
Oestrogenic Regulation of Mitochondrial Dynamics


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
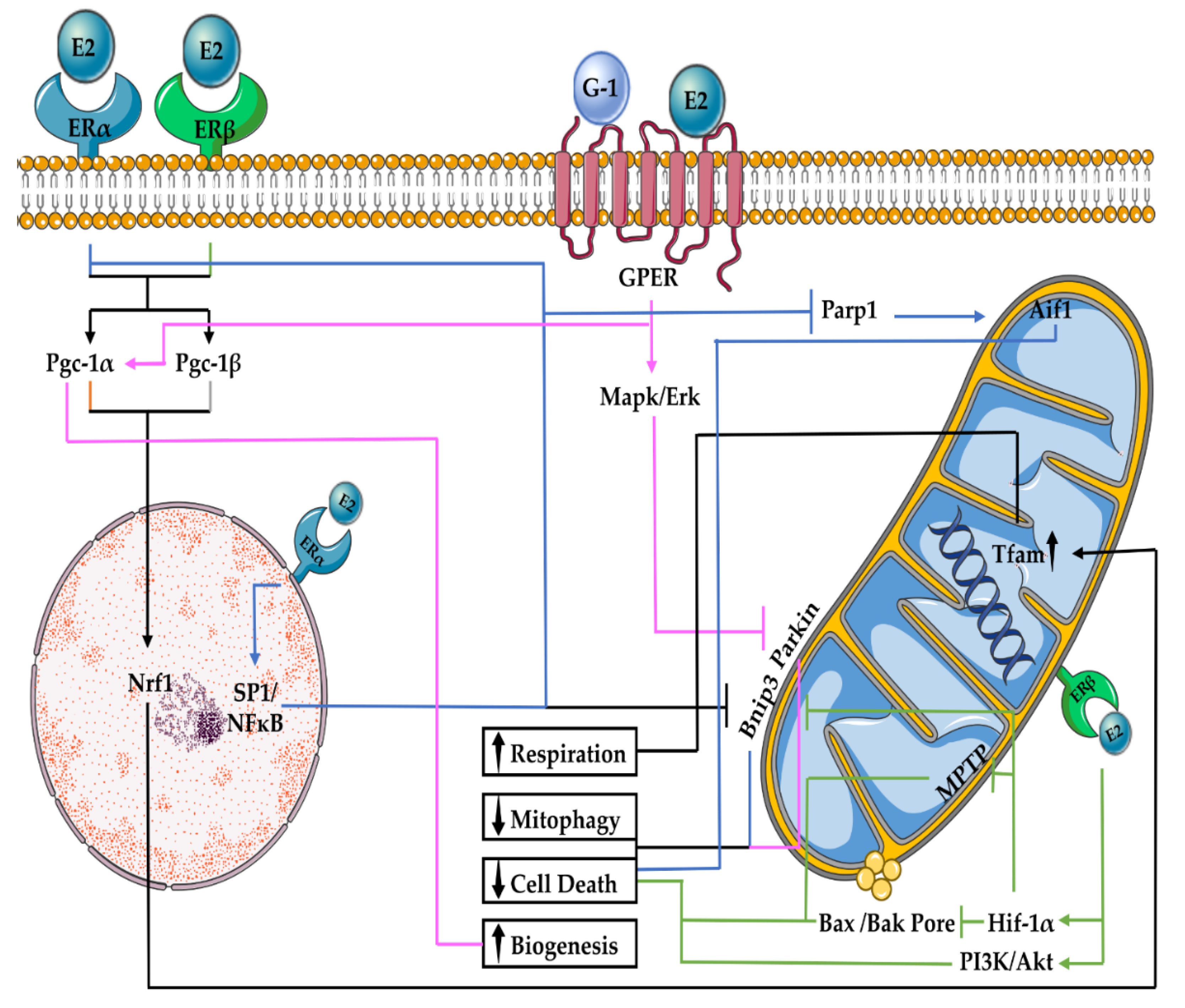
2. E2 Regulation of Mitochondrial Function and Turnover
2.1. E2 Effects on Mitochondrial Respiration
2.2. E2 Effects on Mitophagy
2.3. E2 Effects on Mitochondrial Biogenesis
2.4. E2 Effects on Mitochondrial-Associated Cell Death
3. Mitochondrial Dynamics
3.1. Mitochondrial Fusion
3.2. Mitochondrial Fission
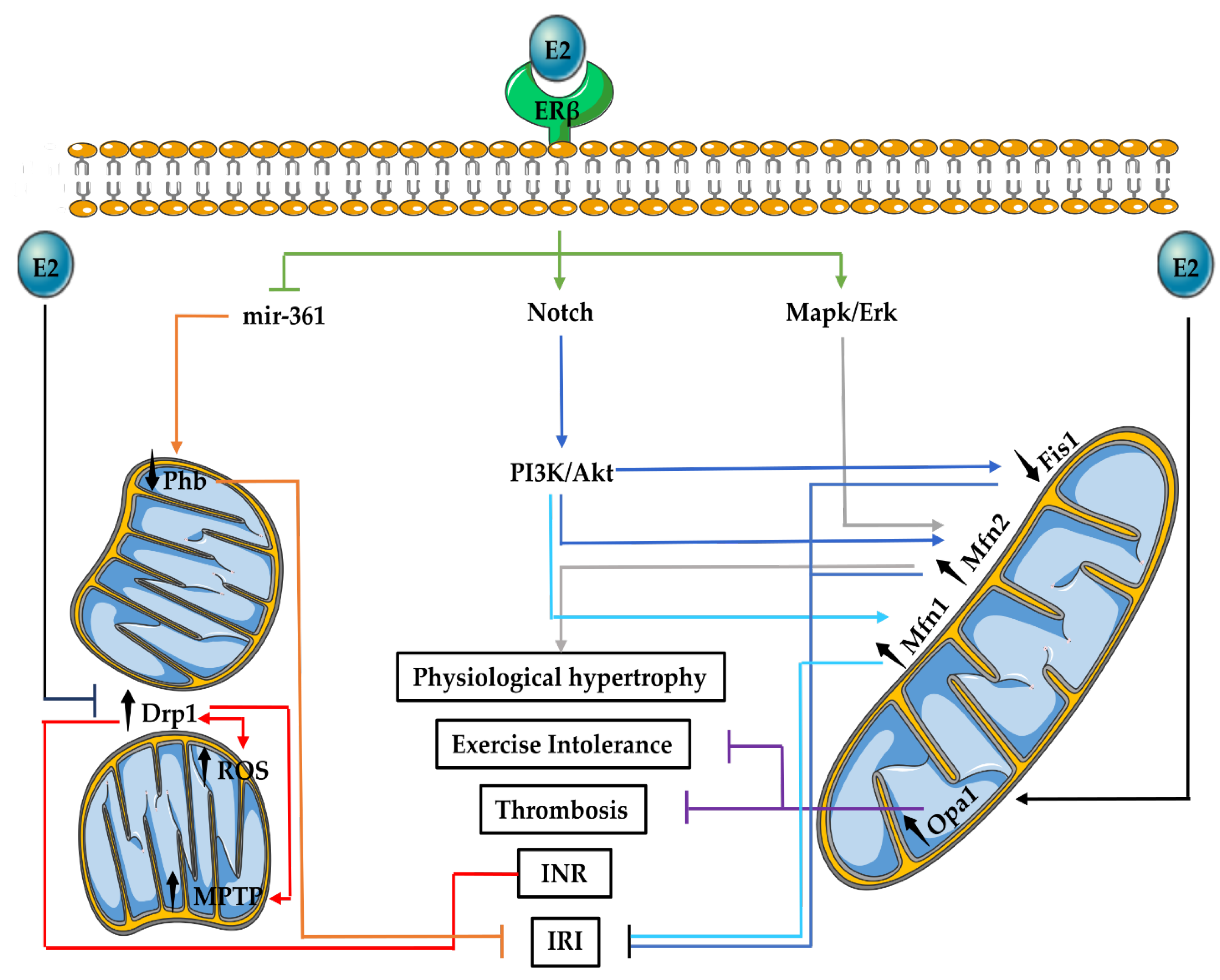
3.3. Mitochondrial Fusion and Fission in the Cardiovascular System
3.4. E2 Effects on Mitochondrial Fusion and Fission in the Cardiovascular System
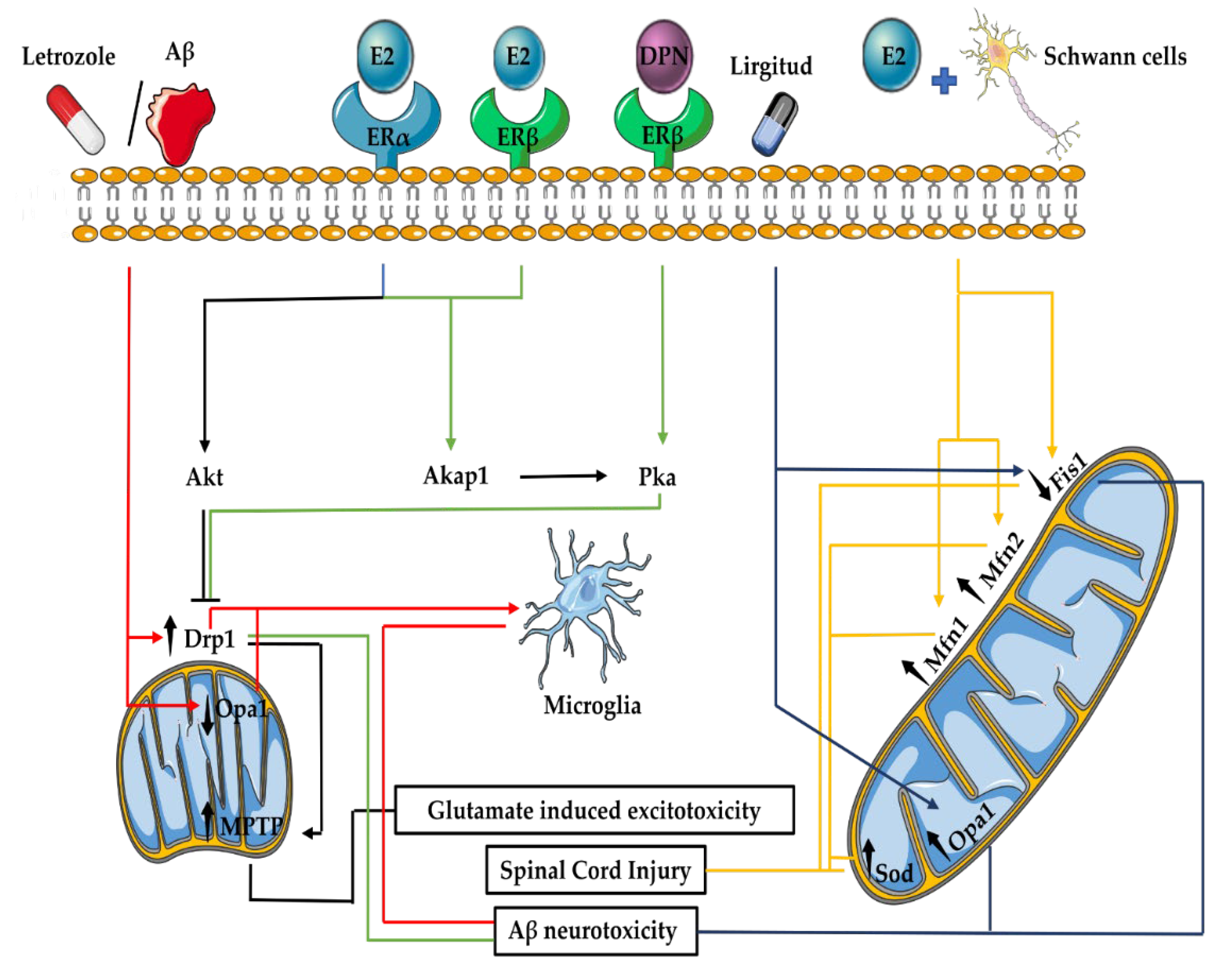
3.5. Mitochondrial Fusion and Fission in the Nervous System
3.6. E2 Effects on Mitochondrial Fusion and Fission in the Nervous System
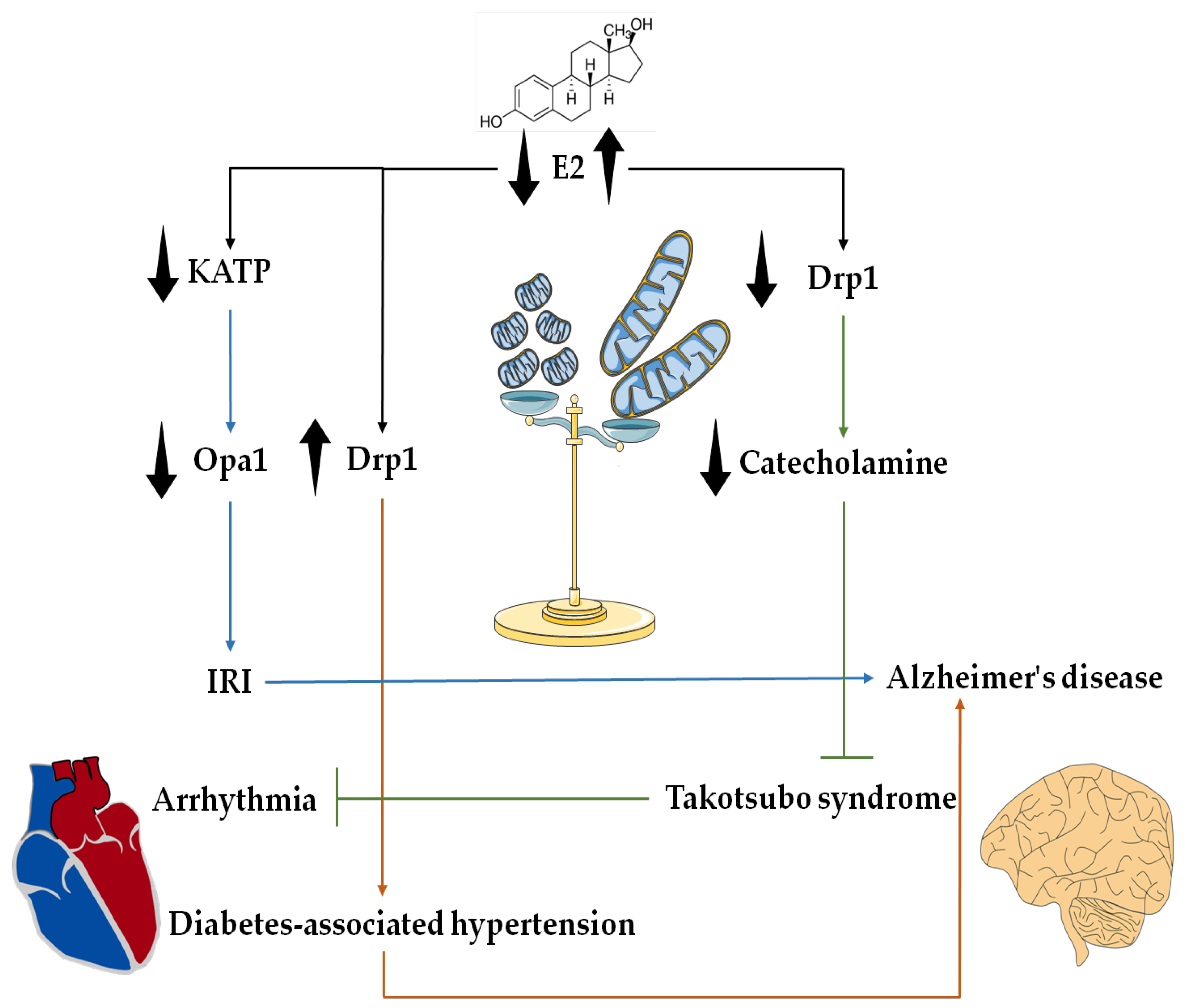
4. Potential Interplay of E2 and Mitochondrial Dynamics in the Heart–Brain Axis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gaignebet, L.; Kararigas, G. En route to precision medicine through the integration of biological sex into pharmacogenomics. Clin. Sci. 2017, 131, 329–342. [Google Scholar] [CrossRef]
- Kararigas, G.; Seeland, U.; de Arellano, M.L.B.; Dworatzek, E.; Regitz-Zagrosek, V. Why the study of the effects of biological sex is important. Commentary. Ann. Ist. Super. Sanita 2016, 52, 149–150. [Google Scholar] [CrossRef]
- Cui, C.; Huang, C.; Liu, K.; Xu, G.; Yang, J.; Zhou, Y.; Feng, Y.; Kararigas, G.; Geng, B.; Cui, Q. Large-scale in silico identification of drugs exerting sex-specific effects in the heart. J. Transl. Med. 2018, 16, 236. [Google Scholar] [CrossRef]
- Ruiz-Meana, M.; Boengler, K.; Garcia-Dorado, D.; Hausenloy, D.J.; Kaambre, T.; Kararigas, G.; Perrino, C.; Schulz, R.; Ytrehus, K. Ageing, sex, and cardioprotection. Br. J. Pharmacol. 2020, 177, 5270–5286. [Google Scholar] [CrossRef]
- Altinbas, L.; Bormann, N.; Lehmann, D.; Jeuthe, S.; Wulsten, D.; Kornak, U.; Robinson, P.N.; Wildemann, B.; Kararigas, G. Assessment of Bones Deficient in Fibrillin-1 Microfibrils Reveals Pronounced Sex Differences. Int. J. Mol. Sci. 2019, 20, 6059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Kararigas, G. Role of Biological Sex in the Cardiovascular-Gut Microbiome Axis. Front. Cardiovasc. Med. 2022, 8, 759735. [Google Scholar] [CrossRef]
- Regitz-Zagrosek, V.; Kararigas, G. Mechanistic Pathways of Sex Differences in Cardiovascular Disease. Physiol. Rev. 2017, 97, 1–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ober, C.; Loisel, D.A.; Gilad, Y. Sex-specific genetic architecture of human disease. Nat. Rev. Genet. 2008, 9, 911–922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCarthy, M.M.; Nugent, B.M.; Lenz, K.M. Neuroimmunology and neuroepigenetics in the establishment of sex differences in the brain. Nat. Rev. Neurosci. 2017, 18, 471–484. [Google Scholar] [CrossRef] [Green Version]
