
Expanding the Therapeutic Window of EGFR-Targeted PE24 Immunotoxin for EGFR-Overexpressing Cancers by Tailoring the EGFR Binding Affinity



Abstract
:
1. Introduction
2. Results
2.1. Construction of ER-PE24 ITs with Different Affinities for EGFR
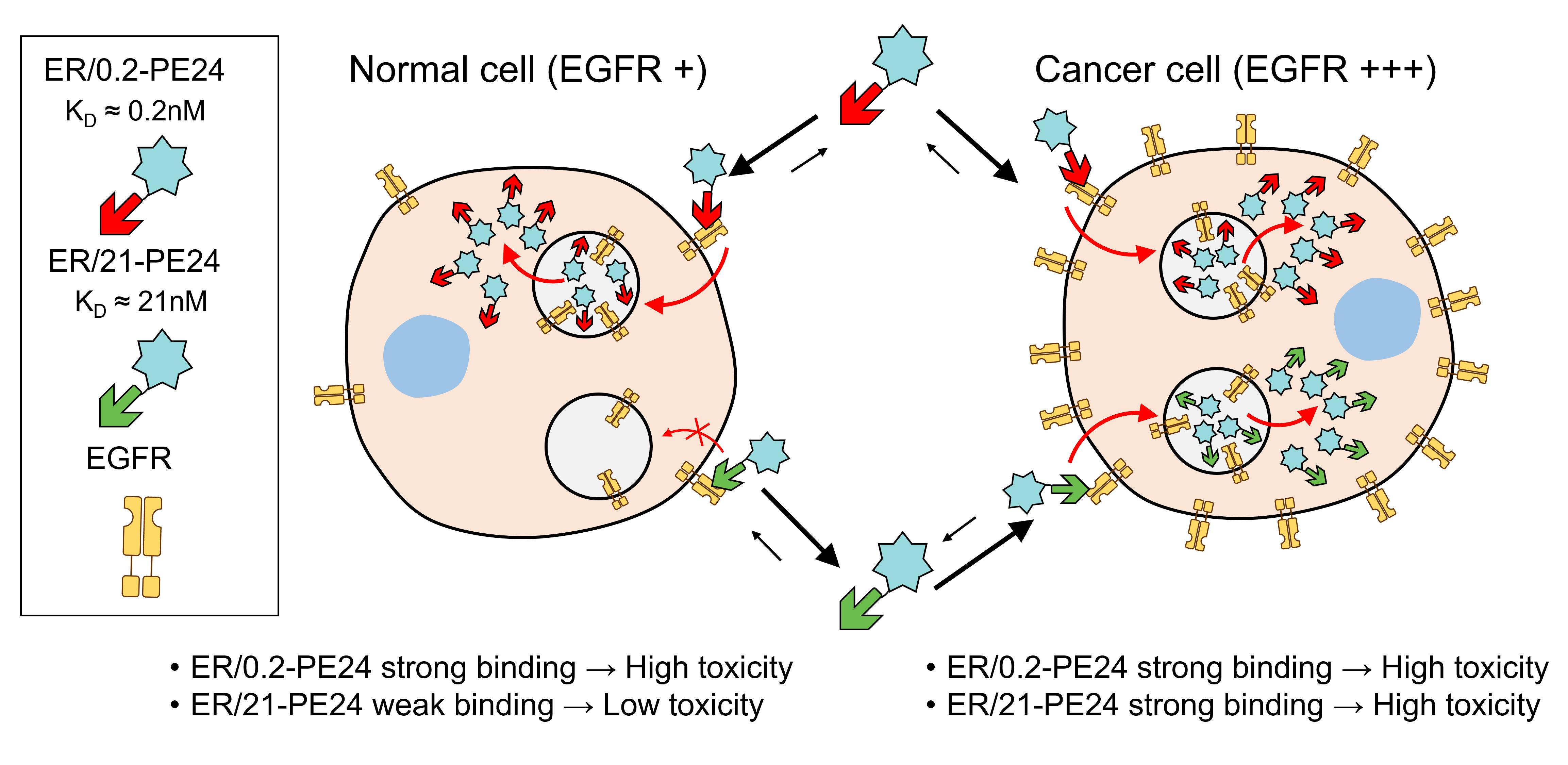
2.2. ER-PE24 IT Binds to EGFR-Expressing Cells According to EGFR Affinity and Cellular EGFR Expression Levels
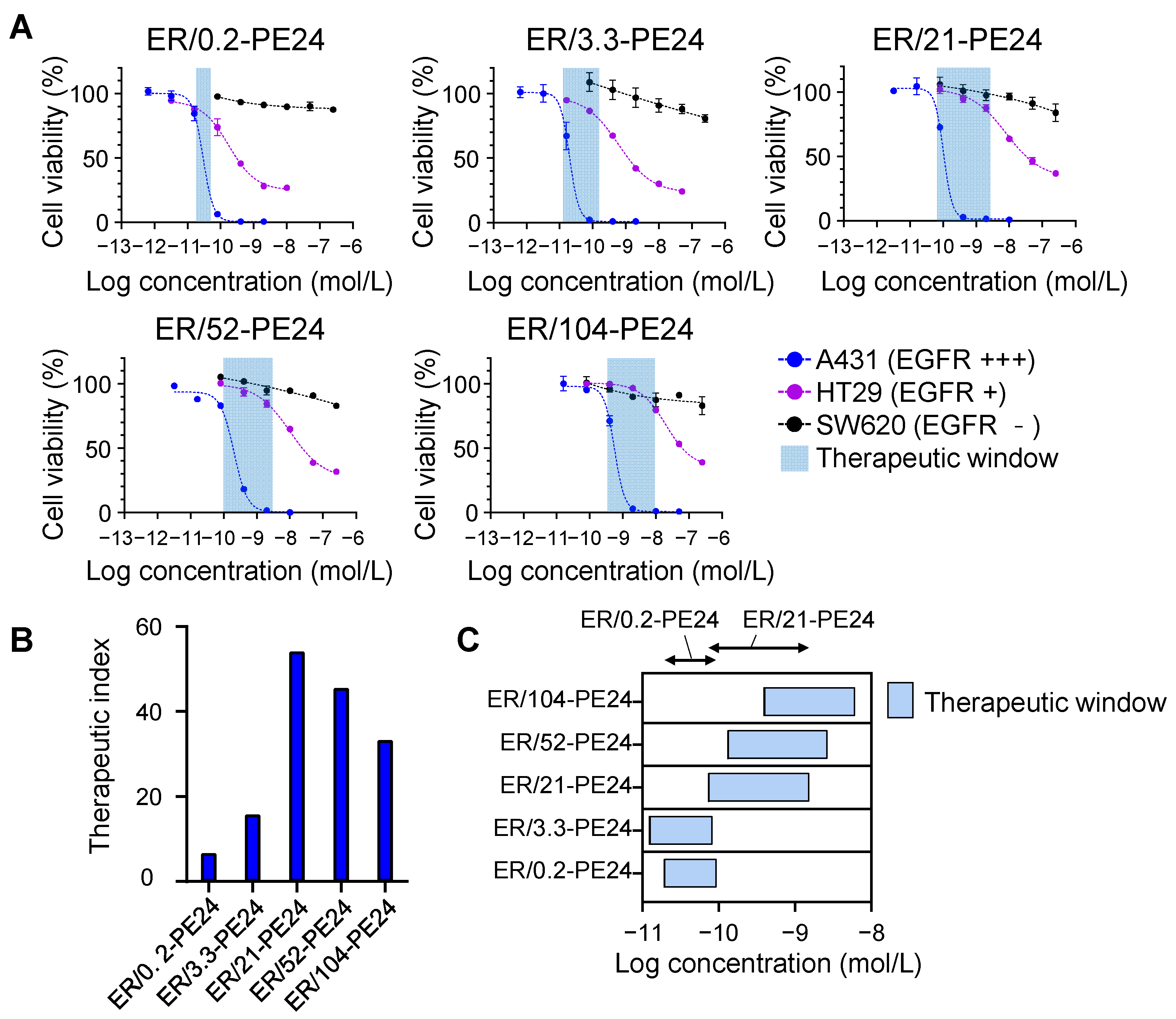
2.3. In Vitro Therapeutic Window of ER-PE24 Varies Depending on the Affinity for EGFR
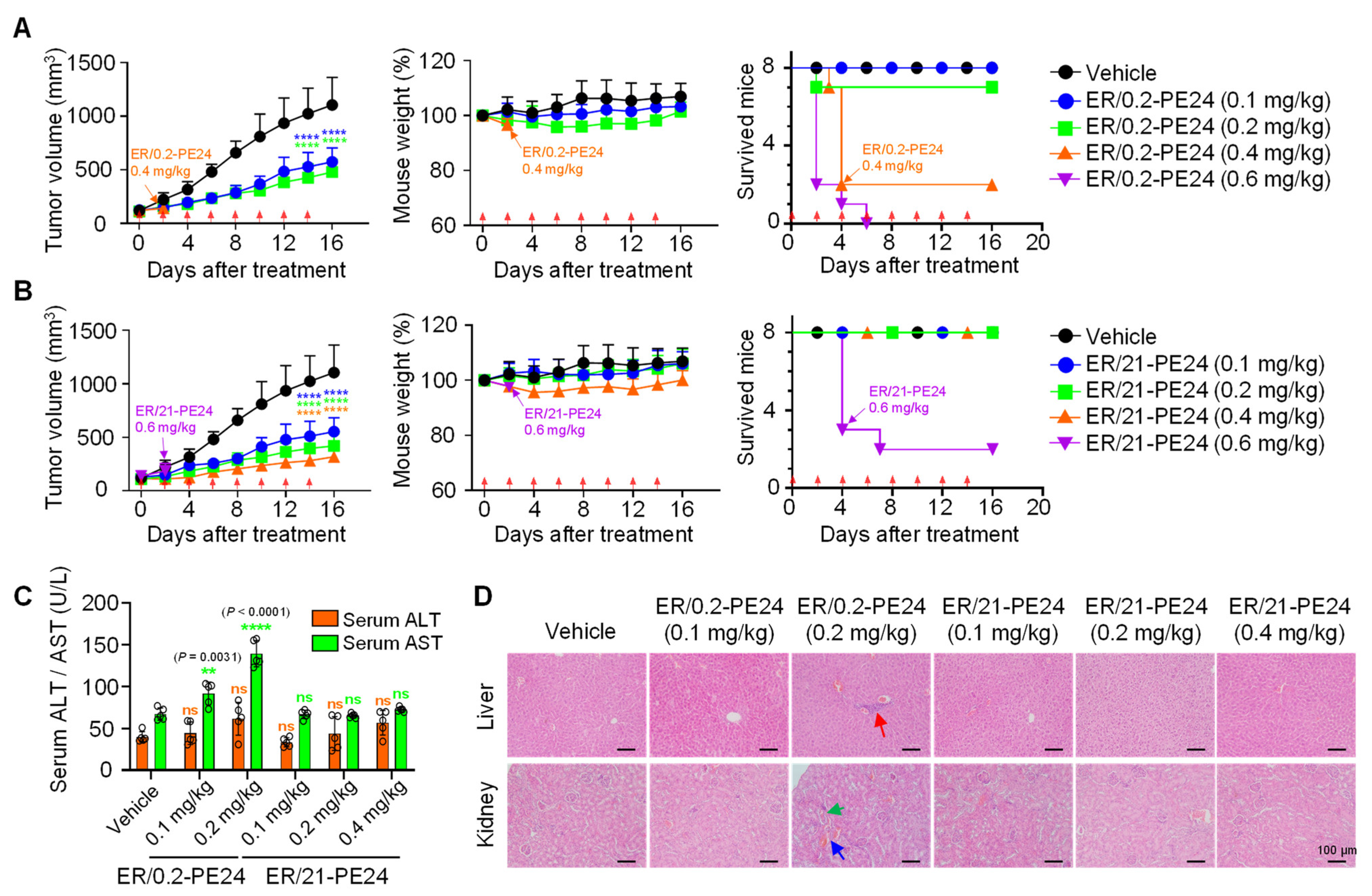
2.4. In Vivo Antitumor Activity and Toxicity of ER/0.2-PE24 and ER/21-PE24 in a Xenograft Model
3. Discussion
4. Materials and Methods
4.1. Cell Lines
4.2. Plasmid Construction
4.3. Protein Expression and Purification
4.4. ELISA
4.5. Bio-Layer Interferometry
4.6. Cell Surface EGFR Binding Assay
4.7. Cell Viability Assay
4.8. Mouse Xenograft Tumor Model
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kim, J.S.; Jun, S.Y.; Kim, Y.S. Critical Issues in the Development of Immunotoxins for Anticancer Therapy. J. Pharm. Sci. 2020, 109, 104–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antignani, A.; Ho, E.C.H.; Bilotta, M.T.; Qiu, R.; Sarnvosky, R.; Fitz Gerald, D.J. Targeting Receptors on Cancer Cells with Protein Toxins. Biomolecules 2020, 10, 1331. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Mei, S.; Yang, Y.; Shen, Y.; Chen, L. Strategies to mitigate the on- and off-target toxicities of recombinant immunotoxins: An antibody engineering perspective. Antib. Ther. 2022, 5, 164–176. [Google Scholar] [CrossRef] [PubMed]
