
Disassembly and Mislocalization of AQP4 in Incipient Scar Formation after Experimental Stroke



Abstract
:1. Introduction
2. Results
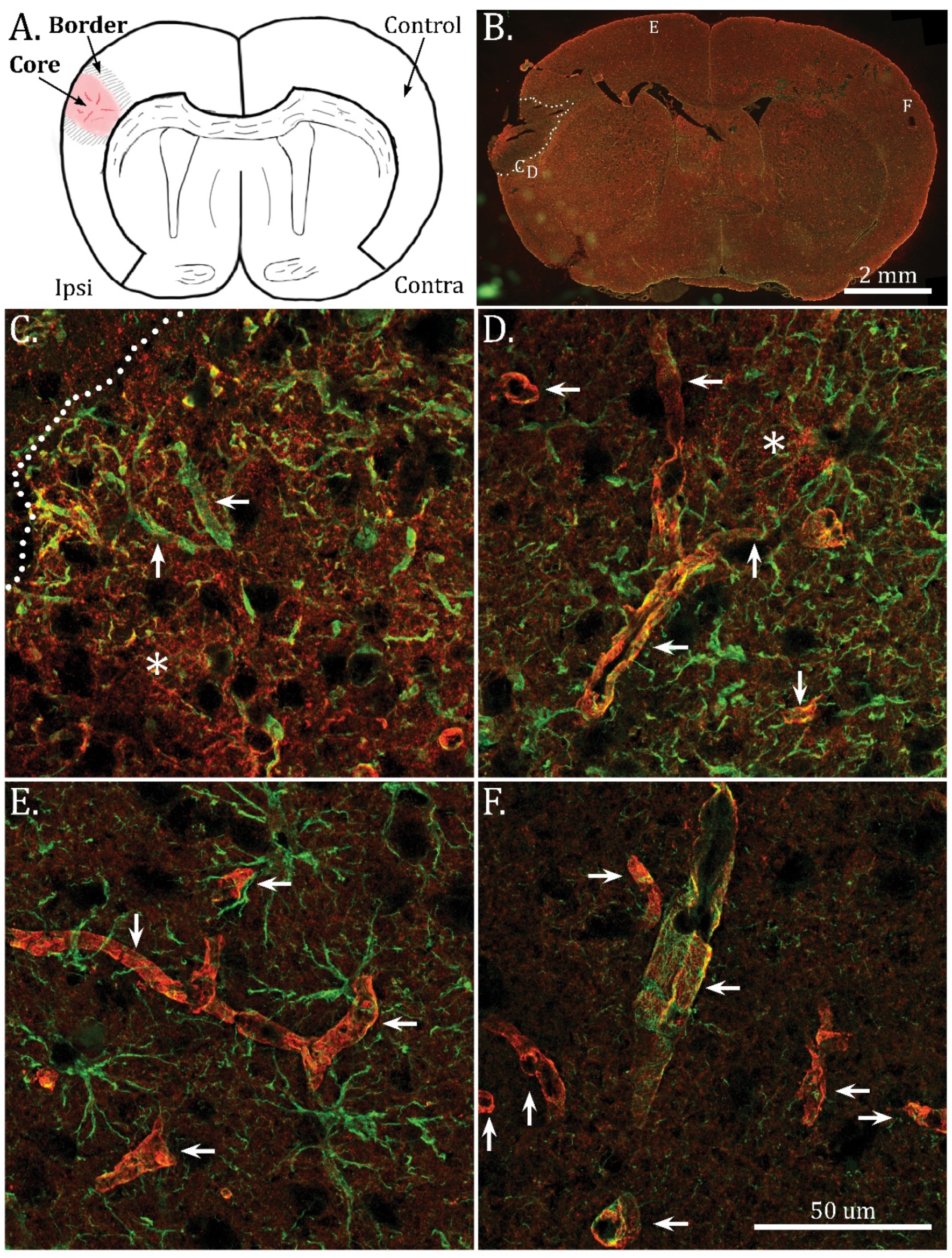
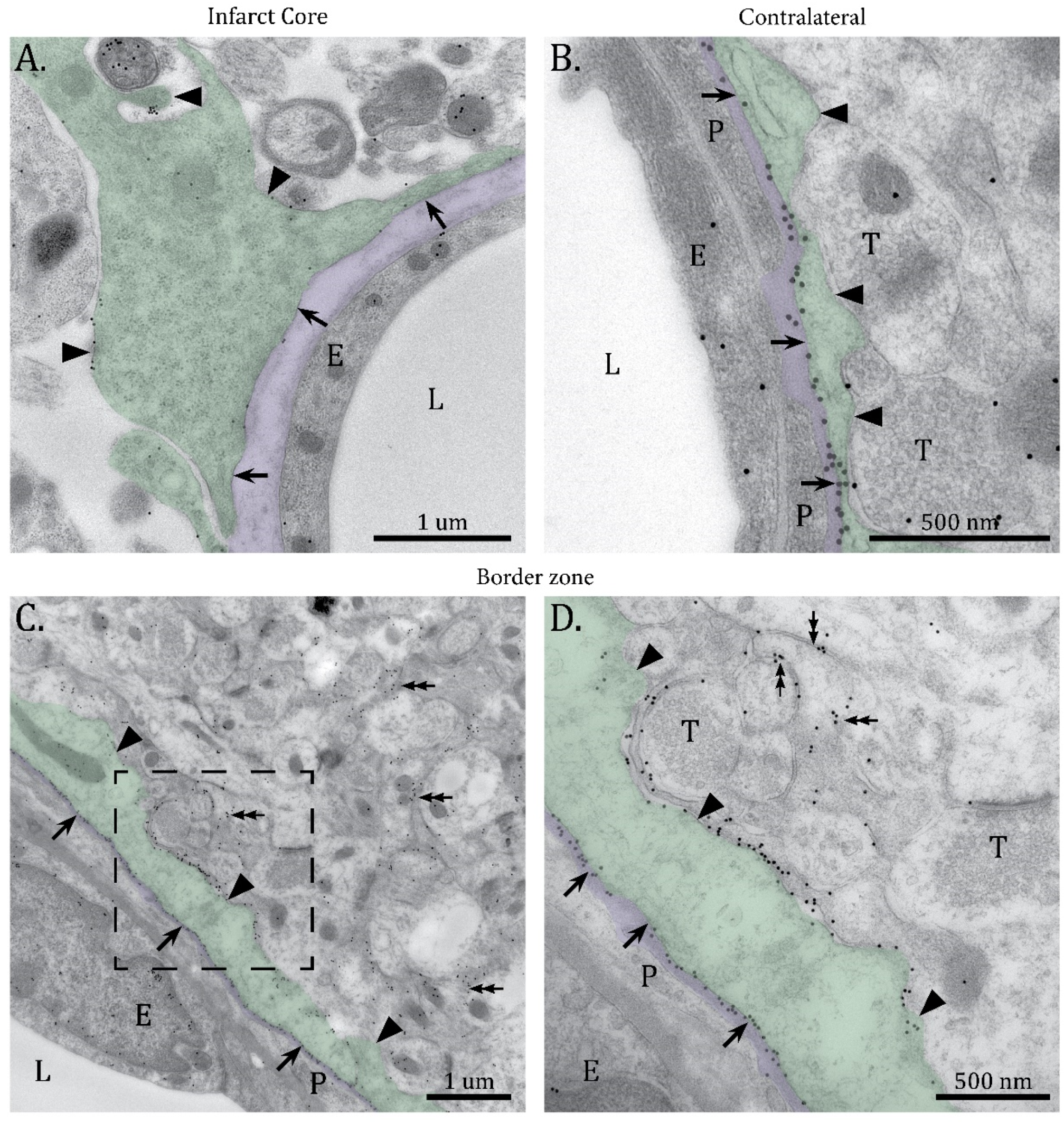
2.1. Mislocalization of AQP4 in the Infarct Border Zone
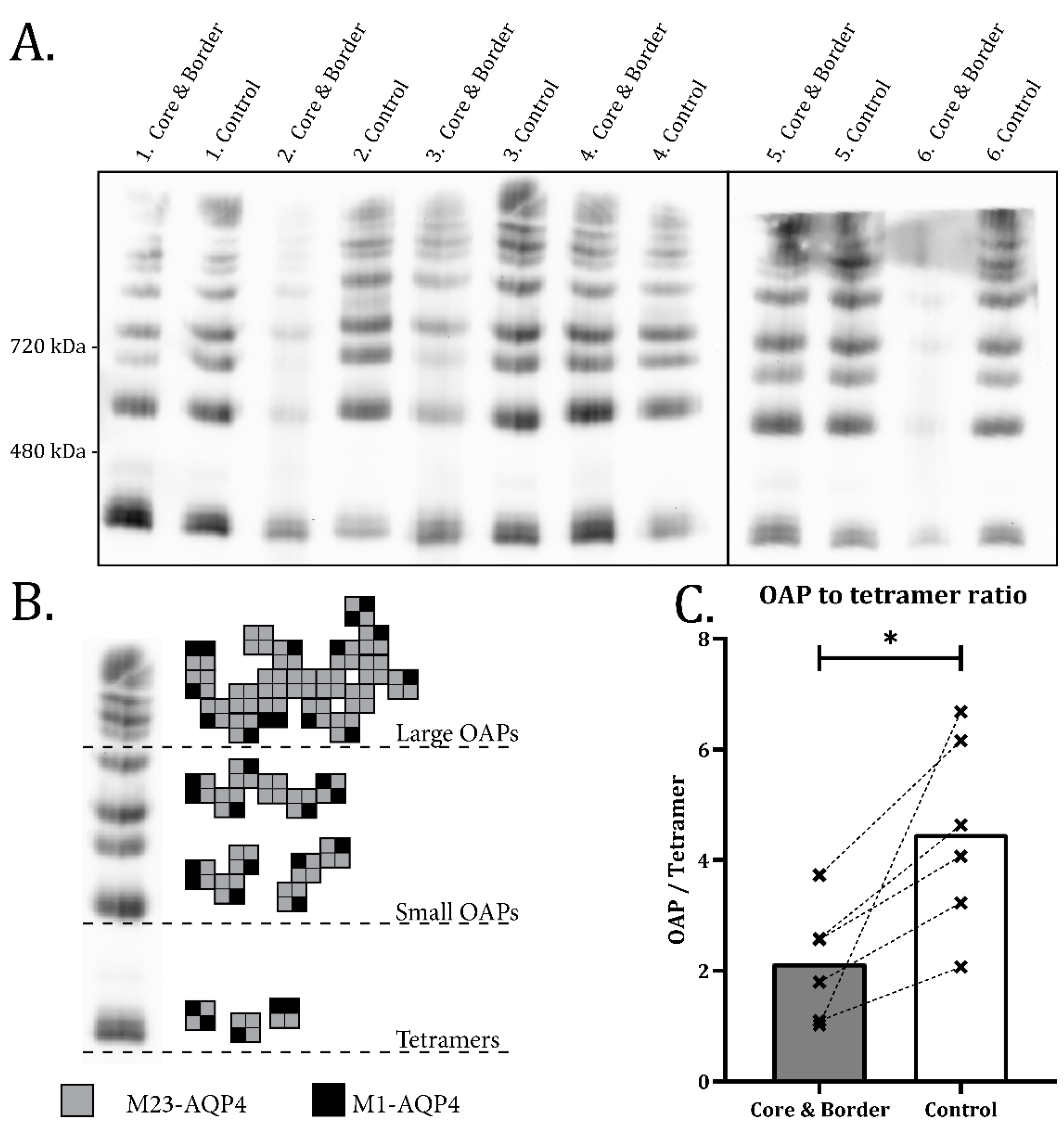
2.2. Disassembly of AQP4 in Infarct Border Zone
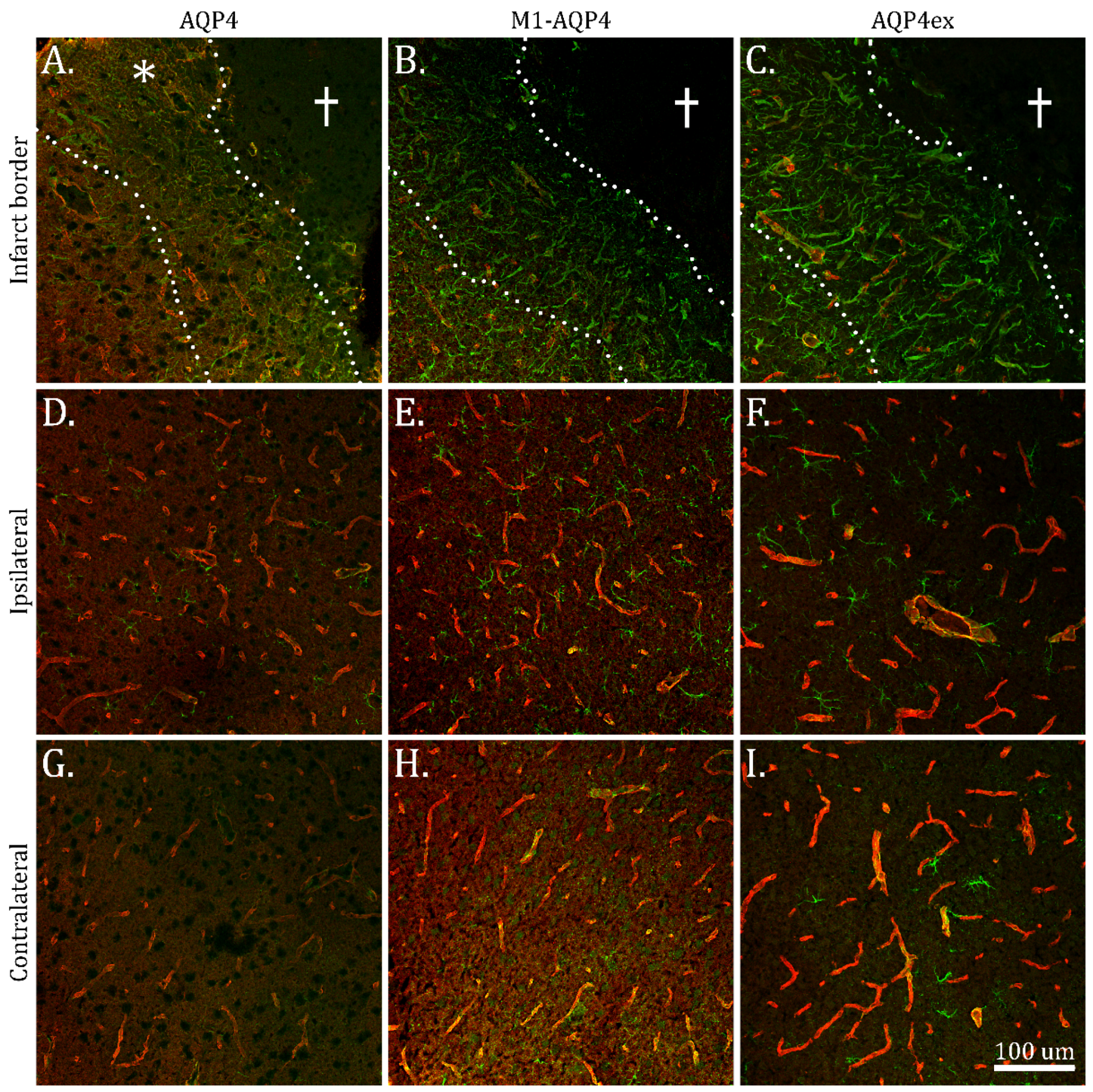
2.3. Reduction in M1-AQP4 and AQP4ex in Border Zone
3. Discussion
4. Materials and Methods
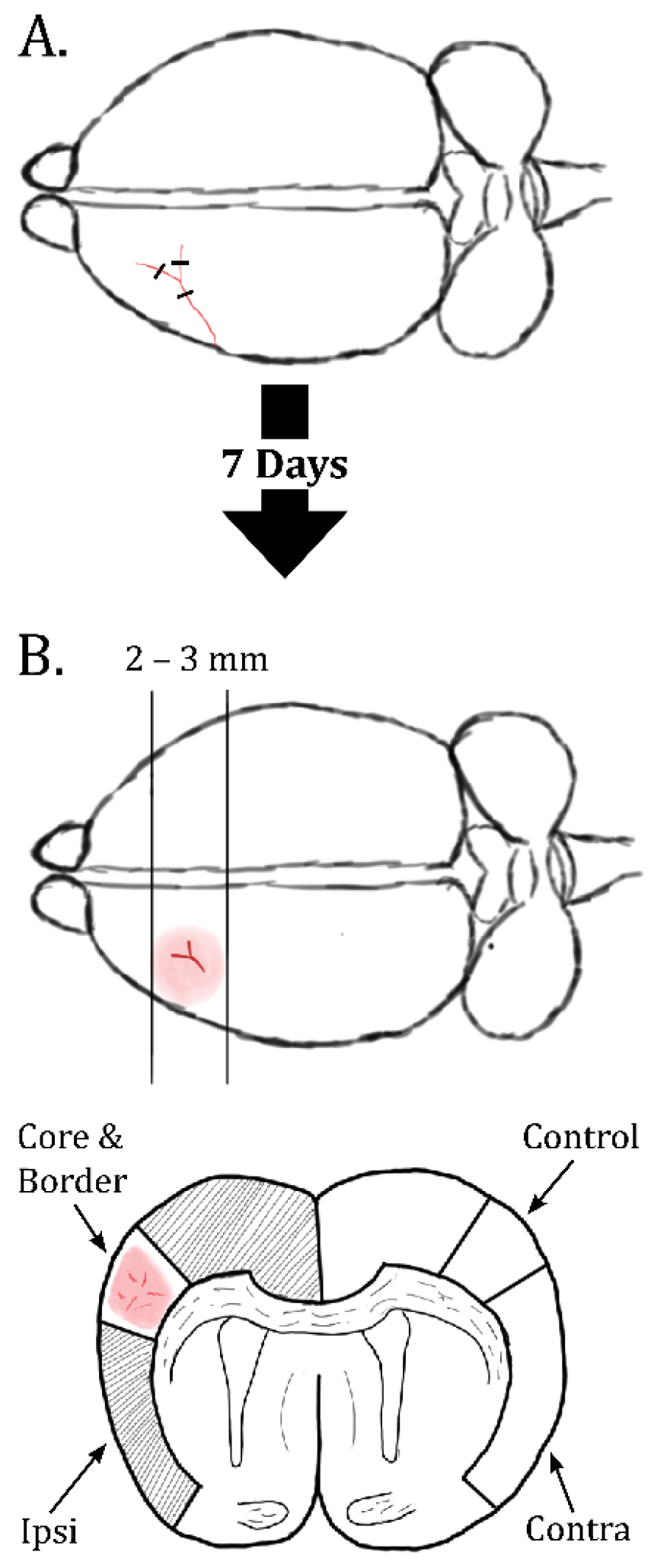
4.1. Animals, Permanent dMCAO and Dissections
4.2. BN-PAGE
4.3. SDS-PAGE on Total Protein Samples
4.4. Western Blot and ECL Signal Acquisition
4.5. Immunogold Transmission Electron Microscopy
4.6. Immunofluorescence Laser Scanning Microscopy
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Choudhury, G.R.; Ding, S. Reactive astrocytes and therapeutic potential in focal ischemic stroke. Neurobiol. Dis. 2016, 85, 234–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pekny, M.; Wilhelmsson, U.; Tatlisumak, T.; Pekna, M. Astrocyte activation and reactive gliosis—A new target in stroke? Neurosci. Lett. 