- Kokras, N.; Hodes, G.E.; Bangasser, D.A.; Dalla, C. Sex differences in the hypothalamic-pituitary-adrenal axis: An obstacle to antidepressant drug development? Br. J. Pharmacol. 2019, 176, 4090–4106. [Google Scholar] [CrossRef] [PubMed]
- Kararigas, G.; Dworatzek, E.; Petrov, G.; Summer, H.; Schulze, T.M.; Baczko, I.; Knosalla, C.; Golz, S.; Hetzer, R.; Regitz-Zagrosek, V. Sex-dependent regulation of fibrosis and inflammation in human left ventricular remodelling under pressure overload. Eur. J. Heart Fail. 2014, 16, 1160–1167. [Google Scholar] [CrossRef] [Green Version]
- Gaignebet, L.; Kandula, M.M.; Lehmann, D.; Knosalla, C.; Kreil, D.P.; Kararigas, G. Sex-Specific Human Cardiomyocyte Gene Regulation in Left Ventricular Pressure Overload. Mayo Clin. Proc. 2020, 95, 688–697. [Google Scholar] [CrossRef]
- Dworatzek, E.; Baczko, I.; Kararigas, G. Effects of aging on cardiac extracellular matrix in men and women. Proteom.-Clin. Appl. 2016, 10, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Petrov, G.; Dworatzek, E.; Schulze, T.M.; Dandel, M.; Kararigas, G.; Mahmoodzadeh, S.; Knosalla, C.; Hetzer, R.; Regitz-Zagrosek, V. Maladaptive remodeling is associated with impaired survival in women but not in men after aortic valve replacement. JACC Cardiovasc. Imaging 2014, 7, 1073–1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pei, J.; Harakalova, M.; Treibel, T.A.; Lumbers, R.T.; Boukens, B.J.; Efimov, I.R.; van Dinter, J.T.; Gonzalez, A.; Lopez, B.; El Azzouzi, H.; et al. H3K27ac acetylome signatures reveal the epigenomic reorganization in remodeled non-failing human hearts. Clin. Epigenet. 2020, 12, 106. [Google Scholar] [CrossRef]
- Iorga, A.; Cunningham, C.M.; Moazeni, S.; Ruffenach, G.; Umar, S.; Eghbali, M. The protective role of estrogen and estrogen receptors in cardiovascular disease and the controversial use of estrogen therapy. Biol. Sex Differ. 2017, 8, 33. [Google Scholar] [CrossRef]
- Murphy, E. Estrogen signaling and cardiovascular disease. Circ. Res. 2011, 109, 687–696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, E.; Steenbergen, C. Estrogen regulation of protein expression and signaling pathways in the heart. Biol. Sex Differ. 2014, 5, 6. [Google Scholar] [CrossRef] [Green Version]
- Menazza, S.; Murphy, E. The Expanding Complexity of Estrogen Receptor Signaling in the Cardiovascular System. Circ. Res. 2016, 118, 994–1007. [Google Scholar] [CrossRef] [PubMed]
- Puglisi, R.; Mattia, G.; Care, A.; Marano, G.; Malorni, W.; Matarrese, P. Non-genomic Effects of Estrogen on Cell Homeostasis and Remodeling With Special Focus on Cardiac Ischemia/Reperfusion Injury. Front. Endocrinol. 2019, 10, 733. [Google Scholar] [CrossRef] [Green Version]
- Lowe, D.A.; Kararigas, G. Editorial: New Insights into Estrogen/Estrogen Receptor Effects in the Cardiac and Skeletal Muscle. Front. Endocrinol. 2020, 11, 141. [Google Scholar] [CrossRef] [PubMed]
- Kararigas, G. Oestrogenic contribution to sex-biased left ventricular remodelling: The male implication. Int. J. Cardiol. 2021, 343, 83–84. [Google Scholar] [CrossRef]
