
GABA Release from Astrocytes in Health and Disease

Abstract
:1. Introduction
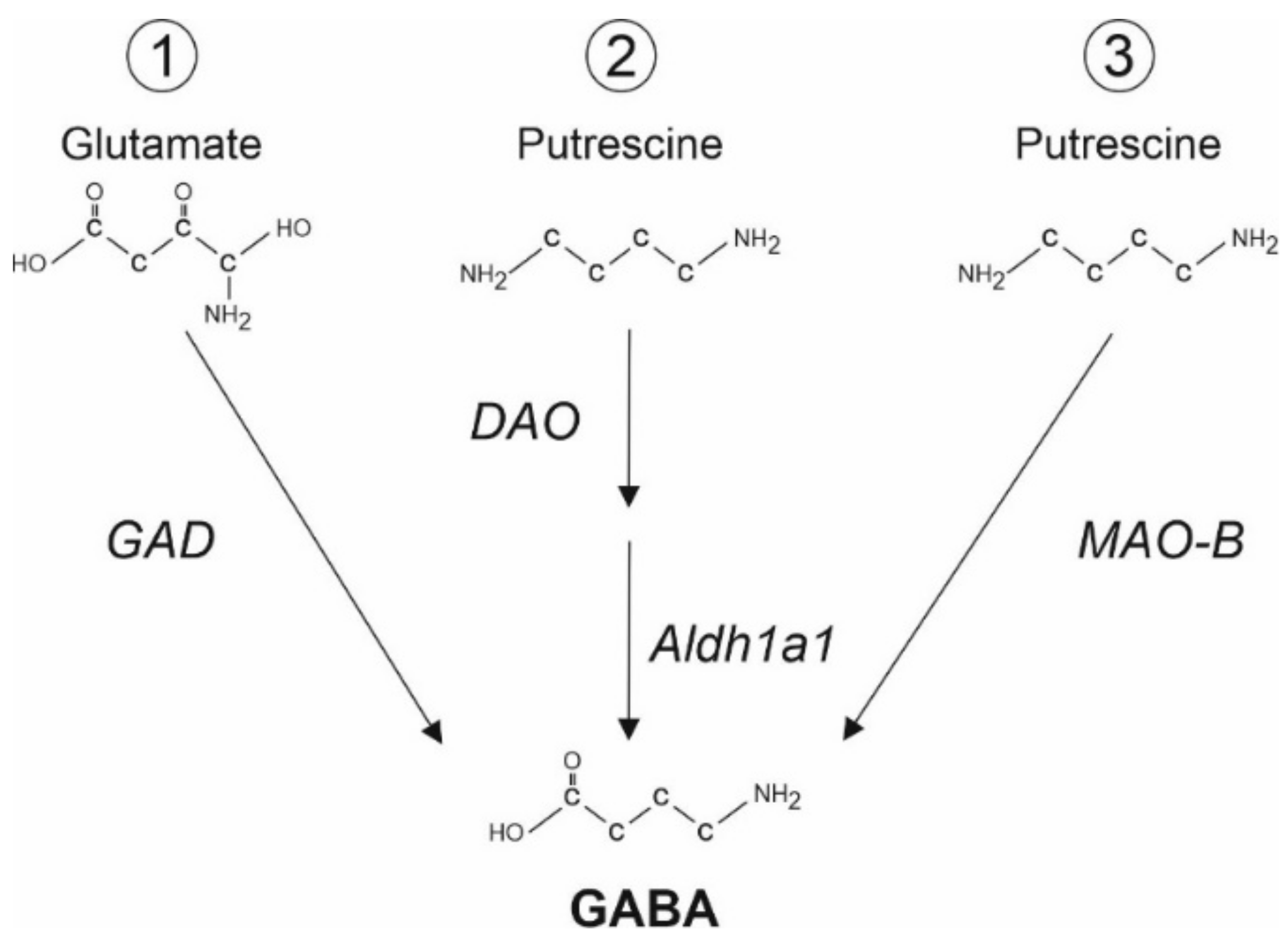
2. GABA Synthesis in Astrocytes
3. GABA Release from Astrocytes in Health
3.1. Volume-Regulated Anionic Channels—Bestrophin1
3.2. GAT-Mediated GABA Release
4. GABA Release from Astrocytes in Disease
4.1. Alzheimer’s Disease
4.2. Huntington’s Disease
4.3. Autism Spectrum Disorder (ASD)
4.4. Epilepsy
5. Summary: Is GABA Release from Astrocytes Mediated by Best1- or GAT3 or Both?
5.1. GAT3-Mediated Release
5.2. Best1-Mediated Release
5.3. Best1-/GAT3-Mediated Release
5.4. Neuroglial Interaction and GABA Release from Astrocytes
6. Conclusions and Perspectives
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Allen, N.J. Astrocyte regulation of synaptic behavior. Annu Rev. Cell Dev. Biol. 2014, 30, 439–463. [Google Scholar] [CrossRef]
- Verkhratsky, A.; Nedergaard, M. Physiology of Astroglia. Physiol. Rev. 2018, 98, 239–389. [Google Scholar] [CrossRef]
- Kofuji, P.; Araque, A. G-Protein-Coupled Receptors in Astrocyte-Neuron Communication. Neuroscience 2021, 456, 71–84. [Google Scholar] [CrossRef]
- Verkhratsky, A.; Chvátal, A. NMDA Receptors in Astrocytes. Neurochem. Res. 2020, 45, 122–133. [Google Scholar] [CrossRef] [Green Version]
- Rose, C.R.; Felix, L.; Zeug, A.; Dietrich, D.; Reiner, A.; Henneberger, C. Astroglial Glutamate Signaling and Uptake in the Hippocampus. Front. Mol. Neurosci. 2017, 10, 451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Feng, X.; Wang, Y.; Xia, X.; Zheng, J.C. Astrocytes: GABAceptive and GABAergic Cells in the Brain. Front. Cell Neurosci. 2022, 16, 892497. [Google Scholar] [CrossRef] [PubMed]
- Rivera, A.; Vanzulli, I.; Butt, A.M. A Central Role for ATP Signalling in Glial Interactions in the CNS. Curr. Drug Targets 2016, 17, 1829–1833. [Google Scholar] [CrossRef]
- Cornell-Bell, A.H.; Finkbeiner, S.M.; Cooper, M.S.; Smith, S.J. Glutamate induces calcium waves in cultured astrocytes: Long-range glial signaling. Science 1990, 247, 470–473. [Google Scholar] [CrossRef]
- Bernardinelli, Y.; Magistretti, P.J.; Chatton, J.Y. Astrocytes generate Na+-mediated metabolic waves. Proc. Natl. Acad. Sci. USA 2004, 101, 14937–14942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parpura, V.; Basarsky, T.A.; Liu, F.; Jeftinija, K.; Jeftinija, S.; Haydon, P.G. Glutamate-mediated astrocyte-neuron signalling. Nature 1994, 369, 744–747. [Google Scholar] [CrossRef] [PubMed]
- Henneberger, C.; Papouin, T.; Oliet, S.H.; Rusakov, D.A. Long-term potentiation depends on release of D-serine from astrocytes. Nature 2010, 463, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Pascual, O.; Casper, K.B.; Kubera, C.; Zhang, J.; Revilla-Sanchez, R.; Sul, J.Y.; Takano, H.; Moss, S.J.; McCarthy, K.; Haydon, P.G. Astrocytic purinergic signaling coordinates synaptic networks. Science 2005, 310, 113–116. [Google Scholar] [CrossRef] [PubMed]
- Montana, V.; Malarkey, E.B.; Verderio, C.; Matteoli, M.; Parpura, V. Vesicular transmitter release from astrocytes. Glia 2006, 54, 700–715. [Google Scholar] [CrossRef] [PubMed]
- Duan, S.; Anderson, C.M.; Keung, E.C.; Chen, Y.; Chen, Y.; Swanson, R.A. P2X7 receptor-mediated release of excitatory amino acids from astrocytes. J. Neurosci. 2003, 23, 1320–1328. [Google Scholar] [CrossRef] [Green Version]
- Ye, Z.C.; Wyeth, M.S.; Baltan-Tekkok, S.; Ransom, B.R. Functional hemichannels in astrocytes: A novel mechanism of glutamate release. J. Neurosci. 2003, 23, 3588–3596. [Google Scholar] [CrossRef] [Green Version]
- Szatkowski, M.; Barbour, B.; Attwell, D. Non-vesicular release of glutamate from glial cells by reversed electrogenic glutamate uptake. Nature 1990, 348, 443–446. [Google Scholar] [CrossRef]
- Attwell, D.; Barbour, B.; Szatkowski, M. Nonvesicular release of neurotransmitter. Neuron 1993, 11, 401–407. [Google Scholar] [CrossRef]
