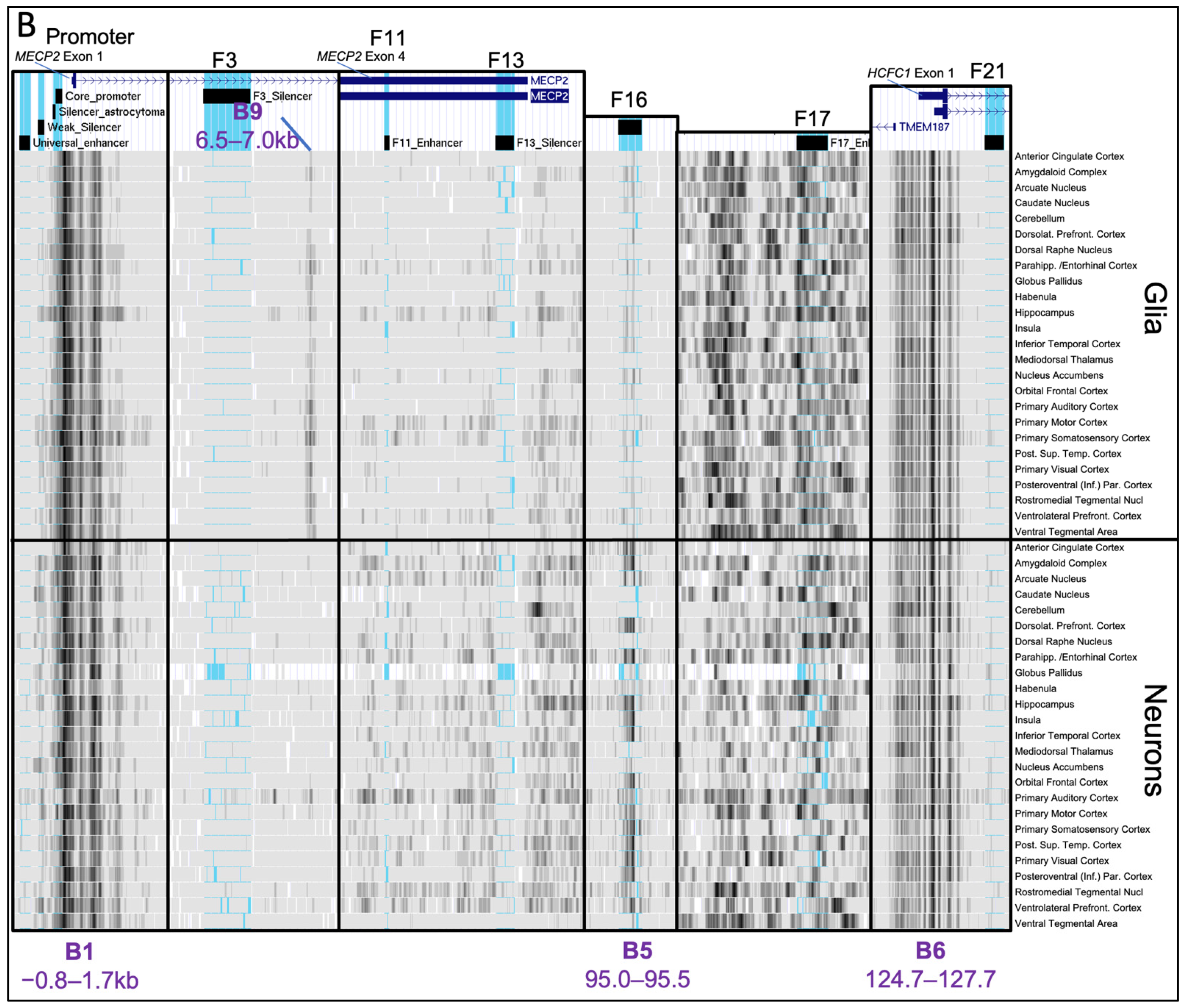
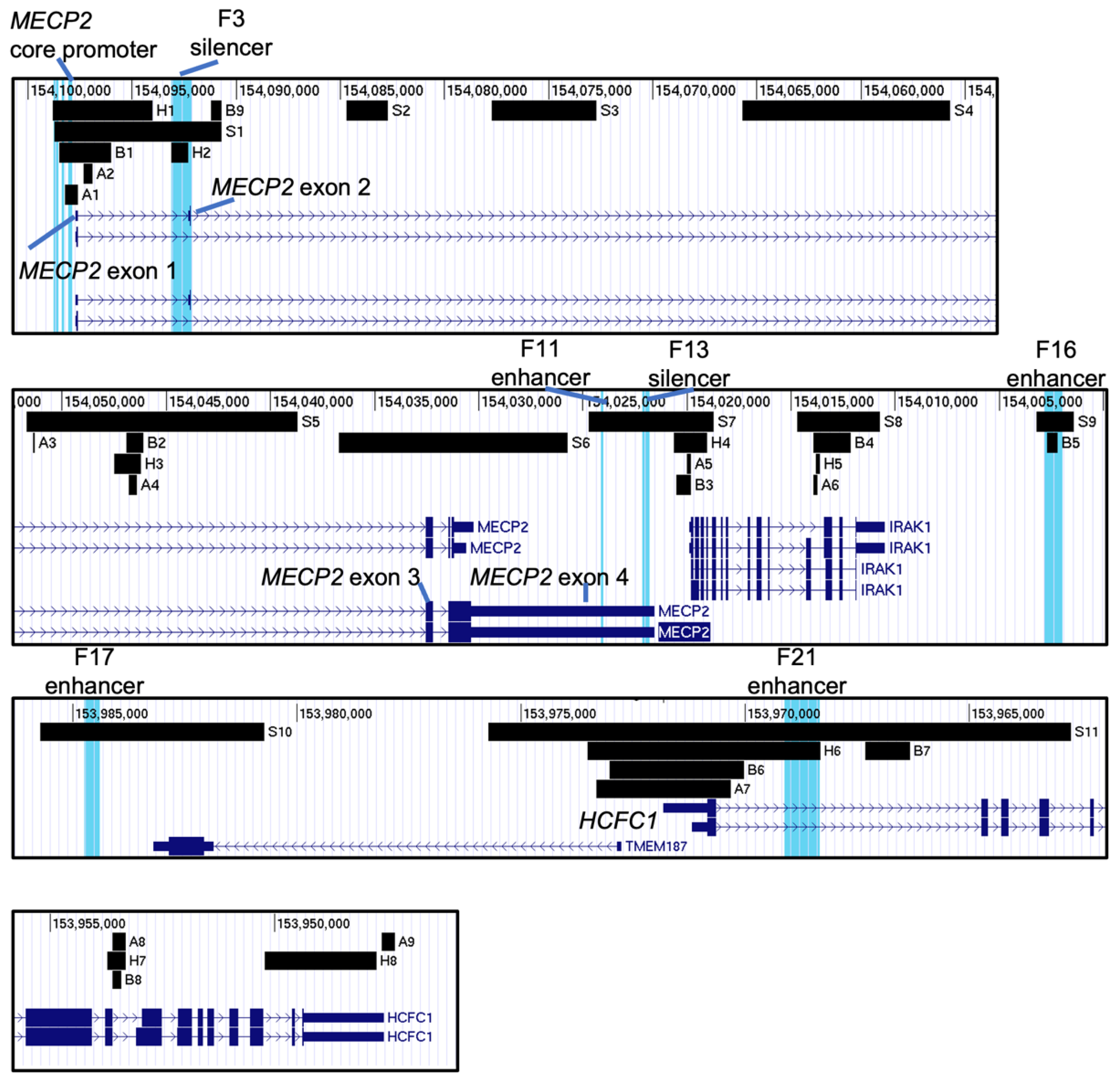
2.2. Histone Post-Translational Modification Profiles at the MECP2 Gene Loci and the Corresponding Cis-Regulatory Elements
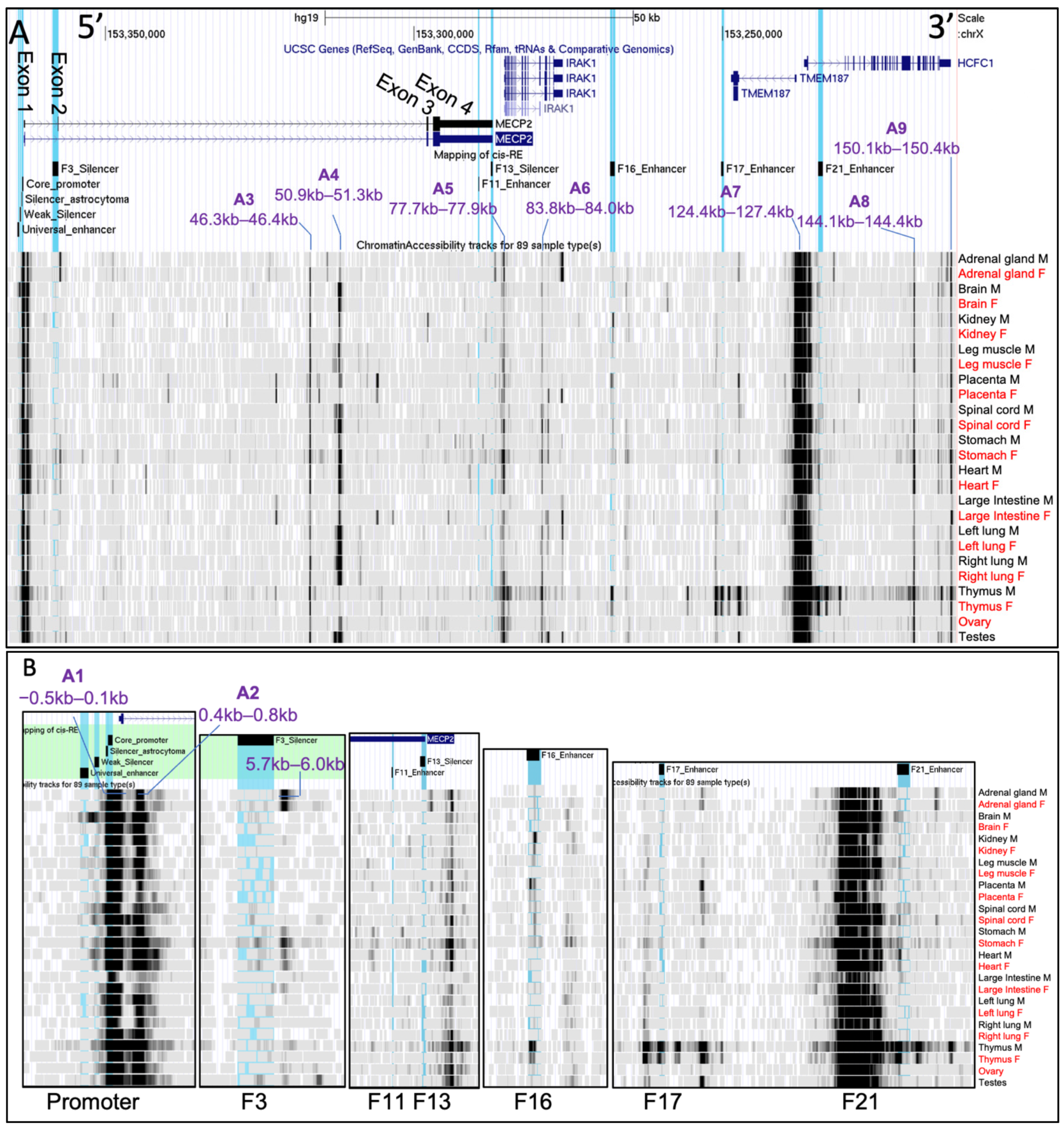
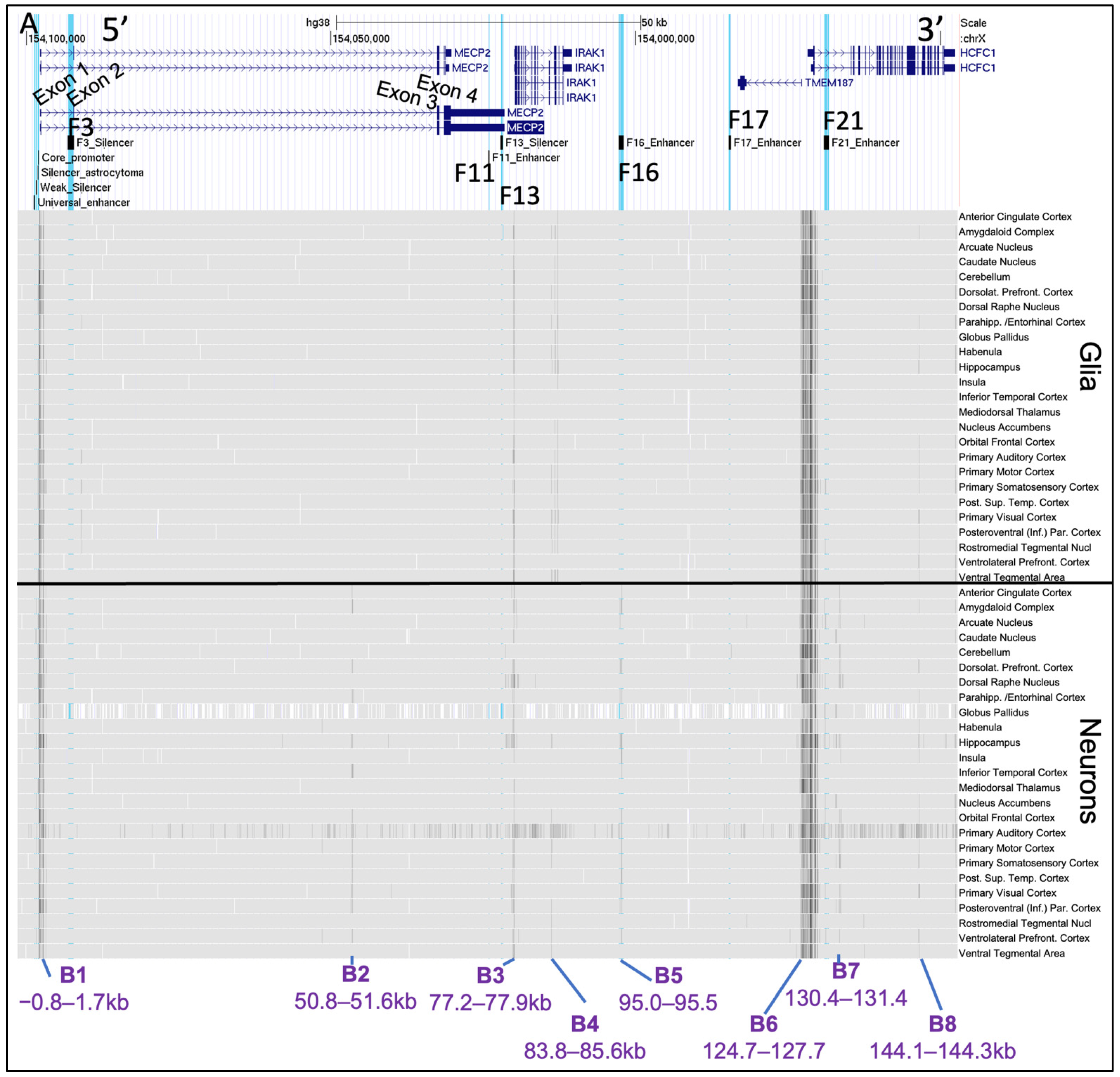
2.3. Chromatin Availability Profiles of the MECP2 Gene and its Cis-Regulatory Elements Characterized by DNase-seq and ATAC-seq in Different Human Tissues and Brain Regions
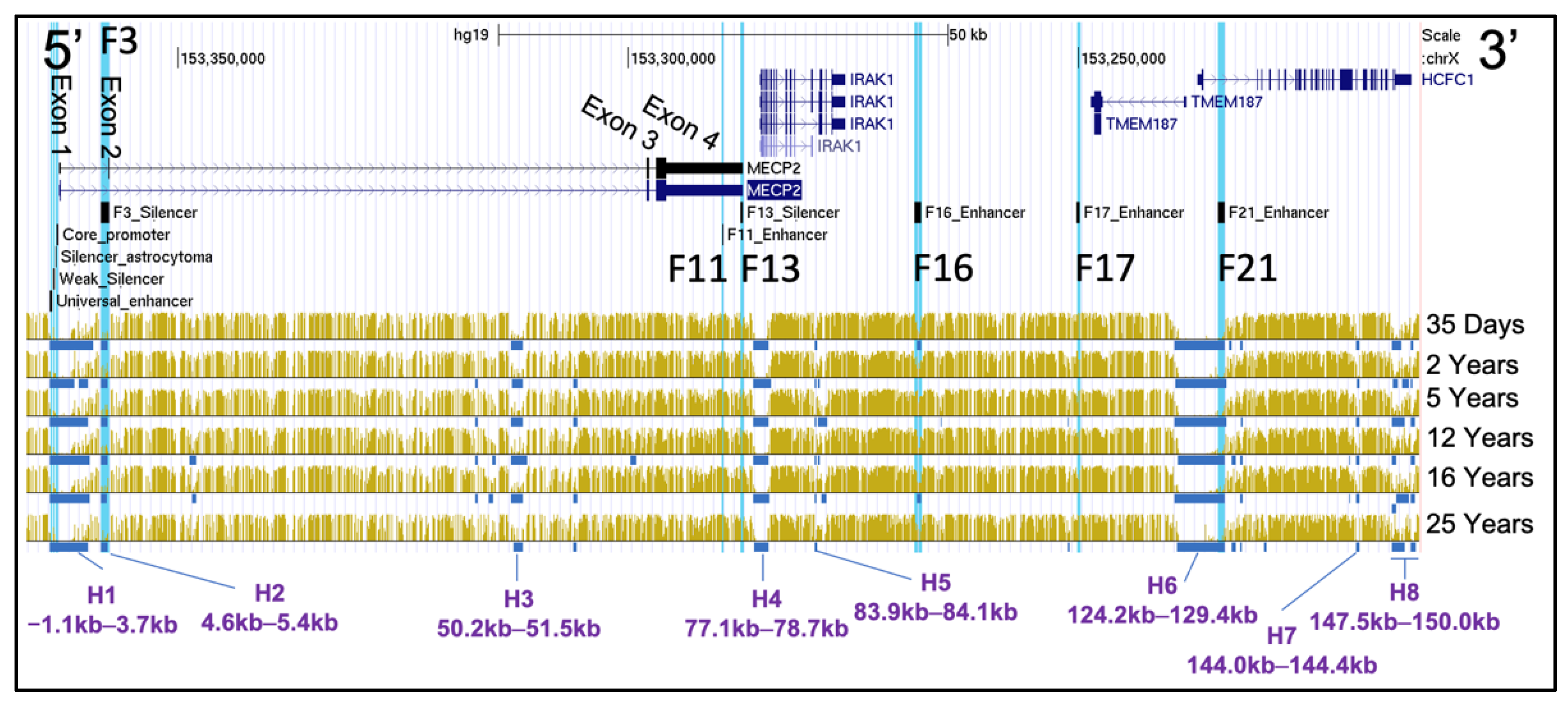
2.4. DNA Methylation of Mid Frontal Gyrus at Different Stages of Human Development
3. Discussion
3.1. Potential MeCP2 Protein Isoforms
3.2. In Silico Analysis of the Human MECP2 Gene
4. Materials and Methods
4.1. Analysis of MECP2 Transcripts and Protein Isoforms from NCBI, UniProt, ENSEMBL
4.2. Locating the MECP2 Gene Regulatory Elements in UCSC
4.3. Selection and Visualization of Datasets in UCSC
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Signor, S.A.; Nuzhdin, S.V. The Evolution of Gene Expression in cis and trans. Trends Genet. 2018, 34, 532–544. [Google Scholar] [CrossRef] [PubMed]
- Carthew, R.W. Gene Regulation and Cellular Metabolism: An Essential Partnership. Trends Genet. 2021, 37, 389–400. [Google Scholar] [CrossRef] [PubMed]
- Bin Akhtar, G.; Buist, M.; Rastegar, M. MeCP2 and transcriptional control of eukaryotic gene expression. Eur. J. Cell Biol. 2022, 101, 151237. [Google Scholar] [CrossRef] [PubMed]
- Riethoven, J.J. Regulatory regions in DNA: Promoters, enhancers, silencers, and insulators. Methods Mol. Biol. 2010, 674, 33–42. [Google Scholar] [CrossRef]
- Rastegar, M.; Szpirer, C.; Rousseau, G.G.; Lemaigre, F.P. Hepatocyte nuclear factor 6: Organization and chromosomal assignment of the rat gene and characterization of its promoter. Biochem. J. 1998, 334 Pt 3, 565–569. [Google Scholar] [CrossRef] [Green Version]
- Kolovos, P.; Knoch, T.A.; Grosveld, F.G.; Cook, P.R.; Papantonis, A. Enhancers and silencers: An integrated and simple model for their function. Epigenetics Chromatin 2012, 5, 1. [Google Scholar] [CrossRef] [Green Version]