2019, 689, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Dias, D.O.; Kalkitsas, J.; Kelahmetoglu, Y.; Estrada, C.P.; Tatarishvili, J.; Holl, D.; Jansson, L.; Banitalebi, S.; Amiry-Moghaddam, M.; Ernst, A.; et al. Pericyte-derived fibrotic scarring is conserved across diverse central nervous system lesions. Nat. Commun. 2021, 12, 5501. [Google Scholar] [CrossRef] [PubMed]
- Pekny, M.; Pekna, M. Reactive gliosis in the pathogenesis of CNS diseases. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2016, 1862, 483–491. [Google Scholar] [CrossRef] [PubMed]
- Nagelhus, E.A.; Ottersen, O.P. Physiological Roles of Aquaporin-4 in Brain. Physiol. Rev. 2013, 93, 1543–1562. [Google Scholar] [CrossRef] [Green Version]
- Nicchia, G.; Rossi, A.; Mola, M.; Pisani, F.; Stigliano, C.; Basco, D.; Mastrototaro, M.; Svelto, M.; Frigeri, A. Higher order structure of aquaporin-4. Neuroscience 2010, 168, 903–914. [Google Scholar] [CrossRef]
- Wolburg, H.; Wolburg-Buchholz, K.; Fallier-Becker, P.; Noell, S.; Mack, A.F. Chapter one-Structure and Functions of Aquaporin-4-Based Orthogonal Arrays of Particles. In International Review of Cell and Molecular Biology; Jeon, K.W., Ed.; Academic Press: Cambridge, MA, USA, 2011; Volume 287, pp. 1–41. [Google Scholar]