- Kimelberg, H.K.; Goderie, S.K.; Higman, S.; Pang, S.; Waniewski, R.A. Swelling-induced release of glutamate, aspartate, and taurine from astrocyte cultures. J. Neurosci. 1990, 10, 1583–1591. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Wang, W.; Diez-Sampedro, A.; Richerson, G.B. Nonvesicular inhibitory neurotransmission via reversal of the GABA transporter GAT-1. Neuron 2007, 56, 851–865. [Google Scholar] [CrossRef] [Green Version]
- Kinney, G.A. GAT-3 transporters regulate inhibition in the neocortex. J. Neurophysiol 2005, 94, 4533–4537. [Google Scholar] [CrossRef]
- Heja, L.; Barabas, P.; Nyitrai, G.; Kekesi, K.A.; Lasztoczi, B.; Toke, O.; Tarkanyi, G.; Madsen, K.; Schousboe, A.; Dobolyi, A.; et al. Glutamate uptake triggers transporter-mediated GABA release from astrocytes. PLoS ONE 2009, 4, e7153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, H.; Tsunenari, T.; Yau, K.W.; Nathans, J. The vitelliform macular dystrophy protein defines a new family of chloride channels. Proc. Natl. Acad. Sci. USA 2002, 99, 4008–4013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Yoon, B.E.; Berglund, K.; Oh, S.J.; Park, H.; Shin, H.S.; Augustine, G.J.; Lee, C.J. Channel-mediated tonic GABA release from glia. Science 2010, 330, 790–796. [Google Scholar] [CrossRef] [PubMed]
- Marmorstein, A.D.; Marmorstein, L.Y.; Rayborn, M.; Wang, X.; Hollyfield, J.G.; Petrukhin, K. Bestrophin, the product of the Best vitelliform macular dystrophy gene (VMD2), localizes to the basolateral plasma membrane of the retinal pigment epithelium. Proc. Natl. Acad. Sci. USA 2000, 97, 12758–12763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, A.A.; Guziewicz, K.E.; Lee, C.J.; Kalathur, R.C.; Pulido, J.S.; Marmorstein, L.Y.; Marmorstein, A.D. Bestrophin 1 and retinal disease. Prog. Retin. Eye Res. 2017, 58, 45–69. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.S.; Choi, J.; Yoon, B.E. Neuron-Glia Interactions in Neurodevelopmental Disorders. Cells 2020, 9, 2176. [Google Scholar] [CrossRef]
- Schousboe, A.; Westergaard, N.; Sonnewald, U.; Petersen, S.B.; Yu, A.C.; Hertz, L. Regulatory role of astrocytes for neuronal biosynthesis and homeostasis of glutamate and GABA. Prog. Brain Res. 1992, 94, 199–211. [Google Scholar]
- Wu, Z.; Guo, Z.; Gearing, M.; Chen, G. Tonic inhibition in dentate gyrus impairs long-term potentiation and memory in an Alzheimer’s disease model. Nat. Commun. 2014, 5, 4159. [Google Scholar] [CrossRef] [Green Version]
- Müller, J.; Timmermann, A.; Henning, L.; Müller, H.; Steinhäuser, C.; Bedner, P. Astrocytic GABA Accumulation in Experimental Temporal Lobe Epilepsy. Front. Neurol. 2020, 11, 614923. [Google Scholar] [CrossRef]
- Otsuka, M.; Obata, K.; Miyata, Y.; Tanaka, Y. Measurement of gamma-aminobutyric acid in isolated nerve cells of cat central nervous system. J. Neurochem 1971, 18, 287–295. [Google Scholar] [CrossRef]
- Sacchettoni, S.A.; Benchaibi, M.; Sindou, M.; Belin, M.F.; Jacquemont, B. Glutamate-modulated production of GABA in immortalized astrocytes transduced by a glutamic acid decarboxylase-expressing retrovirus. Glia 1998, 22, 86–93. [Google Scholar] [CrossRef]
- Seiler, N.; Schmidt-Glenewinkel, T.; Sarhan, S. On the formation of gamma-aminobutyric acid from putrescine in brain. J. Biochem. 1979, 86, 277–278. [Google Scholar]
- Laschet, J.; Grisar, T.; Bureau, M.; Guillaume, D. Characteristics of putrescine uptake and subsequent GABA formation in primary cultured astrocytes from normal C57BL/6J and epileptic DBA/2J mouse brain cortices. Neuroscience 1992, 48, 151–157. [Google Scholar] [CrossRef]
- Kwak, H.; Koh, W.; Kim, S.; Song, K.; Shin, J.I.; Lee, J.M.; Lee, E.H.; Bae, J.Y.; Ha, G.E.; Oh, J.E.; et al. Astrocytes Control Sensory Acuity via Tonic Inhibition in the Thalamus. Neuron 2020, 108, 691–706. [Google Scholar] [CrossRef] [PubMed]
- Jo, S.; Yarishkin, O.; Hwang, Y.J.; Chun, Y.E.; Park, M.; Woo, D.H.; Bae, J.Y.; Kim, T.; Lee, J.; Chun, H.; et al. GABA from reactive astrocytes impairs memory in mouse models of Alzheimer’s disease. Nat. Med. 2014, 20, 886–896. [Google Scholar] [CrossRef] [PubMed]
