
Specific Post-Translational Modifications of VDAC3 in ALS-SOD1 Model Cells Identified by High-Resolution Mass Spectrometry

 , , , ,
, , , ,  , and
, and 
Abstract
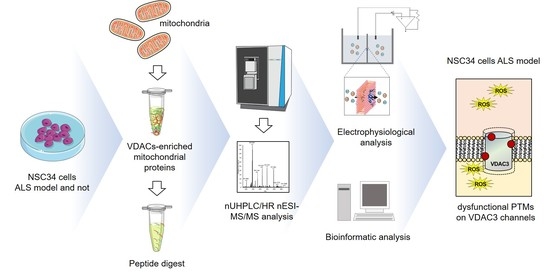
:
1. Introduction
2. Results
2.1. Mass Spectrometry Analysis of VDAC3 from NSC34 Cell Line
2.2. Mass Spectrometry Analysis of VDAC3 from NSC34-SOD1WT Cell Line
2.3. Mass Spectrometry Analysis of VDAC3 from NSC34-SOD1G93A Cell Line
2.4. Other Post-Translational Modifications of VDAC3
2.4.1. Succination, Ubiquitin and Ubiquitination, Phosphorylation, Citrullination, Cysteinylation, and Dioxidation
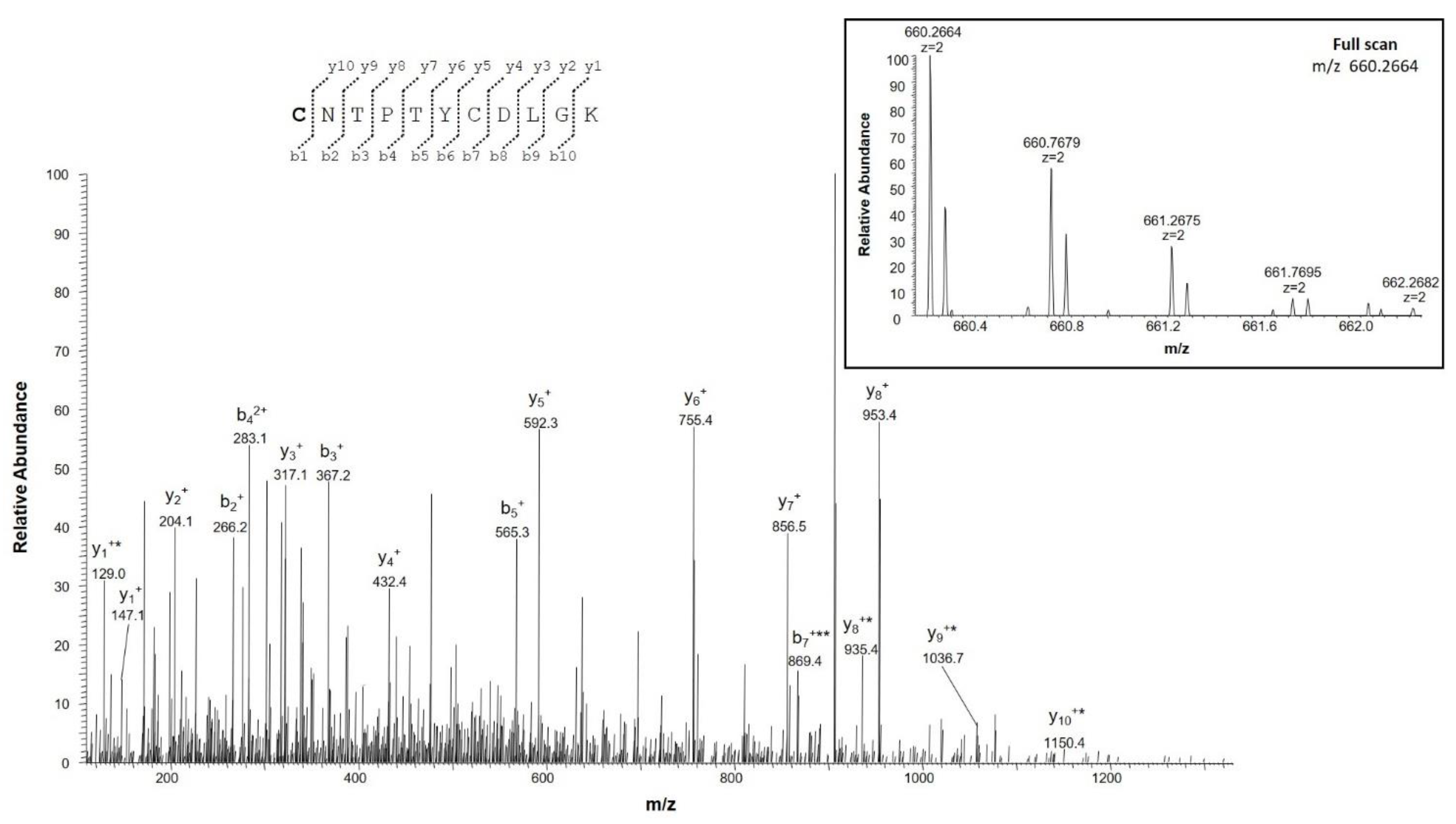
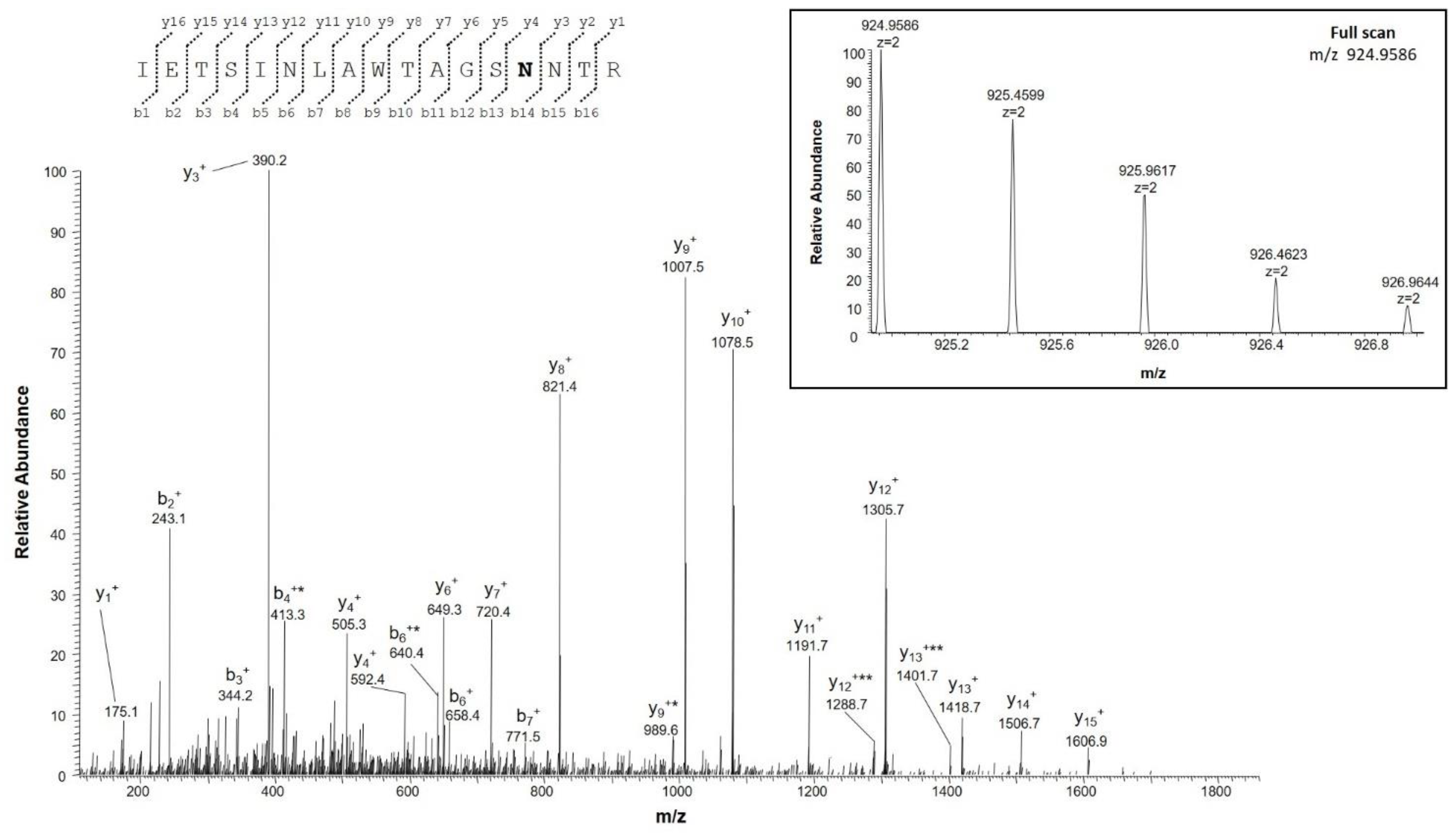
2.4.2. Identification of Deamidation Sites
2.5. Bioinformatic Prediction of VDAC3 N215D Mutant Structure
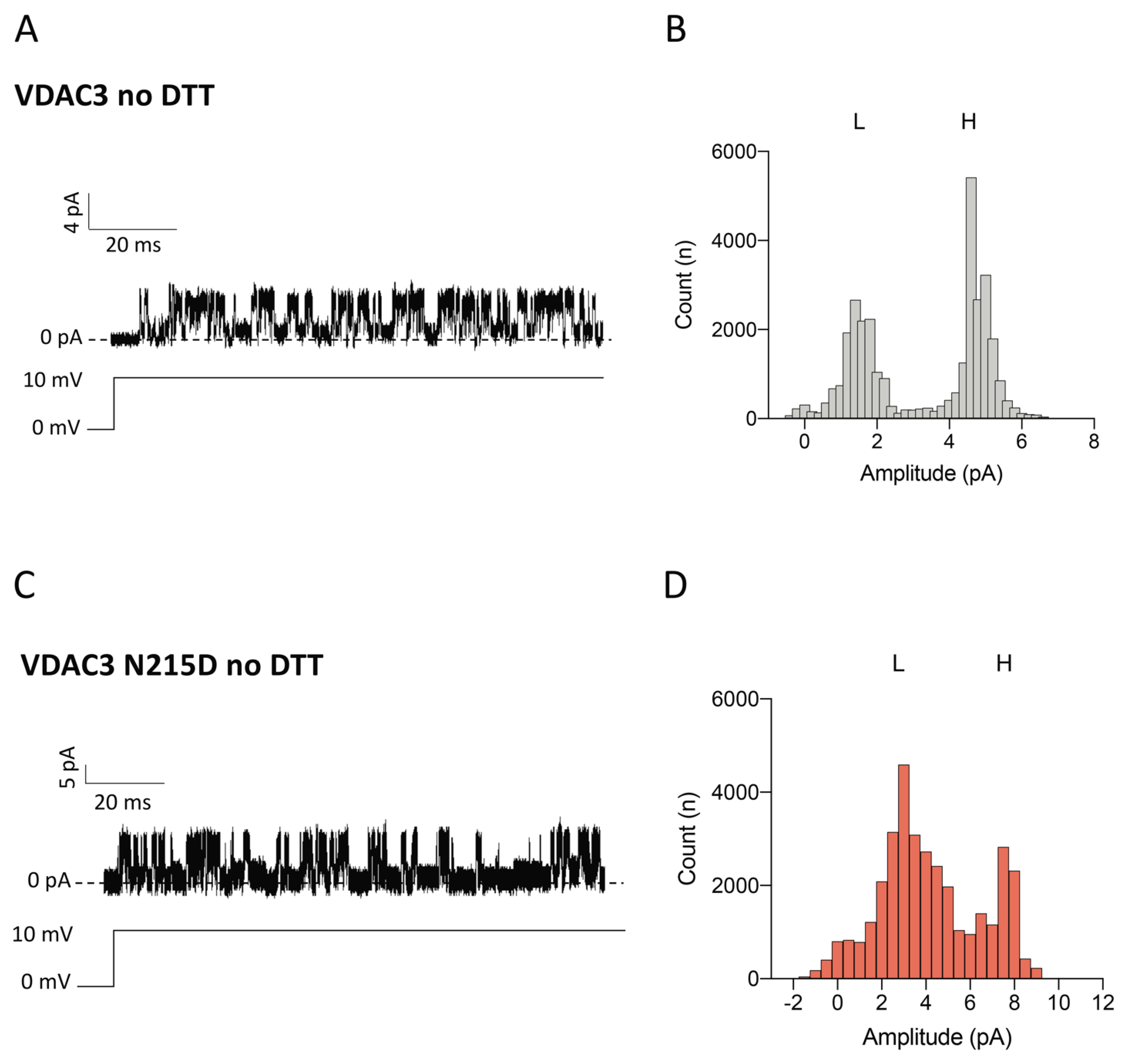
2.6. Electrophysiological Properties of VDAC3 N215D Mutant
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. NSC34 Cell Lines
4.3. Extraction of Mitochondrial Proteins from NSC34 Cells under Reducing Conditions
4.4. Liquid Chromatography and Tandem Mass Spectrometry (LC–MS/MS) Analysis and Database Search
Identification of Deamidation Sites on VDAC3
4.5. Expression, Purification and Refolding of Recombinant VDAC3 Proteins
4.6. Lipid Bilayer Experiments
4.7. Modelling and Bioinformatics Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rowland, L.P.; Shneider, N.A. Amyotrophic Lateral Sclerosis. N. Engl. J. Med. 2001, 344, 1688–1700. [Google Scholar] [CrossRef]
- Pasinelli, P.; Brown, R.H. Molecular biology of amyotrophic lateral sclerosis: Insights from genetics. Nat. Rev. Neurosci. 2006, 7, 710–723. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.H.; Al-Chalabi, A. Amyotrophic Lateral Sclerosis. N. Engl. J. Med. 2017, 377, 162–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaur, S.J.; McKeown, S.R.; Rashid, S. Mutant SOD1 mediated pathogenesis of Amyotrophic Lateral Sclerosis. Gene 2016, 577, 109–118. [Google Scholar] [CrossRef]
- Sanghai, N.; Tranmer, G.K. Hydrogen Peroxide and Amyotrophic Lateral Sclerosis: From Biochemistry to Pathophysiology. Antioxidants 2021, 11, 52. [Google Scholar] [CrossRef]
- Prudencio, M.; Borchelt, D.R. Superoxide dismutase 1 encoding mutations linked to ALS adopts a spectrum of misfolded states. Mol. Neurodegener. 2011, 6, 77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mondola, P.; Damiano, S.; Sassa, A.; Santillo, M. The Cu, Zn Superoxide Dismutase: Not Only a Dismutase Enzyme. Front. Physiol. 2016, 7, 594. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Lu, Y.; Qi, F.; Su, Q.; Wang, L.; You, C.; Che, F.; Yu, J. Effect of the human SOD1-G93A gene on the nrf2/ARE signaling pathway in NSC-34 cells. Mol. Med. Rep. 2014, 9, 2453–2458. [Google Scholar] [CrossRef] [Green Version]