- Schubert, C.; Raparelli, V.; Westphal, C.; Dworatzek, E.; Petrov, G.; Kararigas, G.; Regitz-Zagrosek, V. Reduction of apoptosis and preservation of mitochondrial integrity under ischemia/reperfusion injury is mediated by estrogen receptor beta. Biol. Sex Differ. 2016, 7, 53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahmoodzadeh, S.; Dworatzek, E. The Role of 17beta-Estradiol and Estrogen Receptors in Regulation of Ca(2+) Channels and Mitochondrial Function in Cardiomyocytes. Front. Endocrinol. 2019, 10, 310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sickinghe, A.A.; Korporaal, S.J.A.; den Ruijter, H.M.; Kessler, E.L. Estrogen Contributions to Microvascular Dysfunction Evolving to Heart Failure With Preserved Ejection Fraction. Front. Endocrinol. 2019, 10, 442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ventura-Clapier, R.; Piquereau, J.; Veksler, V.; Garnier, A. Estrogens, Estrogen Receptors Effects on Cardiac and Skeletal Muscle Mitochondria. Front. Endocrinol. 2019, 10, 557. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Miller, V.M.; Miller, J.D. Influences of Sex and Estrogen in Arterial and Valvular Calcification. Front. Endocrinol. 2019, 10, 622. [Google Scholar] [CrossRef]
- Kararigas, G.; Fliegner, D.; Forler, S.; Klein, O.; Schubert, C.; Gustafsson, J.A.; Klose, J.; Regitz-Zagrosek, V. Comparative Proteomic Analysis Reveals Sex and Estrogen Receptor beta Effects in the Pressure Overloaded Heart. J. Proteome Res. 2014, 13, 5829–5836. [Google Scholar] [CrossRef]
- Kararigas, G.; Fliegner, D.; Gustafsson, J.A.; Regitz-Zagrosek, V. Role of the estrogen/estrogen-receptor-beta axis in the genomic response to pressure overload-induced hypertrophy. Physiol. Genom. 2011, 43, 438–446. [Google Scholar] [CrossRef]
- Kararigas, G.; Nguyen, B.T.; Jarry, H. Estrogen modulates cardiac growth through an estrogen receptor alpha-dependent mechanism in healthy ovariectomized mice. Mol. Cell. Endocrinol. 2014, 382, 909–914. [Google Scholar] [CrossRef] [PubMed]
- Kararigas, G.; Nguyen, B.T.; Zelarayan, L.C.; Hassenpflug, M.; Toischer, K.; Sanchez-Ruderisch, H.; Hasenfuss, G.; Bergmann, M.W.; Jarry, H.; Regitz-Zagrosek, V. Genetic background defines the regulation of postnatal cardiac growth by 17beta-estradiol through a beta-catenin mechanism. Endocrinology 2014, 155, 2667–2676. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Ruderisch, H.; Queiros, A.M.; Fliegner, D.; Eschen, C.; Kararigas, G.; Regitz-Zagrosek, V. Sex-specific regulation of cardiac microRNAs targeting mitochondrial proteins in pressure overload. Biol. Sex Differ. 2019, 10, 8. [Google Scholar] [CrossRef] [Green Version]
- Duft, K.; Schanz, M.; Pham, H.; Abdelwahab, A.; Schriever, C.; Kararigas, G.; Dworatzek, E.; Davidson, M.M.; Regitz-Zagrosek, V.; Morano, I.; et al. 17beta-Estradiol-induced interaction of estrogen receptor alpha and human atrial essential myosin light chain modulates cardiac contractile function. Basic Res. Cardiol. 2017, 112, 1. [Google Scholar] [CrossRef] [PubMed]