- Woo, J.; Min, J.O.; Kang, D.S.; Kim, Y.S.; Jung, G.H.; Park, H.J.; Kim, S.; An, H.; Kwon, J.; Kim, J.; et al. Control of motor coordination by astrocytic tonic GABA release through modulation of excitation/inhibition balance in cerebellum. Proc. Natl. Acad. Sci. USA 2018, 115, 5004–5009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.H.; Ju, Y.H.; Choi, J.W.; Song, H.J.; Jang, B.K.; Woo, J.; Chun, H.; Kim, H.J.; Shin, S.J.; Yarishkin, O.; et al. Newly developed reversible MAO-B inhibitor circumvents the shortcomings of irreversible inhibitors in Alzheimer’s disease. Sci. Adv. 2019, 5, eaav0316. [Google Scholar] [CrossRef] [Green Version]
- Yoon, B.E.; Woo, J.; Chun, Y.E.; Chun, H.; Jo, S.; Bae, J.Y.; An, H.; Min, J.O.; Oh, S.J.; Han, K.S.; et al. Glial GABA, synthesized by monoamine oxidase B, mediates tonic inhibition. J. Physiol. 2014, 592, 4951–4968. [Google Scholar] [CrossRef]
- Oakley, H.; Cole, S.L.; Logan, S.; Maus, E.; Shao, P.; Craft, J.; Guillozet-Bongaarts, A.; Ohno, M.; Disterhoft, J.; Van Eldik, L.; et al. Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: Potential factors in amyloid plaque formation. J. Neurosci. 2006, 26, 10129–10140. [Google Scholar] [CrossRef] [Green Version]
- Dvorzhak, A.; Myakhar, O.; Unichenko, P.; Kirmse, K.; Kirischuk, S. Estimation of ambient GABA levels in layer I of the mouse neonatal cortex in brain slices. J. Physiol. 2010, 588, 2351–2360. [Google Scholar] [CrossRef]
- Jensen, K.; Chiu, C.S.; Sokolova, I.; Lester, H.A.; Mody, I. GABA transporter-1 (GAT1)-deficient mice: Differential tonic activation of GABAA versus GABAB receptors in the hippocampus. J. Neurophysiol. 2003, 90, 2690–2701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagelhus, E.A.; Ottersen, O.P. Physiological roles of aquaporin-4 in brain. Physiol. Rev. 2013, 93, 1543–1562. [Google Scholar] [CrossRef] [PubMed]
- Walch, E.; Fiacco, T.A. Honey, I shrunk the extracellular space: Measurements and mechanisms of astrocyte swelling. Glia 2022, 70, 2013–2031. [Google Scholar] [CrossRef] [PubMed]
- Ochoa-de la Paz, L.D.; Gulias-Cañizo, R. Glia as a key factor in cell volume regulation processes of the central nervous system. Front. Cell Neurosci. 2022, 16, 967496. [Google Scholar] [CrossRef]
- Osei-Owusu, J.; Yang, J.; Vitery, M.D.C.; Qiu, Z. Molecular Biology and Physiology of Volume-Regulated Anion Channel (VRAC). Curr. Top. Membr. 2018, 81, 177–203. [Google Scholar]
- Owji, A.P.; Wang, J.; Kittredge, A.; Clark, Z.; Zhang, Y.; Hendrickson, W.A.; Yang, T. Structures and gating mechanisms of human bestrophin anion channels. Nat. Commun. 2022, 13, 3836. [Google Scholar] [CrossRef] [PubMed]
- Qu, Z.; Chien, L.T.; Cui, Y.; Hartzell, H.C. The anion-selective pore of the bestrophins, a family of chloride channels associated with retinal degeneration. J. Neurosci. 2006, 26, 5411–5419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrukhin, K.; Koisti, M.J.; Bakall, B.; Li, W.; Xie, G.; Marknell, T.; Sandgren, O.; Forsman, K.; Holmgren, G.; Andreasson, S.; et al. Identification of the gene responsible for Best macular dystrophy. Nat. Genet. 1998, 19, 241–247. [Google Scholar] [CrossRef]
- Singh Grewal, S.; Smith, J.J.; Carr, A.F. Bestrophinopathies: Perspectives on clinical disease, Bestrophin-1 function and developing therapies. Ther. Adv. Ophthalmol. 2021, 13, 2515841421997191. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Q.; Hartzell, H.C.; Yu, K. Bestrophins and retinopathies. Pflug. Arch. 2010, 460, 559–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qu, Z.; Hartzell, H.C. Bestrophin Cl- channels are highly permeable to HCO3. Am. J. Physiol. Cell Physiol. 2008, 294, C1371-7. [Google Scholar] [CrossRef] [Green Version]