- Marden, J.J.; Harraz, M.M.; Williams, A.J.; Nelson, K.; Luo, M.; Paulson, H.; Engelhardt, J.F. Redox modifier genes in amyotrophic lateral sclerosis in mice. J. Clin. Investig. 2007, 117, 2913–2919. [Google Scholar] [CrossRef]
- Israelson, A.; Arbel, N.; Da Cruz, S.; Ilieva, H.; Yamanaka, K.; Shoshan-Barmatz, V.; Cleveland, D.W. Misfolded Mutant SOD1 Directly Inhibits VDAC1 Conductance in a Mouse Model of Inherited ALS. Neuron 2010, 67, 575–587. [Google Scholar] [CrossRef]
- Shteinfer-Kuzmine, A.; Argueti, S.; Gupta, R.; Shvil, N.; Abu-Hamad, S.; Gropper, Y.; Hoeber, J.; Magrì, A.; Messina, A.; Kozlova, E.N.; et al. A VDAC1-Derived N-Terminal Peptide Inhibits Mutant SOD1-VDAC1 Interactions and Toxicity in the SOD1 Model of ALS. Front. Cell. Neurosci. 2019, 13, 346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shoshan-Barmatz, V.; De Pinto, V.; Zweckstetter, M.; Raviv, Z.; Keinan, N.; Arbel, N. VDAC, a multi-functional mitochondrial protein regulating cell life and death. Mol. Asp. Med. 2010, 31, 227–285. [Google Scholar] [CrossRef] [PubMed]
- Magri, A.; Messina, A. Interactions of VDAC with Proteins Involved in Neurodegenerative Aggregation: An Opportunity for Advancement on Therapeutic Molecules. Curr. Med. Chem. 2017, 24, 4470–4487. [Google Scholar] [CrossRef] [PubMed]
- Shoshan-Barmatz, V.; Ben-Hail, D. VDAC, a multi-functional mitochondrial protein as a pharmacological target. Mitochondrion 2012, 12, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Reina, S.; Guarino, F.; Magrì, A.; De Pinto, V. VDAC3 As a Potential Marker of Mitochondrial Status Is Involved in Cancer and Pathology. Front. Oncol. 2016, 6, 264. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.-C.; Peterson, S.E.; Loring, J.F. Protein post-translational modifications and regulation of pluripotency in human stem cells. Cell Res. 2013, 24, 143–160. [Google Scholar] [CrossRef] [Green Version]
- Duan, G.; Walther, D. The Roles of Post-translational Modifications in the Context of Protein Interaction Networks. PLoS Comput. Biol. 2015, 11, e1004049. [Google Scholar] [CrossRef]
- Reina, S.; Pittalà, M.G.G.; Guarino, F.; Messina, A.; De Pinto, V.; Foti, S.; Saletti, R. Cysteine Oxidations in Mitochondrial Membrane Proteins: The Case of VDAC Isoforms in Mammals. Front. Cell Dev. Biol. 2020, 8, 397. [Google Scholar] [CrossRef]
- Pittalà, M.G.G.; Nibali, S.C.; Reina, S.; Cunsolo, V.; Di Francesco, A.; De Pinto, V.; Messina, A.; Foti, S.; Saletti, R. VDACs Post-Translational Modifications Discovery by Mass Spectrometry: Impact on Their Hub Function. Int. J. Mol. Sci. 2021, 22, 12833. [Google Scholar] [CrossRef]