- Yarden, Y.; Pines, G. The ERBB network: At last, cancer therapy meets systems biology. Nat. Rev. Cancer 2012, 12, 553–563. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.J.; Baek, D.S.; Lee, S.; Park, D.; Kang, H.N.; Cho, B.C.; Kim, Y.S. Dual-targeting of EGFR and Neuropilin-1 attenuates resistance to EGFR-targeted antibody therapy in KRAS-mutant non-small cell lung cancer. Cancer Lett. 2019, 466, 23–34. [Google Scholar] [CrossRef]
- Fischer, A.; Wolf, I.; Fuchs, H.; Masilamani, A.P.; Wolf, P. Pseudomonas Exotoxin A Based Toxins Targeting Epidermal Growth Factor Receptor for the Treatment of Prostate Cancer. Toxins 2020, 12, 753. [Google Scholar] [CrossRef]
- Chaudhary, V.K.; FitzGerald, D.J.; Adhya, S.; Pastan, I. Activity of a recombinant fusion protein between transforming growth factor type alpha and Pseudomonas toxin. Proc. Natl. Acad. Sci. USA 1987, 84, 4538–4542. [Google Scholar] [CrossRef] [Green Version]
- Niesen, J.; Stein, C.; Brehm, H.; Hehmann-Titt, G.; Fendel, R.; Melmer, G.; Fischer, R.; Barth, S. Novel EGFR-specific immunotoxins based on panitumumab and cetuximab show in vitro and ex vivo activity against different tumor entities. J. Cancer Res. Clin. Oncol. 2015, 141, 2079–2095. [Google Scholar] [CrossRef]
- Wu, S.; Deng, C.; Zhang, C.; Xiong, J.; Gu, X.; Wang, Z.; Tu, J.; Xie, J. Preparation of a novel EGFR specific immunotoxin and its efficacy of anti-colorectal cancer in vitro and in vivo. Clin. Transl. Oncol. 2021, 23, 1549–1560. [Google Scholar] [CrossRef]
- Xie, G.; Shan, L.; Liu, Y.; Wu, T.C.; Gu, X. Antitumor Efficacy of EGFR-Targeted Recombinant Immunotoxin in Human Head and Neck Squamous Cell Carcinoma. Biology 2022, 11, 486. [Google Scholar] [CrossRef]