- Calhoun, V.C.; Stathopoulos, A.; Levine, M. Promoter-proximal tethering elements regulate enhancer-promoter specificity in the Drosophila Antennapedia complex. Proc. Natl. Acad. Sci. USA 2002, 99, 9243–9247. [Google Scholar] [CrossRef] [Green Version]
- Delcuve, G.P.; Rastegar, M.; Davie, J.R. Epigenetic control. J. Cell Physiol. 2009, 219, 243–250. [Google Scholar] [CrossRef]
- Liyanage, V.R.; Jarmasz, J.S.; Murugeshan, N.; Del Bigio, M.R.; Rastegar, M.; Davie, J.R. DNA modifications: Function and applications in normal and disease States. Biology 2014, 3, 670–723. [Google Scholar] [CrossRef]
- Rastegar, M.; Yasui, D.H. Editorial: Epigenetic Mechanisms and Their Involvement in Rare Diseases. Front. Genet. 2021, 12, 755076. [Google Scholar] [CrossRef]
- Biswas, S.; Rao, C.M. Epigenetic tools (The Writers, The Readers and The Erasers) and their implications in cancer therapy. Eur. J. Pharmacol. 2018, 837, 8–24. [Google Scholar] [CrossRef] [PubMed]
- Rastegar, M. Epigenetics and Cerebellar Neurodevelopmental Disorders. In Development of the Cerebellum from Molecular Aspects to Diseases; Marzban, H., Ed.; Springer International Publishing: Cham, Switzerland, 2017; pp. 197–218. [Google Scholar]
- Mnatzakanian, G.N.; Lohi, H.; Munteanu, I.; Alfred, S.E.; Yamada, T.; MacLeod, P.J.; Jones, J.R.; Scherer, S.W.; Schanen, N.C.; Friez, M.J.; et al. A previously unidentified MECP2 open reading frame defines a new protein isoform relevant to Rett syndrome. Nat. Genet. 2004, 36, 339–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodrigues, D.C.; Mufteev, M.; Ellis, J. Regulation, diversity and function of MECP2 exon and 3’UTR isoforms. Hum. Mol. Genet. 2020, 29, R89–R99. [Google Scholar] [CrossRef] [PubMed]
- Lewis, J.D.; Meehan, R.R.; Henzel, W.J.; Maurer-Fogy, I.; Jeppesen, P.; Klein, F.; Bird, A. Purification, sequence, and cellular localization of a novel chromosomal protein that binds to methylated DNA. Cell 1992, 69, 905–914. [Google Scholar] [CrossRef]