- Pittalà, M.G.G.; Reina, S.; Cubisino, S.A.M.; Cucina, A.; Formicola, B.; Cunsolo, V.; Foti, S.; Saletti, R.; Messina, A. Post-Translational Modification Analysis of VDAC1 in ALS-SOD1 Model Cells Reveals Specific Asparagine and Glutamine Deamidation. Antioxidants 2020, 9, 1218. [Google Scholar] [CrossRef]
- Magrì, A.; Reina, S.; De Pinto, V. VDAC1 as Pharmacological Target in Cancer and Neurodegeneration: Focus on Its Role in Apoptosis. Front. Chem. 2018, 6, 108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smilansky, S.; Dangoor, L.; Nakdimon, I.; Ben-Hail, D.; Mizrachi, D.; Shosha-Barmatz, V. The Voltage-Dependent Anion Channel 1 mediates amyloid beta toxicity and represents a potential target for Alzheimer’s disease theraphy. JBC 2015, 290, 30670–30683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reina, S.; Checchetto, V.; Saletti, R.; Gupta, A.; Chaturvedi, D.; Guardiani, C.; Guarino, F.; Scorciapino, M.A.; Magrì, A.; Foti, S.; et al. VDAC3 as a sensor of oxidative state of the intermembrane space of mitochondria: The putative role of cysteine residue modifications. Oncotarget 2016, 7, 2249–2268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reina, S.; Nibali, S.C.; Tomasello, M.F.; Magrì, A.; Messina, A.; De Pinto, V. Voltage Dependent Anion Channel 3 (VDAC3) protects mitochondria from oxidative stress. Redox Biol. 2022, 51, 102264. [Google Scholar] [CrossRef] [PubMed]
- Saletti, R.; Reina, S.; Pittalà, M.G.; Belfiore, R.; Cunsolo, V.; Messina, A.; De Pinto, V.; Foti, S. High resolution mass spectrometry characterization of the oxidation pattern of methionine and cysteine residues in rat liver mitochondria voltage-dependent anion selective channel 3 (VDAC3). Biochim. Biophys. Acta (BBA) Biomembr. 2017, 1859, 301–311. [Google Scholar] [CrossRef]
- Pittalà, M.G.G.; Saletti, R.; Reina, S.; Cunsolo, V.; De Pinto, V.; Foti, S. A High Resolution Mass Spectrometry Study Reveals the Potential of Disulfide Formation in Human Mitochondrial Voltage-Dependent Anion Selective Channel Isoforms (hVDACs). Int. J. Mol. Sci. 2020, 21, 1468. [Google Scholar] [CrossRef] [Green Version]
- Guan, Z.; Yates, N.A.; Bakhtiar, R. Detection and characterization of methionine oxidation in peptides by collision-induced dissociation and electron capture dissociation. J. Am. Soc. Mass Spectrom. 2003, 14, 605–613. [Google Scholar] [CrossRef] [Green Version]
- Cournoyer, J.J.; Pittman, J.L.; Ivleva, V.B.; Fallows, E.; Waskell, L.; Costello, C.E.; O’Connor, P.B. Deamidation: Differentiation of aspartyl from isoaspartyl products in peptides by electron capture dissociation. Protein Sci. 2005, 14, 452–463. [Google Scholar] [CrossRef] [Green Version]
- Jové, M.; Pradas, I.; Mota-Martorell, N.; Cabré, R.; Ayala, V.; Ferrer, I.; Pamplona, R. Succination of Protein Thiols in Human Brain Aging. Front. Aging Neurosci. 2020, 12, 52. [Google Scholar] [CrossRef]