- Elorza-Vidal, X.; Gaitán-Peñas, H.; Estévez, R. Chloride Channels in Astrocytes: Structure, Roles in Brain Homeostasis and Implications in Disease. Int. J. Mol. Sci. 2019, 20, 1034. [Google Scholar] [CrossRef] [Green Version]
- Owji, A.P.; Kittredge, A.; Zhang, Y.; Yang, T. Structure and Function of the Bestrophin family of calcium-activated chloride channels. Channels 2021, 15, 604–623. [Google Scholar] [CrossRef]
- Yu, K.; Xiao, Q.; Cui, G.; Lee, A.; Hartzell, H.C. The best disease-linked Cl- channel hBest1 regulates Ca V 1 (L-type) Ca2+ channels via src-homology-binding domains. J. Neurosci. 2008, 28, 5660–5670. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Stanton, J.B.; Wu, J.; Yu, K.; Hartzell, H.C.; Peachey, N.S.; Marmorstein, L.Y.; Marmorstein, A.D. Suppression of Ca2+ signaling in a mouse model of Best disease. Hum. Mol. Genet. 2010, 19, 1108–1118. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.; Rao, Z.; Zhang, Z.; Zhou, J. Function of the GABAergic System in Diabetic Encephalopathy. Cell. Mol. Neurobiol. 2022. [Google Scholar] [CrossRef]
- Park, H.; Oh, S.J.; Han, K.S.; Woo, D.H.; Park, H.; Mannaioni, G.; Traynelis, S.F.; Lee, C.J. Bestrophin-1 encodes for the Ca2+-activated anion channel in hippocampal astrocytes. J. Neurosci. 2009, 29, 13063–13073. [Google Scholar] [CrossRef] [Green Version]
- Le Meur, K.; Mendizabal-Zubiaga, J.; Grandes, P.; Audinat, E. GABA release by hippocampal astrocytes. Front. Comput. Neurosci. 2012, 6, 59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, H.; Han, K.S.; Oh, S.J.; Jo, S.; Woo, J.; Yoon, B.E.; Lee, C.J. High glutamate permeability and distal localization of Best1 channel in CA1 hippocampal astrocyte. Mol. Brain 2013, 6, 54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woo, D.H.; Han, K.S.; Shim, J.W.; Yoon, B.E.; Kim, E.; Bae, J.Y.; Oh, S.J.; Hwang, E.M.; Marmorstein, A.D.; Bae, Y.C.; et al. TREK-1 and Best1 channels mediate fast and slow glutamate release in astrocytes upon GPCR activation. Cell 2012, 151, 25–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, B.E.; Jo, S.; Woo, J.; Lee, J.H.; Kim, T.; Kim, D.; Lee, C.J. The amount of astrocytic GABA positively correlates with the degree of tonic inhibition in hippocampal CA1 and cerebellum. Mol. Brain 2011, 4, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glykys, J.; Mody, I. The main source of ambient GABA responsible for tonic inhibition in the mouse hippocampus. J. Physiol. 2007, 582, 1163–1178. [Google Scholar] [CrossRef] [PubMed]
- Semyanov, A.; Walker, M.C.; Kullmann, D.M. GABA uptake regulates cortical excitability via cell type-specific tonic inhibition. Nat. Neurosci. 2003, 6, 484–490. [Google Scholar] [CrossRef]
- Oh, S.J.; Lee, C.J. Distribution and Function of the Bestrophin-1 (Best1) Channel in the Brain. Exp. Neurobiol. 2017, 26, 113–121. [Google Scholar] [CrossRef] [Green Version]
- An, H.; Koh, W.; Kang, S.; Nam, M.H.; Lee, C.J. Differential Proximity of Perisynaptic Astrocytic Best1 at the Excitatory and Inhibitory Tripartite Synapses in APP/PS1 and MAOB-KO Mice Revealed by Lattice Structured Illumination Microscopy. Exp. Neurobiol. 2021, 30, 213–221. [Google Scholar] [CrossRef]
- Kristensen, A.S.; Andersen, J.; Jorgensen, T.N.; Sorensen, L.; Eriksen, J.; Loland, C.J.; Stromgaard, K.; Gether, U. SLC6 neurotransmitter transporters: Structure, function, and regulation. Pharmacol. Rev. 2011, 63, 585–640. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, E.A. Calcium-independent release of GABA from isolated horizontal cells of the toad retina. J. Physiol. 1982, 323, 211–227. [Google Scholar] [CrossRef] [Green Version]
- Gordon-Weeks, P.R.; Lockerbie, R.O.; Pearce, B.R. Uptake and release of [3H]GABA by growth cones isolated from neonatal rat brain. Neurosci. Lett. 1984, 52, 205–210. [Google Scholar] [CrossRef]
- Gaspary, H.L.; Wang, W.; Richerson, G.B. Carrier-mediated GABA release activates GABA receptors on hippocampal neurons. J. Neurophysiol. 1998, 80, 270–281. [Google Scholar] [CrossRef] [Green Version]