- Fernandes Neto, J.M.; Nadal, E.; Bosdriesz, E.; Ooft, S.N.; Farre, L.; McLean, C.; Klarenbeek, S.; Jurgens, A.; Hagen, H.; Wang, L.; et al. Multiple low dose therapy as an effective strategy to treat EGFR inhibitor-resistant NSCLC tumours. Nat. Commun. 2020, 11, 3157. [Google Scholar] [CrossRef]
- Liu, X.; Jiang, S.; Fang, C.; Yang, S.; Olalere, D.; Pequignot, E.C.; Cogdill, A.P.; Li, N.; Ramones, M.; Granda, B.; et al. Affinity-Tuned ErbB2 or EGFR Chimeric Antigen Receptor T Cells Exhibit an Increased Therapeutic Index against Tumors in Mice. Cancer Res. 2015, 75, 3596–3607. [Google Scholar] [CrossRef] [Green Version]
- Hollevoet, K.; Mason-Osann, E.; Liu, X.F.; Imhof-Jung, S.; Niederfellner, G.; Pastan, I. In vitro and in vivo activity of the low-immunogenic antimesothelin immunotoxin RG7787 in pancreatic cancer. Mol. Cancer Ther. 2014, 13, 2040–2049. [Google Scholar] [CrossRef] [Green Version]
- Mazor, R.; Pastan, I. Immunogenicity of Immunotoxins Containing Pseudomonas Exotoxin A: Causes, Consequences, and Mitigation. Front. Immunol. 2020, 11, 1261. [Google Scholar] [CrossRef]
- Ramamurthy, V.; Krystek, S.R., Jr.; Bush, A.; Wei, A.; Emanuel, S.L.; Das Gupta, R.; Janjua, A.; Cheng, L.; Murdock, M.; Abramczyk, B.; et al. Structures of adnectin/protein complexes reveal an expanded binding footprint. Structure 2012, 20, 259–269. [Google Scholar] [CrossRef] [Green Version]
- Emanuel, S.L.; Engle, L.J.; Chao, G.; Zhu, R.R.; Cao, C.; Lin, Z.; Yamniuk, A.P.; Hosbach, J.; Brown, J.; Fitzpatrick, E. A fibronectin scaffold approach to bispecific inhibitors of epidermal growth factor receptor and insulin-like growth factor-I receptor. mAbs 2011, 3, 38–48. [Google Scholar] [CrossRef]
- Kreitman, R.J.; Pastan, I. Importance of the glutamate residue of KDEL in increasing the cytotoxicity of Pseudomonas exotoxin derivatives and for increased binding to the KDEL receptor. Biochem. J. 1995, 307, 29–37. [Google Scholar] [CrossRef]