- Meehan, R.R.; Lewis, J.D.; McKay, S.; Kleiner, E.L.; Bird, A.P. Identification of a mammalian protein that binds specifically to DNA containing methylated CpGs. Cell 1989, 58, 499–507. [Google Scholar] [CrossRef]
- Mellen, M.; Ayata, P.; Dewell, S.; Kriaucionis, S.; Heintz, N. MeCP2 binds to 5hmC enriched within active genes and accessible chromatin in the nervous system. Cell 2012, 151, 1417–1430. [Google Scholar] [CrossRef] [Green Version]
- Meehan, R.R.; Lewis, J.D.; Bird, A.P. Characterization of MeCP2, a vertebrate DNA binding protein with affinity for methylated DNA. Nucleic Acids Res. 1992, 20, 5085–5092. [Google Scholar] [CrossRef] [Green Version]
- Nan, X.; Campoy, F.J.; Bird, A. MeCP2 is a transcriptional repressor with abundant binding sites in genomic chromatin. Cell 1997, 88, 471–481. [Google Scholar] [CrossRef] [Green Version]
- Yu, F.; Thiesen, J.; Stratling, W.H. Histone deacetylase-independent transcriptional repression by methyl-CpG-binding protein 2. Nucleic Acids Res. 2000, 28, 2201–2206. [Google Scholar] [CrossRef] [Green Version]
- Fuks, F.; Hurd, P.J.; Wolf, D.; Nan, X.; Bird, A.P.; Kouzarides, T. The methyl-CpG-binding protein MeCP2 links DNA methylation to histone methylation. J. Biol. Chem. 2003, 278, 4035–4040. [Google Scholar] [CrossRef]
- Lyst, M.J.; Ekiert, R.; Ebert, D.H.; Merusi, C.; Nowak, J.; Selfridge, J.; Guy, J.; Kastan, N.R.; Robinson, N.D.; de Lima Alves, F.; et al. Rett syndrome mutations abolish the interaction of MeCP2 with the NCoR/SMRT co-repressor. Nat. Neurosci. 2013, 16, 898–902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kokura, K.; Kaul, S.C.; Wadhwa, R.; Nomura, T.; Khan, M.M.; Shinagawa, T.; Yasukawa, T.; Colmenares, C.; Ishii, S. The Ski protein family is required for MeCP2-mediated transcriptional repression. J. Biol. Chem. 2001, 276, 34115–34121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nan, X.; Hou, J.; Maclean, A.; Nasir, J.; Lafuente, M.J.; Shu, X.; Kriaucionis, S.; Bird, A. Interaction