- Kirmse, K.; Kirischuk, S. Ambient GABA constrains the strength of GABAergic synapses at Cajal-Retzius cells in the developing visual cortex. J. Neurosci. 2006, 26, 4216–4227. [Google Scholar] [CrossRef] [Green Version]
- Minelli, A.; Brecha, N.C.; Karschin, C.; DeBiasi, S.; Conti, F. GAT-1, a high-affinity GABA plasma membrane transporter, is localized to neurons and astroglia in the cerebral cortex. J. Neurosci. 1995, 15, 7734–7746. [Google Scholar] [CrossRef]
- Minelli, A.; DeBiasi, S.; Brecha, N.C.; Zuccarello, L.V.; Conti, F. GAT-3, a high-affinity GABA plasma membrane transporter, is localized to astrocytic processes, and it is not confined to the vicinity of GABAergic synapses in the cerebral cortex. J. Neurosci. 1996, 16, 6255–6264. [Google Scholar] [CrossRef] [Green Version]
- Heja, L.; Nyitrai, G.; Kekesi, O.; Dobolyi, A.; Szabo, P.; Fiath, R.; Ulbert, I.; Pal-Szenthe, B.; Palkovits, M.; Kardos, J. Astrocytes convert network excitation to tonic inhibition of neurons. BMC. Biol. 2012, 10, 26. [Google Scholar] [CrossRef]
- Chatton, J.Y.; Pellerin, L.; Magistretti, P.J. GABA uptake into astrocytes is not associated with significant metabolic cost: Implications for brain imaging of inhibitory transmission. Proc. Natl. Acad. Sci. USA 2003, 100, 12456–12461. [Google Scholar] [CrossRef] [Green Version]
- Rose, C.R.; Ransom, B.R. Intracellular sodium homeostasis in rat hippocampal astrocytes. J. Physiol. 1996, 491, 291–305. [Google Scholar] [CrossRef]
- Unichenko, P.; Myakhar, O.; Kirischuk, S. Intracellular Na+ concentration influences short-term plasticity of glutamate transporter-mediated currents in neocortical astrocytes. Glia 2012, 60, 605–614. [Google Scholar] [CrossRef] [PubMed]
- Langer, J.; Rose, C.R. Synaptically induced sodium signals in hippocampal astrocytes in situ. J. Physiol. 2009, 587, 5859–5877. [Google Scholar] [CrossRef] [PubMed]
- Kettenmann, H.; Backus, K.H.; Schachner, M. Gamma-Aminobutyric acid opens Cl-channels in cultured astrocytes. Brain Res. 1987, 404, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Kimelberg, H.K. Active accumulation and exchange transport of chloride in astroglial cells in culture. Biochim. Biophys. Acta 1981, 646, 179–184. [Google Scholar] [CrossRef]
- Unichenko, P.; Dvorzhak, A.; Kirischuk, S. Transporter-mediated replacement of extracellular glutamate for GABA in the developing murine neocortex. Eur. J. Neurosci. 2013, 38, 3580–3588. [Google Scholar] [CrossRef]
- Yu, X.; Taylor, A.M.W.; Nagai, J.; Golshani, P.; Evans, C.J.; Coppola, G.; Khakh, B.S. Reducing Astrocyte Calcium Signaling In Vivo Alters Striatal Microcircuits and Causes Repetitive Behavior. Neuron 2018, 99, 1170–1187. [Google Scholar] [CrossRef] [Green Version]
- Nusser, Z.; Mody, I. Selective modulation of tonic and phasic inhibitions in dentate gyrus granule cells. J. Neurophysiol. 2002, 87, 2624–2628. [Google Scholar] [CrossRef]
- Kersanté, F.; Rowley, S.C.; Pavlov, I.; Gutièrrez-Mecinas, M.; Semyanov, A.; Reul, J.M.; Walker, M.C.; Linthorst, A.C. A functional role for both -aminobutyric acid (GABA) transporter-1 and GABA transporter-3 in the modulation of extracellular GABA and GABAergic tonic conductances in the rat hippocampus. J. Physiol. 2013, 591, 2429–2441. [Google Scholar] [CrossRef]
- Savtchenko, L.; Megalogeni, M.; Rusakov, D.A.; Walker, M.C.; Pavlov, I. Synaptic GABA release prevents GABA transporter type-1 reversal during excessive network activity. Nat. Commun. 2015, 6, 6597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rose, C.R.; Ransom, B.R. Regulation of intracellular sodium in cultured rat hippocampal neurones. J. Physiol. 1997, 499, 573–587. [Google Scholar] [CrossRef]
- Kirmse, K.; Dvorzhak, A.; Kirischuk, S.; Grantyn, R. GABA transporter 1 tunes GABAergic synaptic transmission at output neurons of the mouse neostriatum. J. Physiol. 2008, 586, 5665–5678. [Google Scholar] [CrossRef] [PubMed]