- Pirie, C.M.; Liu, D.V.; Wittrup, K.D. Targeted cytolysins synergistically potentiate cytoplasmic delivery of gelonin immunotoxin. Mol. Cancer Ther. 2013, 12, 1774–1782. [Google Scholar] [CrossRef] [Green Version]
- Wong, O.K.; Tran, T.T.; Ho, W.H.; Casas, M.G.; Au, M.; Bateman, M.; Lindquist, K.C.; Rajpal, A.; Shelton, D.L.; Strop, P.; et al. RN765C, a low affinity EGFR antibody drug conjugate with potent anti-tumor activity in preclinical solid tumor models. Oncotarget 2018, 9, 33446–33458. [Google Scholar] [CrossRef] [Green Version]
- Kohno, M.; Horibe, T.; Haramoto, M.; Yano, Y.; Ohara, K.; Nakajima, O.; Matsuzaki, K.; Kawakami, K. A novel hybrid peptide targeting EGFR-expressing cancers. Eur. J. Cancer 2011, 47, 773–783. [Google Scholar] [CrossRef]
- Shin, S.M.; Kim, J.S.; Park, S.W.; Jun, S.Y.; Kweon, H.J.; Choi, D.K.; Lee, D.; Cho, Y.B.; Kim, Y.S. Direct targeting of oncogenic RAS mutants with a tumor-specific cytosol-penetrating antibody inhibits RAS mutant-driven tumor growth. Sci. Adv. 2020, 6, 2174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Arredondo, A.; Rojas-Molina, A.; Bah, M.; Ibarra-Alvarado, C.; Gallegos-Corona, M.A.; Garcia-Servin, M. Systemic toxic effects induced by the aqueous extract of the fire coral Millepora complanata and partial purification of thermostable neurotoxins with lethal effects in mice. Comp. Biochem. Physiol. Toxicol. Pharmacol. 2015, 169, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Onda, M.; Willingham, M.; Wang, Q.C.; Kreitman, R.J.; Tsutsumi, Y.; Nagata, S.; Pastan, I. Inhibition of TNF-alpha produced by Kupffer cells protects against the nonspecific liver toxicity of immunotoxin anti-Tac (Fv)-PE38, LMB-2. J. Immunol. 