between chromatin proteins MECP2 and ATRX is disrupted by mutations that cause inherited mental retardation. Proc. Natl. Acad. Sci. USA 2007, 104, 2709–2714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaludov, N.K.; Wolffe, A.P. MeCP2 driven transcriptional repression in vitro: Selectivity for methylated DNA, action at a distance and contacts with the basal transcription machinery. Nucleic Acids Res. 2000, 28, 1921–1928. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, H.; Muffat, J.; Cheng, A.W.; Orlando, D.A.; Loven, J.; Kwok, S.M.; Feldman, D.A.; Bateup, H.S.; Gao, Q.; et al. Global transcriptional and translational repression in human-embryonic-stem-cell-derived Rett syndrome neurons. Cell Stem Cell 2013, 13, 446–458. [Google Scholar] [CrossRef] [Green Version]
- Buist, M.; El Tobgy, N.; Shevkoplyas, D.; Genung, M.; Sher, A.A.; Pejhan, S.; Rastegar, M. Differential Sensitivity of the Protein Translation Initiation Machinery and mTOR Signaling to MECP2 Gain- and Loss-of-Function Involves MeCP2 Isoform-Specific Homeostasis in the Brain. Cells 2022, 11, 1442. [Google Scholar] [CrossRef] [PubMed]
- Olson, C.O.; Pejhan, S.; Kroft, D.; Sheikholeslami, K.; Fuss, D.; Buist, M.; Ali Sher, A.; Del Bigio, M.R.; Sztainberg, Y.; Siu, V.M.; et al. MECP2 Mutation Interrupts Nucleolin-mTOR-P70S6K Signaling in Rett Syndrome Patients. Front. Genet. 2018, 9, 635. [Google Scholar] [CrossRef] [Green Version]
- Chahrour, M.; Jung, S.Y.; Shaw, C.; Zhou, X.; Wong, S.T.; Qin, J.; Zoghbi, H.Y. MeCP2, a key contributor to neurological disease, activates and represses transcription. Science 2008, 320, 1224–1229. [Google Scholar] [CrossRef] [Green Version]
- Shahbazian, M.D.; Antalffy, B.; Armstrong, D.L.; Zoghbi, H.Y. Insight into Rett syndrome: MeCP2 levels display tissue- and cell-specific differences and correlate with neuronal maturation. Hum. Mol. Genet. 2002, 11, 115–124. [Google Scholar] [CrossRef]