- Roberts, B.M.; Doig, N.M.; Brimblecombe, K.R.; Lopes, E.F.; Siddorn, R.E.; Threlfell, S.; Connor-Robson, N.; Bengoa-Vergniory, N.; Pasternack, N.; Wade-Martins, R.; et al. GABA uptake transporters support dopamine release in dorsal striatum with maladaptive downregulation in a parkinsonism model. Nat. Commun. 2020, 11, 4958. [Google Scholar] [CrossRef] [PubMed]
- Kirmse, K.; Kirischuk, S.; Grantyn, R. Role of GABA transporter 3 in GABAergic synaptic transmission at striatal output neurons. Synapse 2009, 63, 921–929. [Google Scholar] [CrossRef]
- Reiss, A.B.; Arain, H.A.; Stecker, M.M.; Siegart, N.M.; Kasselman, L.J. Amyloid toxicity in Alzheimer’s disease. Rev. Neurosci. 2018, 29, 613–627. [Google Scholar] [CrossRef]
- Andersen, J.V.; Schousboe, A.; Verkhratsky, A. Astrocyte energy and neurotransmitter metabolism in Alzheimer’s disease: Integration of the glutamate/GABA-glutamine cycle. Prog Neurobiol. 2022, 217, 102331. [Google Scholar] [CrossRef]
- Aldabbagh, Y.; Islam, A.; Zhang, W.; Whiting, P.; Ali, A.B. Alzheimer’s Disease Enhanced Tonic Inhibition is Correlated With Upregulated Astrocyte GABA Transporter-3/4 in a Knock-In APP Mouse Model. Front. Pharmacol. 2022, 13, 822499. [Google Scholar] [CrossRef] [PubMed]
- Yarishkin, O.; Lee, J.; Jo, S.; Hwang, E.M.; Lee, C.J. Disinhibitory Action of Astrocytic GABA at the Perforant Path to Dentate Gyrus Granule Neuron Synapse Reverses to Inhibitory in Alzheimer’s Disease Model. Exp. Neurobiol. 2015, 24, 211–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brawek, B.; Chesters, R.; Klement, D.; Müller, J.; Lerdkrai, C.; Hermes, M.; Garaschuk, O. A bell-shaped dependence between amyloidosis and GABA accumulation in astrocytes in a mouse model of Alzheimer’s disease. Neurobiol. Aging 2018, 61, 187–197. [Google Scholar] [CrossRef]
- Pandit, S.; Neupane, C.; Woo, J.; Sharma, R.; Nam, M.H.; Lee, G.S.; Yi, M.H.; Shin, N.; Kim, D.W.; Cho, H.; et al. Bestrophin1-mediated tonic GABA release from reactive astrocytes prevents the development of seizure-prone network in kainate-injected hippocampi. Glia 2020, 68, 1065–1080. [Google Scholar] [CrossRef] [PubMed]
- Ross, C.A.; Tabrizi, S.J. Huntington’s disease: From molecular pathogenesis to clinical treatment. Lancet Neurol. 2011, 10, 83–98. [Google Scholar] [CrossRef] [PubMed]
- Behrens, P.F.; Franz, P.; Woodman, B.; Lindenberg, K.S.; Landwehrmeyer, G.B. Impaired glutamate transport and glutamate-glutamine cycling: Downstream effects of the Huntington mutation. Brain 2002, 125, 1908–1922. [Google Scholar] [CrossRef] [Green Version]
- Miller, B.R.; Walker, A.G.; Fowler, S.C.; von Hörsten, S.; Riess, O.; Johnson, M.A.; Rebec, G.V. Dysregulation of coordinated neuronal firing patterns in striatum of freely behaving transgenic rats that model Huntington’s disease. Neurobiol. Dis. 2010, 37, 106–113. [Google Scholar] [CrossRef] [Green Version]
- Skotte, N.H.; Andersen, J.V.; Santos, A.; Aldana, B.I.; Willert, C.W.; Nørremølle, A.; Waagepetersen, H.S.; Nielsen, M.L. Integrative Characterization of the R6/2 Mouse Model of Huntington’s Disease Reveals Dysfunctional Astrocyte Metabolism. Cell Rep. 2018, 23, 2211–2224. [Google Scholar] [CrossRef]
- Lee, W.; Reyes, R.C.; Gottipati, M.K.; Lewis, K.; Lesort, M.; Parpura, V.; Gray, M. Enhanced Ca2+-dependent glutamate release from astrocytes of the BACHD Huntington’s disease mouse model. Neurobiol. Dis. 2013, 58, 192–199. [Google Scholar] [CrossRef] [Green Version]
- Hsu, Y.T.; Chang, Y.G.; Chern, Y. Insights into GABA(A)ergic system alteration in Huntington’s disease. Open Biol. 2018, 8, 180165. [Google Scholar] [CrossRef] [Green Version]