- Lai, S.; Collins, B.C.; Colson, B.A.; Kararigas, G.; Lowe, D.A. Estradiol modulates myosin regulatory light chain phosphorylation and contractility in skeletal muscle of female mice. Am. J. Physiol. Endocrinol. Metab. 2016, 310, E724–E733. [Google Scholar] [CrossRef] [Green Version]
- Mahmoodzadeh, S.; Pham, T.H.; Kuehne, A.; Fielitz, B.; Dworatzek, E.; Kararigas, G.; Petrov, G.; Davidson, M.M.; Regitz-Zagrosek, V. 17beta-Estradiol-induced interaction of ERalpha with NPPA regulates gene expression in cardiomyocytes. Cardiovasc. Res. 2012, 96, 411–421. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, B.T.; Kararigas, G.; Jarry, H. Dose-dependent effects of a genistein-enriched diet in the heart of ovariectomized mice. Genes Nutr. 2012, 8, 383–390. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, B.T.; Kararigas, G.; Wuttke, W.; Jarry, H. Long-term treatment of ovariectomized mice with estradiol or phytoestrogens as a new model to study the role of estrogenic substances in the heart. Planta Med. 2012, 78, 6–11. [Google Scholar] [CrossRef] [PubMed]
- Szego, E.M.; Barabas, K.; Balog, J.; Szilagyi, N.; Korach, K.S.; Juhasz, G.; Abraham, I.M. Estrogen induces estrogen receptor alpha-dependent cAMP response element-binding protein phosphorylation via mitogen activated protein kinase pathway in basal forebrain cholinergic neurons in vivo. J. Neurosci. 2006, 26, 4104–4110. [Google Scholar] [CrossRef] [Green Version]
- Sabbatini, A.R.; Kararigas, G. Menopause-Related Estrogen Decrease and the Pathogenesis of HFpEF: JACC Review Topic of the Week. J. Am. Coll. Cardiol. 2020, 75, 1074–1082. [Google Scholar] [CrossRef] [PubMed]
- Sabbatini, A.R.; Kararigas, G. Estrogen-related mechanisms in sex differences of hypertension and target organ damage. Biol. Sex Differ. 2020, 11, 31. [Google Scholar] [CrossRef] [PubMed]
- Kararigas, G.; Becher, E.; Mahmoodzadeh, S.; Knosalla, C.; Hetzer, R.; Regitz-Zagrosek, V. Sex-specific modification of progesterone receptor expression by 17beta-oestradiol in human cardiac tissues. Biol. Sex Differ. 2010, 1, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kararigas, G.; Bito, V.; Tinel, H.; Becher, E.; Baczko, I.; Knosalla, C.; Albrecht-Kupper, B.; Sipido, K.R.; Regitz-Zagrosek, V. Transcriptome characterization of estrogen-treated human myocardium identifies Myosin regulatory light chain interacting protein as a sex-specific element influencing contractile function. J. Am. Coll. Cardiol. 2012, 59, 410–417. [Google Scholar] [CrossRef] [Green Version]
- Hein, S.; Hassel, D.; Kararigas, G. The Zebrafish (Danio rerio) Is a Relevant Model for Studying Sex-Specific Effects of 17beta-Estradiol in the Adult Heart. Int. J. Mol. Sci. 2019, 20, 6287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fliegner, D.; Schubert, C.; Penkalla, A.; Witt, H.; Kararigas, G.; Dworatzek, E.; Staub, E.; Martus, P.; Ruiz Noppinger, P.; Kintscher, U.; et al. Female sex and estrogen receptor-beta attenuate cardiac remodeling and apoptosis in pressure overload. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010, 298, R1597–R1606. [Google Scholar] [CrossRef]