- D’Angelo, S.; Trojsi, F.; Salvatore, A.; Daniele, L.; Raimo, M.; Galletti, P.; Monsurrò, M.R. Accumulation of altered aspartyl residues in erythrocyte membrane proteins from patients with sporadic amyotrophic lateral sclerosis. Neurochem. Int. 2013, 63, 626–634. [Google Scholar] [CrossRef]
- Checchetto, V.; Reina, S.; Magrì, A.; Szabo, I.; De Pinto, V. Recombinant Human Voltage Dependent Anion Selective Channel Isoform 3 (hVDAC3) Forms Pores with a Very Small Conductance. Cell Physiol. Biochem. 2014, 34, 842–853. [Google Scholar] [CrossRef] [PubMed]
- Nibali, S.C.; Di Rosa, M.C.; Rauh, O.; Thiel, G.; Reina, S.; De Pinto, V. Cell-free electrophysiology of human VDACs incorporated into nanodiscs: An improved method. Biophys. Rep. 2021, 1, 100002. [Google Scholar]
- Liddy, K.A.; White, M.Y.; Cordwell, S.J. Functional decorations: Post-translational modifications and heart disease delineated by targeted proteomics. Genome Med. 2013, 5, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomin, T.; Schittmayer, M.; Honeder, S.; Heininger, C.; Birner-Gruenberger, R. Irreversible oxidative post-translational modifications in heart disease. Expert Rev. Proteom. 2019, 16, 681–693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okazaki, M.; Kurabayashi, K.; Asanuma, M.; Saito, Y.; Dodo, K.; Sodeoka, M. VDAC3 gating is activated by suppression of disulfide-bond formation between the N-terminal region and the bottom of the pore. Biochim. Biophys. Acta 2015, 1848, 3188–3196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Messina, A.; Reina, S.; Guarino, F.; Magrì, A.; Tomasello, F.; Clark, R.E.; Ramsay, R.R.; De Pinto, V. Live cell interactome of the human voltage dependent anion channel 3 (VDAC3) revealed in HeLa cells by affinity purification tag technique. Mol. BioSyst. 2014, 10, 2134–2145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravera, S.; Bonifacino, T.; Bartolucci, M.; Milanese, M.; Gallia, E.; Provenzano, F.; Cortese, K.; Panfoli, I.; Bonanno, G. Char-acterization of the Mitochondrial Aerobic Metabolism in the Pre- and Perisynaptic Districts of the SOD1G93A Mouse Model of Amyotrophic Lateral Sclerosis. Mol. Neurobiol. 2018, 55, 9220–9233. [Google Scholar] [CrossRef]
- Hains, P.G.; Truscott, R.J.W. Age-Dependent Deamidation of Life long Proteins in the Human Lens. Investig. Ophthalmol. Vis. Sci. 2010, 51, 3107–3114. [Google Scholar] [CrossRef]
- Hooi, M.Y.S.; Raftery, M.J.; Truscott, R.J.W. Racemization of Two Proteins over Our Lifespan: Deamidation of Asparagine 76 in γS Crystallin Is Greater in Cataract than in Normal Lenses across the Age Range. Investig. Ophthalmol. Vis. Sci. 2012, 53, 3554–3561. [Google Scholar] [CrossRef]
- Truscott, R.J.W. Are Ancient Proteins Responsible for the Age-Related Decline in Health and Fitness? Rejuvenation Res. 2010, 13, 83–89. [Google Scholar] [CrossRef] [Green Version]