- Liyanage, V.R.; Zachariah, R.M.; Davie, J.R.; Rastegar, M. Ethanol deregulates Mecp2/MeCP2 in differentiating neural stem cells via interplay between 5-methylcytosine and 5-hydroxymethylcytosine at the Mecp2 regulatory elements. Exp. Neurol 2015, 265, 102–117. [Google Scholar] [CrossRef]
- Xu, W.; Liyanage, V.R.B.; MacAulay, A.; Levy, R.D.; Curtis, K.; Olson, C.O.; Zachariah, R.M.; Amiri, S.; Buist, M.; Hicks, G.G.; et al. Genome-Wide Transcriptome Landscape of Embryonic Brain-Derived Neural Stem Cells Exposed to Alcohol with Strain-Specific Cross-Examination in BL6 and CD1 Mice. Sci. Rep. 2019, 9, 206. [Google Scholar] [CrossRef] [Green Version]
- Zachariah, R.M.; Olson, C.O.; Ezeonwuka, C.; Rastegar, M. Novel MeCP2 isoform-specific antibody reveals the endogenous MeCP2E1 expression in murine brain, primary neurons and astrocytes. PLoS ONE 2012, 7, e49763. [Google Scholar] [CrossRef] [PubMed]
- Liyanage, V.R.B.; Olson, C.O.; Zachariah, R.M.; Davie, J.R.; Rastegar, M. DNA Methylation Contributes to the Differential Expression Levels of Mecp2 in Male Mice Neurons and Astrocytes. Int. J. Mol. Sci. 2019, 20, 1845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liyanage, V.R.; Zachariah, R.M.; Rastegar, M. Decitabine alters the expression of Mecp2 isoforms via dynamic DNA methylation at the Mecp2 regulatory elements in neural stem cells. Mol. Autism 2013, 4, 46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olson, C.O.; Zachariah, R.M.; Ezeonwuka, C.D.; Liyanage, V.R.; Rastegar, M. Brain region-specific expression of MeCP2 isoforms correlates with DNA methylation within Mecp2 regulatory elements. PLoS ONE 2014, 9, e90645. [Google Scholar] [CrossRef]
- Pejhan, S.; Rastegar, M. Role of DNA Methyl-CpG-Binding Protein MeCP2 in Rett Syndrome Pathobiology and Mechanism of Disease. Biomolecules 2021, 11, 75. [Google Scholar] [CrossRef]
- Pejhan, S.; Del Bigio, M.R.; Rastegar, M. The MeCP2E1/E2-BDNF-miR132 Homeostasis Regulatory Network Is Region-Dependent in the Human Brain and Is Impaired in Rett Syndrome Patients. Front. Cell Dev. Biol. 2020, 8, 763. [Google Scholar] [CrossRef]