- Queiros, A.M.; Eschen, C.; Fliegner, D.; Kararigas, G.; Dworatzek, E.; Westphal, C.; Sanchez Ruderisch, H.; Regitz-Zagrosek, V. Sex- and estrogen-dependent regulation of a miRNA network in the healthy and hypertrophied heart. Int. J. Cardiol. 2013, 169, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Murray, H.E.; Pillai, A.V.; McArthur, S.R.; Razvi, N.; Datla, K.P.; Dexter, D.T.; Gillies, G.E. Dose- and sex-dependent effects of the neurotoxin 6-hydroxydopamine on the nigrostriatal dopaminergic pathway of adult rats: Differential actions of estrogen in males and females. Neuroscience 2003, 116, 213–222. [Google Scholar] [CrossRef]
- Klinge, C.M. Estrogenic control of mitochondrial function. Redox Biol. 2020, 31, 101435. [Google Scholar] [CrossRef]
- Luo, T.; Liu, H.; Kim, J.K. Estrogen Protects the Female Heart from Ischemia/Reperfusion Injury through Manganese Superoxide Dismutase Phosphorylation by Mitochondrial p38beta at Threonine 79 and Serine 106. PLoS ONE 2016, 11, e0167761. [Google Scholar] [CrossRef]
- Tsialtas, I.; Georgantopoulos, A.; Karipidou, M.E.; Kalousi, F.D.; Karra, A.G.; Leonidas, D.D.; Psarra, A.G. Anti-Apoptotic and Antioxidant Activities of the Mitochondrial Estrogen Receptor Beta in N2A Neuroblastoma Cells. Int. J. Mol. Sci. 2021, 22, 7620. [Google Scholar] [CrossRef]
- Zhai, P.; Eurell, T.E.; Cotthaus, R.; Jeffery, E.H.; Bahr, J.M.; Gross, D.R. Effect of estrogen on global myocardial ischemia-reperfusion injury in female rats. Am. J. Physiol. Heart Circ. Physiol. 2000, 279, H2766–H2775. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, Y.; Ikeda, Y.; Uchikado, Y.; Akasaki, Y.; Sadoshima, J.; Ohishi, M. Estrogen Plays a Crucial Role in Rab9-Dependent Mitochondrial Autophagy, Delaying Arterial Senescence. J. Am. Heart Assoc. 2021, 10, e019310. [Google Scholar] [CrossRef]
- Zhao, W.; Hou, Y.; Song, X.; Wang, L.; Zhang, F.; Zhang, H.; Yu, H.; Zhou, Y. Estrogen Deficiency Induces Mitochondrial Damage Prior to Emergence of Cognitive Deficits in a Postmenopausal Mouse Model. Front. Aging Neurosci. 2021, 13, 713819. [Google Scholar] [CrossRef] [PubMed]
- Octavia, Y.; Kararigas, G.; de Boer, M.; Chrifi, I.; Kietadisorn, R.; Swinnen, M.; Duimel, H.; Verheyen, F.K.; Brandt, M.M.; Fliegner, D.; et al. Folic acid reduces doxorubicin-induced cardiomyopathy by modulating endothelial nitric oxide synthase. J. Cell. Mol. Med. 2017, 21, 3277–3287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, R.Z.; Jiang, S.; Zhang, L.; Yu, Z.B. Mitochondrial electron transport chain, ROS generation and uncoupling (Review). Int. J. Mol. Med. 2019, 44, 3–15. [Google Scholar] [CrossRef] [Green Version]