- Lindner, H.; Helliger, W. Age-dependent deamidation of asparagine residues in proteins. Exp. Gerontol. 2001, 36, 1551–1563. [Google Scholar] [CrossRef] [PubMed]
- Robinson, N.E.; Robinson, M.L.; Schulze, S.E.S.; Lai, B.T.; Gray, H.B. Deamidation ofα-synuclein. Protein Sci. 2009, 18, 1766–1773. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, T.; Watanabe, A.; Ogawara, M.; Mori, H.; Shirasawa, T. Isoaspartate Formation and Neurodegeneration in Alzheimer’s Disease. Arch. Biochem. Biophys. 2000, 381, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Vigneswara, V.; Cass, S.; Wayne, D.; Bolt, E.L.; Ray, D.E.; Carter, W.G. Molecular Ageing of Alpha- and Beta-Synucleins: Protein Damage and Repair Mechanisms. PLoS ONE 2013, 8, e61442. [Google Scholar] [CrossRef] [Green Version]
- Bastrup, J.; Kastaniegaard, K.; Asuni, A.A.; Volbracht, C.; Stensballe, A. Proteomicand Unbiased Post-Translational Modification Profiling of Amyloid Plaques and Surrounding Tissue in a Transgenic Mouse Model of Alzheimer’s Disease. J. Alzheimer’s Dis. 2020, 73, 393–411. [Google Scholar] [CrossRef]
- Wilmarth, P.A.; Tanner, S.; Dasari, S.; Nagalla, S.R.; Riviere, M.A.; Bafna, V.; Pevzner, P.A.; David, L.L. Age-related changes in human crystallins determined from comparative analysis of post-translational modifications in young and aged lens: Does deamidation contribute to crystallin insolubility? J. Proteome Res. 2006, 5, 2554–2566. [Google Scholar] [CrossRef] [Green Version]
- Catak, S.; Monard, G.; Aviyente, V.; Ruiz-Lo´pez, M.F. Deamidation of Asparagine Residues: Direct Hydrolysis versus Succinimide-Mediated Deamidation Mechanisms. J. Phys. Chem. 2009, 113, 1111–1120. [Google Scholar] [CrossRef]
- Geiger, T.; Clarke, S. Deamidation, isomerization, and racemization at asparaginyl and aspartyl residues in peptides. Succinimide-linked reactions that contribute to protein degradation. J. Biol. Chem. 1987, 262, 785–794. [Google Scholar] [CrossRef]
- Reissner, K.J.; Aswad, D.W. Deamidation and isoaspartate formation in proteins: Unwanted alterations or surreptitious signals? Cell Mol. Life Sci. 2003, 60, 1281–1295. [Google Scholar] [CrossRef]
- Shimizu, T.; Matsuoka, Y.; Shirasawa, T. Biological Significance of Isoaspartate and Its Repair System. Biol. Pharm. Bull. 2005, 28, 1590–1596. [Google Scholar] [CrossRef] [Green Version]
- Galletti, P.; De Bonis, M.L.; Sorrentino, A.; Raimo, M.; D’Angelo, S.; Scala, I.; Andria, G.; D’Aniello, A.; Ingrosso, D.; Zappia, V. Accumulation of altered aspartyl residues in erythrocyte proteins from patients with Down’s syndrome. FEBS J. 2007, 27, 5263–5277. [Google Scholar] [CrossRef] [PubMed]