2000, 165, 7150–7156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.F.; Wei, J.; Zhou, Q.; Molitoris, B.A.; Sandoval, R.; Kobayashi, H.; Okada, R.; Nagaya, T.; Karim, B.; Butcher, D.; et al. Immunotoxin SS1P is rapidly removed by proximal tubule cells of kidney, whose damage contributes to albumin loss in urine. Proc. Natl. Acad. Sci. USA 2020, 117, 6086–6091. [Google Scholar] [CrossRef] [PubMed]
- Alewine, C.; Hassan, R.; Pastan, I. Advances in anticancer immunotoxin therapy. Oncologist 2015, 20, 176–185. [Google Scholar] [CrossRef] [Green Version]
- Kuan, C.T.; Wikstrand, C.J.; Archer, G.; Beers, R.; Pastan, I.; Zalutsky, M.R.; Bigner, D.D. Increased binding affinity enhances targeting of glioma xenografts by EGFRvIII-specific scFv. Int. J. Cancer 2000, 88, 962–969. [Google Scholar] [CrossRef]
- Simon, N.; Antignani, A.; Sarnovsky, R.; Hewitt, S.M.; FitzGerald, D. Targeting a Cancer-Specific Epitope of the Epidermal Growth Factor Receptor in Triple-Negative Breast Cancer. J. Natl. Cancer Inst. 2016, 108, djw028. [Google Scholar] [CrossRef] [Green Version]
- Chandramohan, V.; Pegram, C.N.; Piao, H.; Szafranski, S.E.; Kuan, C.T.; Pastan, I.H.; Bigner, D.D. Production and quality control assessment of a GLP-grade immunotoxin, D2C7-(scdsFv)-PE38KDEL, for a phase I/II clinical trial. Appl. Microbiol. Biotechnol. 2017, 101, 2747–2766. [Google Scholar] [CrossRef] [Green Version]
- Cao, Y.; Marks, J.D.; Huang, Q.; Rudnick, S.I.; Xiong, C.; Hittelman, W.N.; Wen, X.; Marks, J.W.; Cheung, L.H.; Boland, K.; et al. Single-chain antibody-based immunotoxins targeting Her2/neu: Design optimization and impact of affinity on antitumor efficacy and off-target toxicity. Mol. Cancer Ther. 2012, 11, 143–153. [Google Scholar] [CrossRef] [Green Version]