- Hazell, G.G.; Yao, S.T.; Roper, J.A.; Prossnitz, E.R.; O’Carroll, A.M.; Lolait, S.J. Localisation of GPR30, a novel G protein-coupled oestrogen receptor, suggests multiple functions in rodent brain and peripheral tissues. J. Endocrinol. 2009, 202, 223–236. [Google Scholar] [CrossRef] [PubMed]
- Stirone, C.; Duckles, S.P.; Krause, D.N.; Procaccio, V. Estrogen increases mitochondrial efficiency and reduces oxidative stress in cerebral blood vessels. Mol. Pharmacol. 2005, 68, 959–965. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.H.; Liu, R.; Perez, E.J.; Wen, Y.; Stevens, S.M., Jr.; Valencia, T.; Brun-Zinkernagel, A.M.; Prokai, L.; Will, Y.; Dykens, J.; et al. Mitochondrial localization of estrogen receptor beta. Proc. Natl. Acad. Sci. USA 2004, 101, 4130–4135. [Google Scholar] [CrossRef] [Green Version]
- Pugach, E.K.; Blenck, C.L.; Dragavon, J.M.; Langer, S.J.; Leinwand, L.A. Estrogen receptor profiling and activity in cardiac myocytes. Mol. Cell. Endocrinol. 2016, 431, 62–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, F.; Ru, X.; Wen, T. NRF2, a Transcription Factor for Stress Response and Beyond. Int. J. Mol. Sci. 2020, 21, 4777. [Google Scholar] [CrossRef]
- Maher, A.C.; Akhtar, M.; Tarnopolsky, M.A. Men supplemented with 17beta-estradiol have increased beta-oxidation capacity in skeletal muscle. Physiol. Genom. 2010, 42, 342–347. [Google Scholar] [CrossRef]
- Wu, Z.; Puigserver, P.; Andersson, U.; Zhang, C.; Adelmant, G.; Mootha, V.; Troy, A.; Cinti, S.; Lowell, B.; Scarpulla, R.C.; et al. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell 1999, 98, 115–124. [Google Scholar] [CrossRef] [Green Version]
- Rius-Perez, S.; Torres-Cuevas, I.; Millan, I.; Ortega, A.L.; Perez, S. PGC-1alpha, Inflammation, and Oxidative Stress: An Integrative View in Metabolism. Oxid. Med. Cell Longev. 2020, 2020, 1452696. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.; Kroemer, G.; Kepp, O. Mitophagy: An Emerging Role in Aging and Age-Associated Diseases. Front. Cell Dev. Biol. 2020, 8, 200. [Google Scholar] [CrossRef] [Green Version]
- Marinkovic, M.; Sprung, M.; Novak, I. Dimerization of mitophagy receptor BNIP3L/NIX is essential for recruitment of autophagic machinery. Autophagy 2021, 17, 1232–1243. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Feng, D.; Chen, G.; Chen, M.; Zheng, Q.; Song, P.; Ma, Q.; Zhu, C.; Wang, R.; Qi, W.; et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat. Cell Biol. 2012, 14, 177–185. [Google Scholar] [CrossRef]
- Chen, G.; Han, Z.; Feng, D.; Chen, Y.; Chen, L.; Wu, H.; Huang, L.; Zhou, C.; Cai, X.; Fu, C.; et al. A regulatory signaling loop comprising the PGAM5 phosphatase and CK2 controls receptor-mediated mitophagy. Mol. Cell 2014, 54, 362–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiba-Fukushima, K.; Imai, Y.; Yoshida, S.; Ishihama, Y.; Kanao, T.; Sato, S.; Hattori, N. PINK1-mediated phosphorylation of the Parkin ubiquitin-like domain primes mitochondrial translocation of Parkin and regulates mitophagy. Sci. Rep. 2012, 2, 1002. [Google Scholar] [CrossRef] [PubMed]