- Alderson, N.L.; Wang, Y.; Blatnik, M.; Frizzell, N.; Walla, M.D.; Lyons, T.J.; Alt, N.; Carson, J.A.; Nagai, R.; Thorpe, S.R.; et al. S-(2-Succinyl)cysteine: A novel chemical modification of tissue proteins by a Krebs cycle intermediate. Arch. Biochem. Biophys. 2006, 450, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Saletti, R.; Reina, S.; Pittalà, M.G.; Magrì, A.; Cunsolo, V.; Foti, S.; De Pinto, V. Post-translational modifications of VDAC1 and VDAC2 cysteines from rat liver mitochondria. Biochim. Biophys. Acta Bioenerg. 2018, 1859, 806–816. [Google Scholar] [CrossRef] [PubMed]
- Guan, R.; Wang, J.; Cai, Z.; Li, Z.; Wang, L.; Li, Y.; Xu, J.; Li, D.; Yao, H.; Liu, W.; et al. Hydrogen sulfide attenuates cigarette smoke-induced airway remodeling by upregulating SIRT1 signaling pathway. Redox Biol. 2020, 28, 101356. [Google Scholar] [CrossRef]
- Merkley, E.D.; Metz, T.O.; Smith, R.D.; Baynes, J.W.; Frizzell, N. The succinated proteome. Mass Spectrom. Rev. 2014, 32, 98–109. [Google Scholar] [CrossRef] [Green Version]
- Manuel, A.M.; Walla, M.D.; Faccenda, A.; Martin, S.L.; Tanis, R.M.; Piroli, G.G.; Adam, J.; Kantor, B.; Mutus, B.; Townsend, D.M.; et al. Succination of Protein Disulfide Isomerase Links Mitochondrial Stress and Endoplasmic Reticulum Stress in the Adipocyte During Diabetes. Antioxid. Redox Signal 2017, 27, 1281–1296. [Google Scholar] [CrossRef]
- Piroli, G.G.; Manuel, A.M.; Walla, M.D.; Jepson, M.J.; Brock, J.W.C.; Rajesh, M.P.; Tanis, R.M.; Cotham, W.E.; Frizzell, N. Identification of protein succination as a novel modification of tubulin. Biochem. J. 2014, 462, 231–245. [Google Scholar] [CrossRef] [Green Version]
- Twelvetrees, A.E. The lifecycle of the neuronal microtubule transport machinery. Semin. Cell Dev. Biol. 2020, 107, 74–81. [Google Scholar] [CrossRef]
- Ferri, A.; Cozzolino, M.; Crosio, C.; Nencini, M.; Casciati, A.; Gralla, E.D.; Rotilio, G.; Valentine, J.S.; Carrì, M.T. Familial ALS-superoxide dismutases associate with mitochondria and shift their redox potentials. Proc. Natl. Acad. Sci. USA 2006, 103, 13860–13865. [Google Scholar] [CrossRef] [Green Version]
- Magrì, A.; Belfiore, R.; Reina, S.; Tomasello, M.F.; Di Rosa, M.C.; Guarino, F.; Leggio, L.; De Pinto, V.; Messina, A. Hexokinase I N-terminal based peptide prevents the VDAC1-SOD1 G93A interaction and re-establishes ALS cell viability. Sci. Rep. 2016, 6, 34802. [Google Scholar] [CrossRef] [Green Version]
- Aiello, R.; Messina, A.; Schiffler, B.; Benz, R.; Tasco, G.; Casadio, R.; De Pinto, V. Functional characterization of a second porin isoform in Drosophila melanogaster–DmPorin2 forms voltage-independent cation-selective pores. J. Biol. Chem. 2004, 279, 25364–25373. [Google Scholar] [CrossRef] [PubMed]