- Purde, V.; Kudryashova, E.; Heisler, D.B.; Shakya, R.; Kudryashov, D.S. Intein-mediated cytoplasmic reconstitution of a split toxin enables selective cell ablation in mixed populations and tumor xenografts. Proc. Natl. Acad. Sci. USA 2020, 117, 22090–22100. [Google Scholar] [CrossRef]
- Zheng, Z.; Okada, R.; Kobayashi, H.; Nagaya, T.; Wei, J.; Zhou, Q.; Lee, F.; Bera, T.K.; Gao, Y.; Kuhlman, W.; et al. Site-Specific PEGylation of Anti-Mesothelin Recombinant Immunotoxins Increases Half-life and Antitumor Activity. Mol. Cancer Ther. 2020, 19, 812–821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, K.; Yoo, S.; Kim, J.-E.; Kim, W.; Kim, Y.-S. Improved intratumoral penetration of IL12 immunocytokine enhances the antitumor efficacy. Front. Immunol. 2022, 13, 1034774. [Google Scholar] [CrossRef] [PubMed]
- Kriegs, M.; Clauditz, T.S.; Hoffer, K.; Bartels, J.; Buhs, S.; Gerull, H.; Zech, H.B.; Bussmann, L.; Struve, N.; Rieckmann, T.; et al. Analyzing expression and phosphorylation of the EGF receptor in HNSCC. Sci. Rep. 2019, 9, 13564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baek, D.S.; Kim, J.H.; Kim, Y.J.; Kim, Y.S. Immunoglobulin Fc-Fused Peptide without C-Terminal Arg or Lys Residue Augments Neuropilin-1-Dependent Tumor Vascular Permeability. Mol. Pharm. 2018, 15, 394–402. [Google Scholar] [CrossRef]
- Jung, K.; Son, M.J.; Lee, S.Y.; Kim, J.A.; Ko, D.H.; Yoo, S.; Kim, C.H.; Kim, Y.S. Antibody-mediated delivery of a viral MHC-I epitope into the cytosol of target tumor cells repurposes virus-specific CD8(+) T cells for cancer immunotherapy. Mol. Cancer 2022, 21, 102. [Google Scholar] [CrossRef]