- Rossi, A.; Moritz, T.J.; Ratelade, J.; Verkman, A.S. Super-resolution imaging of aquaporin-4 orthogonal arrays of particles in cell membranes. J. Cell Sci. 2012, 125, 4405–4412. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, H.; Nishikawa, K.; Hiroaki, Y.; Fujiyoshi, Y. Formation of aquaporin-4 arrays is inhibited by palmitoylation of N-terminal cysteine residues. Biochim. Biophys. Acta (BBA)-Biomembr. 2008, 1778, 1181–1189. [Google Scholar] [CrossRef] [Green Version]
- De Bellis, M.; Cibelli, A.; Mola, M.G.; Pisani, F.; Barile, B.; Mastrodonato, M.; Banitalebi, S.; Amiry-Mogghaddam, M.; Abbrescia, P.; Frigeri, A.; et al. Orthogonal arrays of particle assembly are essential for normal aquaporin-4 expression level in the brain. Glia 2021, 69, 473–488. [Google Scholar] [CrossRef]
- Jorgačevski, J.; Zorec, R.; Potokar, M. Insights into Cell Surface Expression, Supramolecular Organization, and Functions of Aquaporin 4 Isoforms in Astrocytes. Cells 2020, 9, 2622. [Google Scholar] [CrossRef]