- Zachariah, R.M.; Rastegar, M. Linking epigenetics to human disease and Rett syndrome: The emerging novel and challenging concepts in MeCP2 research. Neural Plast. 2012, 2012, 415825. [Google Scholar] [CrossRef] [Green Version]
- Buist, M.; Fuss, D.; Rastegar, M. Transcriptional Regulation of MECP2E1-E2 Isoforms and BDNF by Metformin and Simvastatin through Analyzing Nascent RNA Synthesis in a Human Brain Cell Line. Biomolecules 2021, 11, 1253. [Google Scholar] [CrossRef]
- Amir, R.E.; Van den Veyver, I.B.; Wan, M.; Tran, C.Q.; Francke, U.; Zoghbi, H.Y. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 1999, 23, 185–188. [Google Scholar] [CrossRef]
- Liyanage, V.R.; Rastegar, M. Rett syndrome and MeCP2. Neuromol. Med. 2014, 16, 231–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheikh, T.I.; Ausio, J.; Faghfoury, H.; Silver, J.; Lane, J.B.; Eubanks, J.H.; MacLeod, P.; Percy, A.K.; Vincent, J.B. From Function to Phenotype: Impaired DNA Binding and Clustering Correlates with Clinical Severity in Males with Missense Mutations in MECP2. Sci. Rep. 2016, 6, 38590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pejhan, S.; Siu, V.M.; Ang, L.C.; Del Bigio, M.R.; Rastegar, M. Differential brain region-specific expression of MeCP2 and BDNF in Rett Syndrome patients: A distinct grey-white matter variation. Neuropathol. Appl. Neurobiol. 2020, 46, 735–750. [Google Scholar] [CrossRef] [PubMed]
- Hagberg, B.; Aicardi, J.; Dias, K.; Ramos, O. A progressive syndrome of autism, dementia, ataxia, and loss of purposeful hand use in girls: Rett’s syndrome: Report of 35 cases. Ann. Neurol 1983, 14, 471–479. [Google Scholar] [CrossRef]
- Ramocki, M.B.; Tavyev, Y.J.; Peters, S.U. The MECP2 duplication syndrome. Am. J. Med. Genet. A 2010, 152, 1079–1088. [Google Scholar] [CrossRef] [Green Version]
- The NCBI Handbook [Internet]; 2nd ed.; National Center for Biotechnology Information (US): Bethesda (MD, USA). 2013. Available online: https://www.ncbi.nlm.nih.gov/books/NBK143764/ (accessed on 1 October 2022).