- Kim, J.E.; Lee, D.H.; Jung, K.; Kim, E.J.; Choi, Y.; Park, H.S.; Kim, Y.S. Engineering of Humanized Antibodies Against Human Interleukin 5 Receptor Alpha Subunit That Cause Potent Antibody-Dependent Cell-Mediated Cytotoxicity. Front. Immunol. 2020, 11, 593748. [Google Scholar] [CrossRef]
- Jung, K.; Kim, J.A.; Kim, Y.J.; Lee, H.W.; Kim, C.H.; Haam, S.; Kim, Y.S. A Neuropilin-1 Antagonist Exerts Antitumor Immunity by Inhibiting the Suppressive Function of Intratumoral Regulatory T Cells. Cancer Immunol. Res. 2020, 8, 46–56. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variants | Mutations in Anti-EGFR Monobody | Binding Kinetic Parameters (a) | ||
|---|---|---|---|---|
| KD (nM) | kon (M−1s−1, ×104) | koff (s−1, ×10−4) | ||
| ER/0.2 (b) | T49I | 0.11 ± 0.03 | 14.0 ± 0.2 | 0.16 ± 0.04 |
| ER/0.2-PE24 | 0.24 ± 0.05 | 24.3 ± 0.9 | 0.59 ± 0.12 | |
| ER/3.3 (b) | Wild type | 3.62 ± 0.08 | 7.44 ± 0.08 | 2.69 ± 0.05 |
| ER/3.3-PE24 | 3.27 ± 0.14 | 9.82 ± 0.24 | 3.21 ± 0.12 | |
| ER/21 (b) | Y29H, T49I, V54H, Y83H | 41.5 ± 0.6 | 6.93 ± 0.09 | 28.7 ± 0.1 |
| ER/21-PE24 | 21.3 ± 0.7 | 12.0 ± 0.4 | 25.5 ± 0.4 | |
| ER/52 (b) | T49H, K79H | 59.5 ± 1.2 | 5.43 ± 0.11 | 32.3 ± 0.2 |
| ER/52-PE24 | 52.0 ± 2.6 | 7.57 ± 0.28 | 39.3 ± 1.28 | |
| ER/104 (b) | Q30H | 139 ± 3 | 7.71 ± 0.14 | 107 ± 1 |
| ER/104-PE24 | 104 ± 4 | 6.41 ± 0.24 | 66.7 ± 1.4 | |
| ITs | IC50 (b) | Therapeutic Index (c) | Therapeutic Window (nM) (c) | |
|---|---|---|---|---|
| A431 (nM) | HT29 (nM) | |||
| ER/0.2-PE24 | 0.0305 ± 0.0019 | 0.202 ± 0.020 | 6.6 | (0.0192 ± 0.0022)–(0.0928 ± 0.0016) |
| ER/3.3-PE24 | 0.0186 ± 0.0018 | 0.294 ± 0.088 | 15.8 | (0.0123 ± 0.0005)–(0.0823 ± 0.0320) |