- Verkman, A.; Rossi, A.; Crane, J.M. Live-Cell Imaging of Aquaporin-4 Supramolecular Assembly and Diffusion. Imaging Spectrosc. Anal. Living Cells-Opt. Spectrosc. Tech. 2012, 504, 341–354. [Google Scholar] [CrossRef] [Green Version]
- Saadoun, S.; Papadopoulos, M.; Watanabe, H.; Yan, D.; Manley, G.T.; Verkman, A.S. Involvement of aquaporin-4 in astroglial cell migration and glial scar formation. J. Cell Sci. 2005, 118, 5691–5698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Auguste, K.I.; Jin, S.; Uchida, K.; Yan, D.; Manley, G.T.; Papadopoulos, M.; Verkman, A.S. Greatly impaired migration of implanted aquaporin-4-deficient astroglial cells in mouse brain toward a site of injury. FASEB J. 2006, 21, 108–116. [Google Scholar] [CrossRef] [PubMed]
- De Bellis, M.; Pisani, F.; Mola, M.G.; Rosito, S.; Simone, L.; Buccoliero, C.; Trojano, M.; Nicchia, G.P.; Svelto, M.; Frigeri, A. Translational readthrough generates new astrocyte AQP4 isoforms that modulate supramolecular clustering, glial endfeet localization, and water transport. Glia 2017, 65, 790–803. [Google Scholar] [CrossRef]