- Yates, A.D.; Achuthan, P.; Akanni, W.; Allen, J.; Allen, J.; Alvarez-Jarreta, J.; Amode, M.R.; Armean, I.M.; Azov, A.G.; Bennett, R.; et al. Ensembl 2020. Nucleic Acids Res. 2020, 48, D682–D688. [Google Scholar] [CrossRef]
- UniProt Consortium, T. UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2018, 46, 2699. [Google Scholar] [CrossRef] [Green Version]
- Kent, W.J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, D. The human genome browser at UCSC. Genome Res. 2002, 12, 996–1006. [Google Scholar] [CrossRef] [Green Version]
- Robinson, J.T.; Thorvaldsdottir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [Green Version]
- Kriaucionis, S.; Bird, A. The major form of MeCP2 has a novel N-terminus generated by alternative splicing. Nucleic Acids Res. 2004, 32, 1818–1823. [Google Scholar] [CrossRef]
- Yasui, D.H.; Gonzales, M.L.; Aflatooni, J.O.; Crary, F.K.; Hu, D.J.; Gavino, B.J.; Golub, M.S.; Vincent, J.B.; Carolyn Schanen, N.; Olson, C.O.; et al. Mice with an isoform-ablating Mecp2 exon 1 mutation recapitulate the neurologic deficits of Rett syndrome. Hum. Mol. Genet. 2014, 23, 2447–2458. [Google Scholar] [CrossRef] [PubMed]
- Martinez de Paz, A.; Khajavi, L.; Martin, H.; Claveria-Gimeno, R.; Tom Dieck, S.; Cheema, M.S.; Sanchez-Mut, J.V.; Moksa, M.M.; Carles, A.; Brodie, N.I.; et al. MeCP2-E1 isoform is a dynamically expressed, weakly DNA-bound protein with different protein and DNA interactions compared to MeCP2-E2. Epigenetics Chromatin 2019, 12, 63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Francke, U. Identification of cis-regulatory elements for MECP2 expression. Hum. Mol. Genet. 2006, 15, 1769–1782. [Google Scholar] [CrossRef] [Green Version]
- Itoh, M.; Tahimic, C.G.; Ide, S.; Otsuki, A.; Sasaoka, T.; Noguchi, S.; Oshimura, M.; Goto, Y.; Kurimasa, A. Methyl CpG-binding protein isoform MeCP2_e2 is dispensable for Rett syndrome phenotypes but essential for embryo viability and placenta development. J. Biol. Chem. 2012, 287, 13859–13867. [Google Scholar] [CrossRef] [Green Version]
- Creyghton, M.P.; Cheng, A.W.; Welstead, G.G.; Kooistra, T.; Carey, B.W.; Steine, E.J.; Hanna, J.; Lodato, M.A.; Frampton, G.M.; Sharp, P.A.; et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci. USA 2010, 107, 21931–21936. [Google Scholar] [CrossRef] [Green Version]
- Schneider, R.; Bannister, A.J.; Myers, F.A.; Thorne, A.W.; Crane-Robinson, C.; Kouzarides, T. Histone H3 lysine 4 methylation patterns in higher eukaryotic genes. Nat. Cell Biol. 2004, 6, 73–77. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, S.; Meshorer, E.; Ast, G. Chromatin organization marks exon-intron structure. Nat. Struct. Mol. Biol. 2009, 16, 990–995. [Google Scholar] [CrossRef] [PubMed]
- Cao, R.; Wang, L.; Wang, H.; Xia, L.; Erdjument-Bromage, H.; Tempst, P.; Jones, R.S.; Zhang, Y. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science 2002, 298, 1039–1043. [Google Scholar] [CrossRef] [Green Version]
- Giglio, M.; Tauber, R.; Nadendla, S.; Munro, J.; Olley, D.; Ball, S.; Mitraka, E.; Schriml, L.M.; Gaudet, P.; Hobbs, E.T.; et al. ECO, the Evidence & Conclusion Ontology: Community standard for evidence information. Nucleic Acids Res. 2019, 47, D1186–D1194. [Google Scholar] [CrossRef] [Green Version]
- McWilliam, H.; Li, W.; Uludag, M.; Squizzato, S.; Park, Y.M.; Buso, N.; Cowley, A.P.; Lopez, R. Analysis Tool Web Services from the EMBL-EBI. Nucleic Acids Res. 2013, 41, W597-600. [Google Scholar] [CrossRef]
- Gasteiger, E.; Gattiker, A.; Hoogland, C.; Ivanyi, I.; Appel, R.D.; Bairoch, A. ExPASy: The proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res. 2003, 31, 3784–3788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nan, X.; Meehan, R.R.; Bird, A. Dissection of the methyl-CpG binding domain from the chromosomal protein MeCP2. Nucleic Acids Res. 1993, 21, 4886–4892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, V.H.; McBryant, S.J.; Wade, P.A.; Woodcock, C.L.; Hansen, J.C. Intrinsic disorder and autonomous domain function in the multifunctional nuclear protein, MeCP2. J. Biol. Chem. 2007, 282, 15057–15064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kent, W.J. BLAT--the BLAST-like alignment tool. Genome Res. 2002, 12, 656–664. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef] [Green Version]
- Consortium, E.P. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef] [Green Version]
- Davis, C.A.; Hitz, B.C.; Sloan, C.A.; Chan, E.T.; Davidson, J.M.; Gabdank, I.; Hilton, J.A.; Jain, K.; Baymuradov, U.K.; Narayanan, A.K.; et al. The Encyclopedia of DNA elements (ENCODE): Data portal update. Nucleic Acids Res. 2018, 46, D794–D801. [Google Scholar] [CrossRef] [Green Version]
- Lister, R.; Mukamel, E.A.; Nery, J.R.; Urich, M.; Puddifoot, C.A.; Johnson, N.D.; Lucero, J.; Huang, Y.; Dwork, A.J.; Schultz, M.D.; et al. Global epigenomic reconfiguration during mammalian brain development. Science 2013, 341, 1237905. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Accession Number | Transcript Variant | Protein Isoform Encoded |