| ER/21-PE24 | 0.112 ± 0.015 | 6.06 ± 1.48 | 54.1 | (0.0732 ± 0.0060)–(1.52 ± 0.14) |
| ER/52-PE24 | 0.296 ± 0.005 | 10.3 ± 1.1 | 45.5 | (0.130 ± 0.018)–(2.65 ± 0.08) |
| ER/104-PE24 | 0.562 ± 0.001 | 18.7 ± 2.4 | 33.3 | (0.389 ± 0.064)–(6.07 ± 2.88) |
| ITs | Dose | Weight Loss (%) | Death/Total Mice |
|---|---|---|---|
| ER/0.2-PE24 | 0.6 mg/kg × 2 | - | 8/8 |
| 0.4 mg/kg × 2 | 3.4 | 6/8 | |
| 0.2 mg/kg × 8 | 0 | 1/8 | |
| 0.1 mg/kg × 8 | 0 | 0/8 | |
| ER/21-PE24 | 0.6 mg/kg × 2 | 2.2 | 6/8 |
| 0.4 mg/kg × 8 | 0 | 0/8 | |
| 0.2 mg/kg × 8 | 0 | 0/8 | |
| 0.1 mg/kg × 8 | 0 | 0/8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jun, S.-Y.; Kim, D.-S.; Kim, Y.-S. Expanding the Therapeutic Window of EGFR-Targeted PE24 Immunotoxin for EGFR-Overexpressing Cancers by Tailoring the EGFR Binding Affinity. Int. J. Mol. Sci. 2022, 23, 15820. https://doi.org/10.3390/ijms232415820
Jun S-Y, Kim D-S, Kim Y-S. Expanding the Therapeutic Window of EGFR-Targeted PE24 Immunotoxin for EGFR-Overexpressing Cancers by Tailoring the EGFR Binding Affinity. International Journal of Molecular Sciences. 2022; 23(24):15820. https://doi.org/10.3390/ijms232415820
Chicago/Turabian StyleJun, Sei-Yong, Dae-Seong Kim, and Yong-Sung Kim. 2022. "Expanding the Therapeutic Window of EGFR-Targeted PE24 Immunotoxin for EGFR-Overexpressing Cancers by Tailoring the EGFR Binding Affinity" International Journal of Molecular Sciences 23, no. 24: 15820. https://doi.org/10.3390/ijms232415820
APA StyleJun, S. -Y., Kim, D. -S., & Kim, Y. -S. (2022). Expanding the Therapeutic Window of EGFR-Targeted PE24 Immunotoxin for EGFR-Overexpressing Cancers by Tailoring the EGFR Binding Affinity. International Journal of Molecular Sciences, 23(24), 15820. https://doi.org/10.3390/ijms232415820