- Palazzo, C.; Buccoliero, C.; Mola, M.G.; Abbrescia, P.; Nicchia, G.P.; Trojano, M.; Frigeri, A. AQP4ex is crucial for the anchoring of AQP4 at the astrocyte end-feet and for neuromyelitis optica antibody binding. Acta Neuropathol. Commun. 2019, 7, 1–15. [Google Scholar] [CrossRef]
- Palazzo, C.; Abbrescia, P.; Valente, O.; Nicchia, G.P.; Banitalebi, S.; Amiry-Moghaddam, M.; Trojano, M.; Frigeri, A. Tissue Distribution of the Readthrough Isoform of AQP4 Reveals a Dual Role of AQP4ex Limited to CNS. Int. J. Mol. Sci. 2020, 21, 1531. [Google Scholar] [CrossRef] [Green Version]
- Rash, J.E.; Yasumura, T.; Hudson, C.S.; Agre, P.; Nielsen, S. Direct immunogold labeling of aquaporin-4 in square arrays of astrocyte and ependymocyte plasma membranes in rat brain and spinal cord. Proc. Natl. Acad. Sci. USA 1998, 95, 11981–11986. [Google Scholar] [CrossRef] [Green Version]
- Neely, J.D.; Amiry-Moghaddam, M.; Ottersen, O.P.; Froehner, S.C.; Agre, P.; Adams, M.E. Syntrophin-dependent expression and localization of Aquaporin-4 water channel protein. Proc. Natl. Acad. Sci. USA 2001, 98, 14108–14113. [Google Scholar] [CrossRef] [Green Version]
- Sofroniew, M.V. Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci. 2009, 32, 638–647. [Google Scholar] [CrossRef] [Green Version]