- Reina, S.; Magrì, A.; Lolicato, M.; Guarino, F.; Impellizzeri, A.; Maier, E.; Benz, R.; Ceccarelli, M.; De Pinto, V.; Messina, A. Deletion of β-strands 9 and 10 converts VDAC1 voltage-dependence in an asymmetrical process. Biochim. Biophys. Acta Bioenerg. 2013, 1827, 793–805. [Google Scholar] [CrossRef] [PubMed]
- Guardiani, C.; Magrì, A.; Karachitos, A.; Di Rosa, M.C.; Reina, S.; Bodrenko, I.; Messina, A.; Kmita, H.; Ceccarelli, M.; De Pinto, V. yVDAC2, the second mitochondrial porin isoform of Saccharomyces cerevisiae. Biochim. Biophys. Acta Bioenerg. 2018, 1859, 270–279. [Google Scholar] [CrossRef] [PubMed]
- Webb, B.; Sali, A. Comparative Protein Structure Modeling Using MODELLER. Curr. Protoc. 2016, 54, 5–6. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide | Position in the Sequence | Calculated Monoisotopic m/z | Cell Lines | Ratio Ox/Red | |
|---|---|---|---|---|---|
| Avg | Dev St | ||||
| GYGFGMVK | 21–28 | 437.7103 (2+) | NSC34 | 10.1 | 0.85 |
| GYGFGMVK | 429.7128 (2+) | NSC34-SOD1G93A | - Met26 totally oxidized | - | |
| SCSGVEFSTSGHAYTDTGK | 35–53 | 991.4079 (2+) | NSC34 | 0.08 | 0.01 |
| NSC34-SOD1WT | 0.05 | 0.01 | |||
| SCSGVEFSTSGHAYTDTGK | 995.9260 (2+) | ||||
| NSC34-SOD1G93A | 0.05 | 0.01 | |||
| YKVCNYGLTFTQK | 62–74 | 806.8877 (2+) | NSC34 | 0.87 | 0.06 |
| NSC34-SOD1WT | 0.6 | 0.10 | |||
| YKVCNYGLTFTQK | 811.4060 (2+) | ||||
| NSC34-SOD1G93A | 0.67 | 0.12 | |||
| DCFSLGSNVDIDFSGPTIYGWAVLAFEGWLAGYQMSFDTAK | 121–161 | 1518.0315 (3+) | NSC34-SOD1WT | 6.63 | 1.33 |
| DCFSLGSNVDIDFSGPTIYGWAVLAFEGWLAGYQMSFDTAK | 1512.6995 (3+) | NSC34-SOD1G93A | - Met155 totally oxidized | - | |
| Peptide | Position in the Sequence | Calculated Monoisotopic m/z | Cell Lines | Ratio Ox/Red | |
|---|---|---|---|---|---|
| Avg | Dev St | ||||
| GFGMVKIDL | 23–31 | 498.2657 (2+) | NSC34 | 11.73 | 2.23 |
| NSC34-SOD1WT | 12.07 | 1.27 | |||
| GFGMVKIDL | 490.2682 (2+) | ||||
| NSC34-SOD1G93A | - Met26 totally oxidized | - | |||
| QMSFDTAKSKL | 154–164 | 636.3192 (2+) | NSC34 | 0.50 | 0.30 |
| QMSFDTAKSKL | 628.3217 (2+) | ||||
| KLDCRTSL | 226–233 | 492.2455 (2+) | NSC34 | 0.10 | 0 |
| NSC34-SOD1WT | 0.53 | 0.15 | |||
| KLDCRTSL | 496.7638 (2+) | ||||
| NSC34-SOD1G93A | 0.33 | 0.15 | |||
| Peptide | Position in the Sequence | Calculated Monoisotopic m/z | Cell Lines | Ratio Succinated/Red | |
|---|---|---|---|---|---|
| Avg | Dev St | ||||
| YKVNYGLTFTQK | 62–74 | 560.9367 (3+) | NSC34 | 0.3 | 0 |
| NSC34-SOD1WT | 0.40 | 0.10 | |||
| YKVCNYGLTFTQK | 811.4060 (2+) | ||||
| NSC34-SOD1G93A | 1.07 | 0.12 | |||
| Peptide | Position in the Sequence | Calculated Monoisotopic m/z | Ratio Deam/Norm | |
|---|---|---|---|---|
| Avg | Dev St | |||
| WNTDNTLGTEISWENK | 75–90 | 954.9344 (2+) | 0.003 | 0 |
| WNTDNTLGTEISWENK | 954.4424 (2+) | |||
| LTLDTIFVPNTGK | 97–109 | 710.3904 (2+) | 0.003 | 0 |
| LTLDTIFVPNTGK | 709.8984 (2+) | |||