|---|---|---|
| NM_004992.4 | 1 | Translation initiates in exon 2, producing protein isoform 1 with a distinct N-terminus |
| NM_001110792.2 | 2 | Translation initiates in exon 1, producing protein isoform 2 with a distinct N-terminus |
| NM_001316337.2 | 3 | Product: Protein isoform 3; it has a shorter N-terminus compared to isoform 1 |
| NM_001369391.2 | 4 | |
| NM_001369392.2 | 5 | |
| NM_001369393.2 | 6 | |
| NM_001369394.2 | 7 | |
| NM_001386137.1 | 8 | Product: Protein isoform 4 |
| NM_001386138.1 | 9 | |
| NM_001386139.1 | 10 |
| UniProt Data | Ensembl Data | |||||
|---|---|---|---|---|---|---|
| UniProt Identifier | Status | Evidence | Ensembl Transcript ID | Biotype | Transcript Support Level (TSL) | Incomplete |
| P51608-1 | Reviewed | Evidence at protein level | ENST00000303391.11 | Protein coding | TSL:1 | - |
| P51608-2 | Reviewed | Evidence at protein level | ENST00000453960.7 | Protein coding | TSL:1 | - |
| B5MCB4 | Unreviewed | Evidence at protein level | ENST00000407218.5 | Protein coding | TSL:5 | - |
| A0A0D9SFX7 | Unreviewed | Evidence at protein level | ENST00000628176.2 | Protein coding | TSL:3 | - |
| C9JH89 | Unreviewed | Evidence at protein level | ENST00000415944.3 | Protein coding | TSL:5 | CDS 3’ incomplete |
| A0A1B0GTV0 | Unreviewed | Evidence at protein level | ENST00000637917.1 | Protein coding | TSL:5 | CDS 5’ and 3’ incomplete |
| A0A0D9SEX1 | Unreviewed | Evidence at protein level | ENST00000630151.2 | Protein coding | TSL:5 | CDS 3’ incomplete |
| H7BY72 | Unreviewed | Protein predicted | ENST00000369957.5 | Nonsense mediated decay | TSL:3 | - |
| A0A6Q8PHQ3 | Unreviewed | Protein predicted | ENST00000674996.1 | Nonsense mediated decay | - | - |
| A0A6Q8PF93 | Unreviewed | Protein predicted | ENST00000675526.1 | Nonsense mediated decay | - | - |
| NCBI | ||||
|---|---|---|---|---|
| Isoform 1 | Isoform 2 | Isoform 3 | Isoform 4 | |
| Exon 1 | - | ✓ | - | - |
| Exon 2 | ✓ | - | - | - |
| Exon 3 | ✓ | ✓ | N-term. incompl. Starts at alternative M in exon 3 | - |
| Exon 4 | ✓ | ✓ | ✓ | N-term. incompl. Starts at alternative M in exon 4 |
| HMGD1 | ✓ | Possible different N-terminus | - | - |
| HMGD2 | ✓ | ✓ | ✓ | - |
| MBD | ✓ | ✓ | N-term. incompl. | - |
| aDBD | ✓ | ✓ | ✓ | N-term. incompl. |
| TRD | ✓ | ✓ | ✓ | N-term. incompl. |
| NLS1 | ✓ | ✓ | ✓ | - |
| NLS2 | ✓ | ✓ | ✓ | ✓ |
| AT hook 1 | ✓ | ✓ | ✓ | - |
| AT hook 2 | ✓ | ✓ | ✓ | ✓ |
| H3-M-T IS | ✓ | ✓ | N-term. incompl. | - |
| NID | ✓ | ✓ | ✓ | ✓ |
| TBL1XR1 IS | ✓ | ✓ | ✓ | ✓ |
| WW-2 IS | ✓ | ✓ | ✓ | ✓ |
| CTD⍺ | ✓ | ✓ | ✓ | ✓ |
| CTDβ | ✓ | ✓ | ✓ | ✓ |
| UniProt | ||||||||
|---|---|---|---|---|---|---|---|---|
| B5MCB4 | A0A0D 9SFX7 | C9JH89 | A0A1B0 GTV0 | A0A0D 9SEX1 | H7BY72 | A0A6Q 8PHQ3 | A0A6Q 8PF93 | |
| Exon 1 | ✓ | - | - | - | - | - | - | - |
| Exon 2 | - | ✓ | ✓ | - | ✓ | C-term. incompl. | ✓ | ✓ |
| Exon 3 | ✓ | ✓ | C-term. incompl. | - | C-term. incompl. | - | - | - |
| Exon 4 | C-term. incompl. | C-term. incompl. | - | C-term. incompl, missing fragment inside | - | - | - | - |
| HMGD1 | Possible different N-terminus | ✓ | C-term. incompl. | - | C-term. incompl. | C-term. incompl. | C-term. incompl. | C-term. incompl. |
| HMGD2 | - | C-term. incompl. | - | - | - | - | - | - |
| MBD | C-term. incompl. | C-term. incompl. | - | N-term. and C-term. Incompl. | - | - | - | - |
| aDBD | - | - | - | N-term. incompl. | - | - | - | - |
| TRD | - | - | - | N-term. incompl. | - | - | - | - |
| H3-M-T IS | ✓ | ✓ | - | - | - | - | - | - |
| NID | - | - | - | N-term. incompl. | - | - | - | - |
| TBL1XR1 IS | - | - | - | N-term. incompl. | - | - | - | - |
| WW-2 IS | - | - | - | - | - | - | - | - |
| CTDα | - | - | - | C-term. incompl. | - | - | - | - |
| CTDβ | - | - | - | - | - | - | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shevkoplyas, D.; Vuu, Y.M.; Davie, J.R.; Rastegar, M. The Chromatin Structure at the MECP2 Gene and In Silico Prediction of Potential Coding and Non-Coding MECP2 Splice Variants. Int. J. Mol. Sci. 2022, 23, 15643. https://doi.org/10.3390/ijms232415643
Shevkoplyas D, Vuu YM, Davie JR, Rastegar M. The Chromatin Structure at the MECP2 Gene and In Silico Prediction of Potential Coding and Non-Coding MECP2 Splice Variants. International Journal of Molecular Sciences. 2022; 23(24):15643. https://doi.org/10.3390/ijms232415643
Chicago/Turabian StyleShevkoplyas, Danilo, Yen My Vuu, James R. Davie, and Mojgan Rastegar. 2022. "The Chromatin Structure at the MECP2 Gene and In Silico Prediction of Potential Coding and Non-Coding MECP2 Splice Variants" International Journal of Molecular Sciences 23, no. 24: 15643. https://doi.org/10.3390/ijms232415643
APA StyleShevkoplyas, D., Vuu, Y. M., Davie, J. R., & Rastegar, M. (2022). The Chromatin Structure at the MECP2 Gene and In Silico Prediction of Potential Coding and Non-Coding MECP2 Splice Variants. International Journal of Molecular Sciences, 23(24), 15643. https://doi.org/10.3390/ijms232415643