- Engel, A.; Fujiyoshi, Y.; Gonen, T.; Walz, T. Junction-forming aquaporins. Curr. Opin. Struct. Biol. 2008, 18, 229–235. [Google Scholar] [CrossRef] [Green Version]
- Amiry-Moghaddam, M.; Ottersen, O.P. The molecular basis of water transport in the brain. Nat. Rev. Neurosci. 2003, 4, 991–1001. [Google Scholar] [CrossRef]
- Amiry-Moghaddam, M.; Otsuka, T.; Hurn, P.D.; Traystman, R.J.; Haug, F.-M.; Froehner, S.C.; Adams, M.E.; Neely, J.D.; Agre, P.; Ottersen, O.P.; et al. An-syntrophin-dependent pool of AQP4 in astroglial end-feet confers bidirectional water flow between blood and brain. Proc. Natl. Acad. Sci. USA 2003, 100, 2106–2111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verbavatz, J.-M.; Ma, T.; Gobin, R.; Verkman, A. Absence of orthogonal arrays in kidney, brain and muscle from transgenic knockout mice lacking water channel aquaporin-4. J. Cell Sci. 1997, 110, 2855–2860. [Google Scholar] [CrossRef] [PubMed]
- Simone, L.; Pisani, F.; Mola, M.G.; De Bellis, M.; Merla, G.; Micale, L.; Frigeri, A.; Vescovi, A.L.; Svelto, M.; Nicchia, G.P. AQP4 Aggregation State Is a Determinant for Glioma Cell Fate. Cancer Res. 2019, 79, 2182–2194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amiry-Moghaddam, M.; Frydenlund, D.; Ottersen, O. Anchoring of aquaporin-4 in brain: Molecular mechanisms and implications for the physiology and pathophysiology of water transport. Neuroscience 2004, 129, 997–1008. [Google Scholar] [CrossRef] [PubMed]
- Frydenlund, D.S.; Bhardwaj, A.; Otsuka, T.; Mylonakou, M.N.; Yasumura, T.; Davidson, K.G.V.; Zeynalov, E.; Skare, Ø.; Laake, P.; Haug, F.-M.; et al. Temporary loss of perivascular aquaporin-4 in neocortex after transient middle cerebral artery occlusion in mice. Proc. Natl. Acad. Sci. USA 2006, 103, 13532–13536. [Google Scholar] [CrossRef] [Green Version]
- Llovera, G.; Roth, S.; Plesnila, N.; Veltkamp, R.; Liesz, A. Modeling Stroke in Mice: Permanent Coagulation of the Distal Middle Cerebral Artery. J. Vis. Exp. 2014, 89, e51729. [Google Scholar] [CrossRef] [Green Version]
- Life Technologies. NativePAGE™ Novex® Bis-Tris Gel System. 2012. Available online: https://assets.thermofisher.com/TFS-Assets/LSG/manuals/nativepage_man.pdf (accessed on 14 September 2021).