| LSQNNFALGYK | 164–174 | 628.3197 (2+) | 0.01 | 0 |
| LSQNNFALGYK | ||||
| LSQNNFALGYK | 627.8277 (2+) | 0.003 | 0 | |
| LSQNNFALGYK | ||||
| IETSINLAWTAGSNNTR | 202–218 | 924.9582 (2+) | 0.1 | 0 |
| IETSINLAWTAGSNNTR | 924.4662 (2+) | |||
| VNNASLIGLGYTQTLRPGVK | 237–256 | 701.3920 (3+) | 0.01 | 0 |
| VNNASLIGLGYTQTLRPGVK | ||||
| VNNASLIGLGYTQTLRPGVK | 701.0640 (3+) | 0.01 | 0 | |
| VNNASLIGLGYTQTLRPGVK | ||||
| Peptide | Position in the Sequence | Calculated Monoisotopic m/z | Cell Lines | Ratio Succinimide/Norm | |
|---|---|---|---|---|---|
| Avg | Dev St | ||||
| LSQFALGYK | 164–174 | 619.3144 (2+) | NSC34 | 0.002 | 0 |
| NSC34-SOD1WT | 0.003 | 0 | |||
| LSQNNFALGYK | 627.8277 (2+) | ||||
| NSC34-SOD1G93A | 0.01 | 0 | |||
| VNASLIGLGYTQTLRPGVK | 237–256 | 1042.5788 (2+) | NSC34-SOD1WT | 0.003 | 0 |
| VNNASLIGLGYTQTLRPGVK | 1051.0924 (2+) | ||||
| Cell Lines | Peptide | Position in the Sequence | Rt (min) | Calculated Monoisotopic m/z | Absolute Intensity |
|---|---|---|---|---|---|
| NSC34 | WTDTLGTEISWENK | 75–90 | 52.07 | 953.4408 (2+) | 6.7 · 103 |
| 51.55 | 3.9 · 103 | ||||
| NSC34-SOD1WT | 51.56 | 1.1 · 105 | |||
| 51.32 | 1.3 · 105 | ||||
| 51.04 | 6.1 · 104 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pittalà, M.G.G.; Reina, S.; Nibali, S.C.; Cucina, A.; Cubisino, S.A.M.; Cunsolo, V.; Amodeo, G.F.; Foti, S.; De Pinto, V.; Saletti, R.; et al. Specific Post-Translational Modifications of VDAC3 in ALS-SOD1 Model Cells Identified by High-Resolution Mass Spectrometry. Int. J. Mol. Sci. 2022, 23, 15853. https://doi.org/10.3390/ijms232415853
Pittalà MGG, Reina S, Nibali SC, Cucina A, Cubisino SAM, Cunsolo V, Amodeo GF, Foti S, De Pinto V, Saletti R, et al. Specific Post-Translational Modifications of VDAC3 in ALS-SOD1 Model Cells Identified by High-Resolution Mass Spectrometry. International Journal of Molecular Sciences. 2022; 23(24):15853. https://doi.org/10.3390/ijms232415853
Chicago/Turabian StylePittalà, Maria Gaetana Giovanna, Simona Reina, Stefano Conti Nibali, Annamaria Cucina, Salvatore Antonio Maria Cubisino, Vincenzo Cunsolo, Giuseppe Federico Amodeo, Salvatore Foti, Vito De Pinto, Rosaria Saletti, and et al. 2022. "Specific Post-Translational Modifications of VDAC3 in ALS-SOD1 Model Cells Identified by High-Resolution Mass Spectrometry" International Journal of Molecular Sciences 23, no. 24: 15853. https://doi.org/10.3390/ijms232415853
APA StylePittalà, M. G. G., Reina, S., Nibali, S. C., Cucina, A., Cubisino, S. A. M., Cunsolo, V., Amodeo, G. F., Foti, S., De Pinto, V., Saletti, R., & Messina, A. (2022). Specific Post-Translational Modifications of VDAC3 in ALS-SOD1 Model Cells Identified by High-Resolution Mass Spectrometry. International Journal of Molecular Sciences, 23(24), 15853. https://doi.org/10.3390/ijms232415853