- Wojtowicz, A.M.; Dvorzhak, A.; Semtner, M.; Grantyn, R. Reduced tonic inhibition in striatal output neurons from Huntington mice due to loss of astrocytic GABA release through GAT-3. Front. Neural. Circuits 2013, 7, 188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, R.; Diaz-Castro, B.; Looger, L.L.; Khakh, B.S. Dysfunctional Calcium and Glutamate Signaling in Striatal Astrocytes from Huntington’s Disease Model Mice. J. Neurosci. 2016, 36, 3453–3470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steele, A.D.; Jackson, W.S.; King, O.D.; Lindquist, S. The power of automated high-resolution behavior analysis revealed by its application to mouse models of Huntington’s and prion diseases. Proc. Natl. Acad. Sci. USA 2007, 104, 1983–1988. [Google Scholar] [CrossRef] [PubMed]
- Canitano, R.; Palumbi, R. Excitation/Inhibition Modulators in Autism Spectrum Disorder: Current Clinical Research. Front. Neurosci. 2021, 15, 753274. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Mao, X.; Zhu, C.; Zou, X.; Peng, F.; Yang, W.; Li, B.; Li, G.; Ge, T.; Cui, R. GABAergic System Dysfunction in Autism Spectrum Disorders. Front. Cell Dev. Biol. 2021, 9, 781327. [Google Scholar] [CrossRef] [PubMed]
- Yizhar, O.; Fenno, L.E.; Prigge, M.; Schneider, F.; Davidson, T.J.; O’Shea, D.J.; Sohal, V.S.; Goshen, I.; Finkelstein, J.; Paz, J.T.; et al. Neocortical excitation/inhibition balance in information processing and social dysfunction. Nature 2011, 477, 171–178. [Google Scholar] [CrossRef] [Green Version]
- Rubenstein, J.L.; Merzenich, M.M. Model of autism: Increased ratio of excitation/inhibition in key neural systems. Genes Brain Behav. 2003, 2, 255–267. [Google Scholar] [CrossRef]
- Tanaka, K.; Watase, K.; Manabe, T.; Yamada, K.; Watanabe, M.; Takahashi, K.; Iwama, H.; Nishikawa, T.; Ichihara, N.; Kikuchi, T.; et al. Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1. Science 1997, 276, 1699–1702. [Google Scholar] [CrossRef]
- Aida, T.; Yoshida, J.; Nomura, M.; Tanimura, A.; Iino, Y.; Soma, M.; Bai, N.; Ito, Y.; Cui, W.; Aizawa, H.; et al. Astroglial glutamate transporter deficiency increases synaptic excitability and leads to pathological repetitive behaviors in mice. Neuropsychopharmacology 2015, 40, 1569–1579. [Google Scholar] [CrossRef]
- Kirischuk, S.; Kettenmann, H.; Verkhratsky, A. Na+/Ca2+ exchanger modulates kainate-triggered Ca2+ signaling in Bergmann glial cells in situ. FASEB J. 1997, 11, 566–572. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, K. Astroglia and Obsessive Compulsive Disorder. Adv. Neurobiol. 2021, 26, 139–149. [Google Scholar] [PubMed]
- Treiman, D.M. GABAergic mechanisms in epilepsy. Epilepsia 2001, 42 (Suppl. S3), 8–12. [Google Scholar] [CrossRef] [PubMed]
- Akyuz, E.; Polat, A.K.; Eroglu, E.; Kullu, I.; Angelopoulou, E.; Paudel, Y.N. Revisiting the role of neurotransmitters in epilepsy: An updated review. Life Sci. 2021, 265, 118826. [Google Scholar] [CrossRef] [PubMed]
- Richter, D.; Luhmann, H.J.; Kilb, W. Intrinsic activation of GABA(A) receptors suppresses epileptiform activity in the cerebral cortex of immature mice. Epilepsia 2010, 51, 1483–1492. [Google Scholar] [CrossRef] [PubMed]
- Chuang, S.H.; Reddy, D.S. Genetic and Molecular Regulation of Extrasynaptic GABA-A Receptors in the Brain: Therapeutic Insights for Epilepsy. J. Pharm. Exp. Ther. 2018, 364, 180–197. [Google Scholar] [CrossRef] [PubMed]
- Maljevic, S.; Møller, R.S.; Reid, C.A.; Pérez-Palma, E.; Lal, D.; May, P.; Lerche, H. Spectrum of GABAA receptor variants in epilepsy. Curr. Opin. Neurol. 2019, 32, 183–190. [Google Scholar] [CrossRef]
- Lie, M.E.K.; Al-Khawaja, A.; Damgaard, M.; Haugaard, A.S.; Schousboe, A.; Clarkson, A.N.; Wellendorph, P. Glial GABA Transporters as Modulators of Inhibitory Signalling in Epilepsy and Stroke. Adv. Neurobiol. 2017, 16, 137–167. [Google Scholar]
- Kovács, Z.; Skatchkov, S.N.; Veh, R.W.; Szabó, Z.; Németh, K.; Szabó, P.T.; Kardos, J.; Héja, L. Critical Role of Astrocytic Polyamine and GABA Metabolism in Epileptogenesis. Front. Cell Neurosci. 2021, 15, 787319. [Google Scholar] [CrossRef]