- Campagne, M.V.L.; Oestreicher, A.B.; Van Der Krift, T.P.; Gispen, W.H.; Verkleij, A.J. Freeze-substitution and Lowicryl HM20 embedding of fixed rat brain: Suitability for immunogold ultrastructural localization of neural antigens. J. Histochem. Cytochem. 1991, 39, 1267–1279. [Google Scholar] [CrossRef] [Green Version]
- Prydz, A.; Stahl, K.; Puchades, M.; Davarpaneh, N.; Nadeem, M.; Ottersen, O.P.; Gundersen, V.; Amiry-Moghaddam, M. Subcellular expression of aquaporin-4 in substantia nigra of normal and MPTP-treated mice. Neuroscience 2017, 359, 258–266. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | Primary Antibody | Secondary Antibody |
|---|---|---|
| Western Blot (SDS and BN-PAGE) | Anti-AQP4 (rabbit), Sigma Cat# A5971, RRID: AB_258270, 1:5000 | ECL anti-rabbit (donkey), GE Healthcare Cat# NA934, RRID: AB_772206, 1:25,000 dilution |
| Anti-M1-AQP4 (rabbit), A. Frigeri 1, 1:2000 | ECL anti-rabbit (donkey), GE Healthcare Cat# NA934, RRID: AB_772206, 1:25,000 dilution | |
| Anti-AQP4ex (rabbit), A. Frigeri 1, 1:2000 | ECL anti-rabbit (donkey), GE Healthcare Cat# NA934, RRID: AB_772206, 1:25,000 dilution | |
| Anti-α-tubulin (rabbit), Abcam Cat# ab4074, RRID: AB_2288001, 1:5000 dilution | ECL anti-rabbit (donkey), GE Healthcare Cat# NA934, RRID: AB_772206, 1:25,000 dilution | |
| Immunofluorescence LSM | Anti-AQP4 (rabbit), Sigma Cat# A5971, RRID: AB_258270, 1:1200 | Anti-rabbit Cy3 (donkey), Jackson ImmunoResearch Labs Cat# 711-165-152, RRID: AB_2307443, 1:250 dilution |
| Anti-M1-AQP4 (rabbit), A. Frigeri 1, 1:2000 dilution | Anti-rabbit Cy3 (donkey), Jackson ImmunoResearch Labs Cat# 711-165-152, RRID: AB_2307443, 1:250 dilution | |
| Anti-AQP4ex (rabbit), A. Frigeri 1, 1:2000 dilution | Anti-rabbit Cy3 (donkey), Jackson ImmunoResearch Labs Cat# 711-165-152, RRID: AB_2307443, 1:250 dilution | |
| Anti-GFAP (chicken), BioLegend Cat# 829401, RRID:AB_2564929, 1:400 dilution | Anti-chicken Cy2 (donkey), Jackson ImmunoResearch Labs Cat# 703-225-155, RRID: AB_2340370, 1:250 dilution | |
| Immunogold TEM | Anti-AQP4 (rabbit), Sigma Cat# A5971, RRID: AB_258270, 1:400 dilution | Anti-rabbit (Goat) 15nm gold conjugated, Abcam Cat# ab27236, RRID: AB_954457, 1:20 dilution |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Banitalebi, S.; Skauli, N.; Geiseler, S.; Ottersen, O.P.; Amiry-Moghaddam, M. Disassembly and Mislocalization of AQP4 in Incipient Scar Formation after Experimental Stroke. Int. J. Mol. Sci. 2022, 23, 1117. https://doi.org/10.3390/ijms23031117
Banitalebi S, Skauli N, Geiseler S, Ottersen OP, Amiry-Moghaddam M. Disassembly and Mislocalization of AQP4 in Incipient Scar Formation after Experimental Stroke. International Journal of Molecular Sciences. 2022; 23(3):1117. https://doi.org/10.3390/ijms23031117
Chicago/Turabian StyleBanitalebi, Shervin, Nadia Skauli, Samuel Geiseler, Ole Petter Ottersen, and Mahmood Amiry-Moghaddam. 2022. "Disassembly and Mislocalization of AQP4 in Incipient Scar Formation after Experimental Stroke" International Journal of Molecular Sciences 23, no. 3: 1117. https://doi.org/10.3390/ijms23031117
APA StyleBanitalebi, S., Skauli, N., Geiseler, S., Ottersen, O. P., & Amiry-Moghaddam, M. (2022). Disassembly and Mislocalization of AQP4 in Incipient Scar Formation after Experimental Stroke. International Journal of Molecular Sciences, 23(3), 1117. https://doi.org/10.3390/ijms23031117