- Burton, T.R.; Gibson, S.B. The role of Bcl-2 family member BNIP3 in cell death and disease: NIPping at the heels of cell death. Cell Death Differ. 2009, 16, 515–523. [Google Scholar] [CrossRef] [Green Version]
- Chen, B.C.; Weng, Y.J.; Shibu, M.A.; Han, C.K.; Chen, Y.S.; Shen, C.Y.; Lin, Y.M.; Viswanadha, V.P.; Liang, H.Y.; Huang, C.Y. Estrogen and/or Estrogen Receptor alpha Inhibits BNIP3-Induced Apoptosis and Autophagy in H9c2 Cardiomyoblast Cells. Int. J. Mol. Sci. 2018, 19, 1298. [Google Scholar] [CrossRef] [Green Version]
- Yao, M.; Nguyen, T.V.; Pike, C.J. Estrogen regulates Bcl-w and Bim expression: Role in protection against beta-amyloid peptide-induced neuronal death. J. Neurosci. 2007, 27, 1422–1433. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, D.J.; Kuo, W.W.; Lai, Y.P.; Shibu, M.A.; Shen, C.Y.; Pai, P.; Yeh, Y.L.; Lin, J.Y.; Viswanadha, V.P.; Huang, C.Y. 17beta-Estradiol and/or Estrogen Receptor beta Attenuate the Autophagic and Apoptotic Effects Induced by Prolonged Hypoxia Through HIF-1alpha-Mediated BNIP3 and IGFBP-3 Signaling Blockage. Cell Physiol. Biochem. 2015, 36, 274–284. [Google Scholar] [CrossRef]
- Feng, Y.; Madungwe, N.B.; da Cruz Junho, C.V.; Bopassa, J.C. Activation of G protein-coupled oestrogen receptor 1 at the onset of reperfusion protects the myocardium against ischemia/reperfusion injury by reducing mitochondrial dysfunction and mitophagy. Br. J. Pharmacol. 2017, 174, 4329–4344. [Google Scholar] [CrossRef] [PubMed]
- Gureev, A.P.; Shaforostova, E.A.; Popov, V.N. Regulation of Mitochondrial Biogenesis as a Way for Active Longevity: Interaction Between the Nrf2 and PGC-1alpha Signaling Pathways. Front. Genet. 2019, 10, 435. [Google Scholar] [CrossRef] [Green Version]
- Capllonch-Amer, G.; Sbert-Roig, M.; Galmes-Pascual, B.M.; Proenza, A.M.; Llado, I.; Gianotti, M.; Garcia-Palmer, F.J. Estradiol stimulates mitochondrial biogenesis and adiponectin expression in skeletal muscle. J. Endocrinol. 2014, 221, 391–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kemper, M.F.; Stirone, C.; Krause, D.N.; Duckles, S.P.; Procaccio, V. Genomic and non-genomic regulation of PGC1 isoforms by estrogen to increase cerebral vascular mitochondrial biogenesis and reactive oxygen species protection. Eur. J. Pharmacol. 2014, 723, 322–329. [Google Scholar] [CrossRef] [Green Version]
- Galmes-Pascual, B.M.; Nadal-Casellas, A.; Bauza-Thorbrugge, M.; Sbert-Roig, M.; Garcia-Palmer, F.J.; Proenza, A.M.; Gianotti, M.; Llado, I. 17beta-estradiol improves hepatic mitochondrial biogenesis and function through PGC1B. J. Endocrinol. 2017, 232, 297–308. [Google Scholar] [CrossRef] [Green Version]