- Kovács, Z.; Skatchkov, S.N.; Szabó, Z.; Qahtan, S.; Méndez-González, M.P.; Malpica-Nieves, C.J.; Eaton, M.J.; Kardos, J.; Héja, L. Putrescine Intensifies Glu/GABA Exchange Mechanism and Promotes Early Termination of Seizures. Int. J. Mol. Sci. 2022, 23, 8191. [Google Scholar] [CrossRef]
- de Lanerolle, N.C.; Kim, J.H.; Robbins, R.J.; Spencer, D.D. Hippocampal interneuron loss and plasticity in human temporal lobe epilepsy. Brain Res. 1989, 495, 387–395. [Google Scholar] [CrossRef]
- Buckmaster, P.S.; Abrams, E.; Wen, X. Seizure frequency correlates with loss of dentate gyrus GABAergic neurons in a mouse model of temporal lobe epilepsy. J. Comp. Neurol. 2017, 525, 2592–2610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Righes Marafiga, J.; Vendramin Pasquetti, M.; Calcagnotto, M.E. GABAergic interneurons in epilepsy: More than a simple change in inhibition. Epilepsy Behav. 2021, 121, 106935. [Google Scholar] [CrossRef] [PubMed]
- Kirischuk, S.; Parpura, V.; Verkhratsky, A. Sodium dynamics: Another key to astroglial excitability? Trends Neurosci. 2012, 35, 497–506. [Google Scholar] [CrossRef]
- Felix, L.; Delekate, A.; Petzold, G.C.; Rose, C.R. Sodium Fluctuations in Astroglia and Their Potential Impact on Astrocyte Function. Front. Physiol. 2020, 11, 871. [Google Scholar] [CrossRef]
- Chatton, J.Y.; Magistretti, P.J.; Barros, L.F. Sodium signaling and astrocyte energy metabolism. Glia 2016, 64, 1667–1676. [Google Scholar] [CrossRef]
- Boddum, K.; Jensen, T.P.; Magloire, V.; Kristiansen, U.; Rusakov, D.A.; Pavlov, I.; Walker, M.C. Astrocytic GABA transporter activity modulates excitatory neurotransmission. Nat. Commun. 2016, 7, 13572. [Google Scholar] [CrossRef]
- Kirischuk, S.; Moller, T.; Voitenko, N.; Kettenmann, H.; Verkhratsky, A. ATP-induced cytoplasmic calcium mobilization in Bergmann glial cells. J. Neurosci. 1995, 15, 7861–7871. [Google Scholar] [CrossRef] [PubMed]
- King, C.M.; Bohmbach, K.; Minge, D.; Delekate, A.; Zheng, K.; Reynolds, J.; Rakers, C.; Zeug, A.; Petzold, G.C.; Rusakov, D.A.; et al. Local Resting Ca2+ Controls the Scale of Astroglial Ca2+ Signals. Cell Rep. 2020, 30, 3466–3477. [Google Scholar] [CrossRef] [Green Version]
- Barakat, L.; Bordey, A. GAT-1 and reversible GABA transport in Bergmann glia in slices. J. Neurophysiol. 2002, 88, 1407–1419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, D.; Semyanov, A.; Genazzani, A.; Verkhratsky, A. Calcium signaling in neuroglia. Int. Rev. Cell Mol. Biol. 2021, 362, 1–53. [Google Scholar]
- Bazargani, N.; Attwell, D. Astrocyte calcium signaling: The third wave. Nat. Neurosci. 2016, 19, 182–189. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Brain Region | Disease Model | Best1 | GAT3 | References |
|---|---|---|---|---|
| Cerebellum | WT | + | [23] | |
| Thalamus | WT | + | [34] | |
| Cerebral Cortex | WT | + | [20,76] | |
| Striatum | WT | + | [101] | |
| Hippocampus | 5xFAD (AD) | [28] | ||
| Hippocampus | APPNL−F/NL−F knock-in (AD) | + | [91] | |
| Hippocampus | APP/PS1 (AD) | + | [35] | |
| Hippocampus | APP/PS1 (AD) | + | [35] | |
| Striatum | Z-Q175-KI and R2/6 (HD) | + (reduced) | [101] | |
| Hippocampus | Kainate model (epilepsy) | + | [94] | |
| Hippocampus | WAG/Rij (epilepsy) | + | [118] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kilb, W.; Kirischuk, S. GABA Release from Astrocytes in Health and Disease. Int. J. Mol. Sci. 2022, 23, 15859. https://doi.org/10.3390/ijms232415859
Kilb W, Kirischuk S. GABA Release from Astrocytes in Health and Disease. International Journal of Molecular Sciences. 2022; 23(24):15859. https://doi.org/10.3390/ijms232415859
Chicago/Turabian StyleKilb, Werner, and Sergei Kirischuk. 2022. "GABA Release from Astrocytes in Health and Disease" International Journal of Molecular Sciences 23, no. 24: 15859. https://doi.org/10.3390/ijms232415859
APA StyleKilb, W., & Kirischuk, S. (2022). GABA Release from Astrocytes in Health and Disease. International Journal of Molecular Sciences, 23(24), 15859. https://doi.org/10.3390/ijms232415859