- Sbert-Roig, M.; Bauza-Thorbrugge, M.; Galmes-Pascual, B.M.; Capllonch-Amer, G.; Garcia-Palmer, F.J.; Llado, I.; Proenza, A.M.; Gianotti, M. GPER mediates the effects of 17beta-estradiol in cardiac mitochondrial biogenesis and function. Mol. Cell. Endocrinol. 2016, 420, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Tait, S.W.; Green, D.R. Mitochondrial regulation of cell death. Cold Spring Harb. Perspect. Biol. 2013, 5, a008706. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Yu, S.W.; Koh, D.W.; Lew, J.; Coombs, C.; Bowers, W.; Federoff, H.J.; Poirier, G.G.; Dawson, T.M.; Dawson, V.L. Apoptosis-inducing factor substitutes for caspase executioners in NMDA-triggered excitotoxic neuronal death. J. Neurosci. 2004, 24, 10963–10973. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.; Ding, W.; Tariq, M.A.; Wang, Y.; Wan, Q.; Li, M.; Wang, J. Molecular mechanism and therapy application of necrosis during myocardial injury. J. Cell. Mol. Med. 2018, 22, 2547–2557. [Google Scholar] [CrossRef] [PubMed]
- Milerova, M.; Drahota, Z.; Chytilova, A.; Tauchmannova, K.; Houstek, J.; Ostadal, B. Sex difference in the sensitivity of cardiac mitochondrial permeability transition pore to calcium load. Mol. Cell Biochem. 2016, 412, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Mendelowitsch, A.; Ritz, M.F.; Ros, J.; Langemann, H.; Gratzl, O. 17beta-Estradiol reduces cortical lesion size in the glutamate excitotoxicity model by enhancing extracellular lactate: A new neuroprotective pathway. Brain Res. 2001, 901, 230–236. [Google Scholar] [CrossRef]
- Jover-Mengual, T.; Miyawaki, T.; Latuszek, A.; Alborch, E.; Zukin, R.S.; Etgen, A.M. Acute estradiol protects CA1 neurons from ischemia-induced apoptotic cell death via the PI3K/Akt pathway. Brain Res. 2010, 1321, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patten, R.D.; Pourati, I.; Aronovitz, M.J.; Baur, J.; Celestin, F.; Chen, X.; Michael, A.; Haq, S.; Nuedling, S.; Grohe, C.; et al. 17beta-estradiol reduces cardiomyocyte apoptosis in vivo and in vitro via activation of phospho-inositide-3 kinase/Akt signaling. Circ. Res. 2004, 95, 692–699. [Google Scholar] [CrossRef] [Green Version]
- Pelzer, T.; Neumann, M.; de Jager, T.; Jazbutyte, V.; Neyses, L. Estrogen effects in the myocardium: Inhibition of NF-kappaB DNA binding by estrogen receptor-alpha and -beta. Biochem. Biophys. Res. Commun. 2001, 286, 1153–1157. [Google Scholar] [CrossRef] [PubMed]
- Waters, E.M.; Simerly, R.B. Estrogen induces caspase-dependent cell death during hypothalamic development. J. Neurosci. 2009, 29, 9714–9718. [Google Scholar] [CrossRef] [Green Version]
- Batnasan, E.; Wang, R.; Wen, J.; Ke, Y.; Li, X.; Bohio, A.A.; Zeng, X.; Huo, H.; Han, L.; Boldogh, I.; et al. 17-beta estradiol inhibits oxidative stress-induced accumulation of AIF into nucleolus and PARP1-dependent cell death via estrogen receptor alpha. Toxicol. Lett. 2015, 232, 1–9. [Google Scholar] [CrossRef]
- Kalkhoran, S.B.; Hernandez-Resendiz, S.; Ong, S.G.; Ramachandra, C.J.A.; Hausenloy, D.J. Mitochondrial shaping proteins as novel treatment targets for cardiomyopathies. Cond. Med. 2020, 3, 216–226. [Google Scholar]
- Chen, H.; Vermulst, M.; Wang, Y.E.; Chomyn, A.; Prolla, T.A.; McCaffery, J.M.; Chan, D.C. Mitochondrial fusion is required for mtDNA stability in skeletal muscle and tolerance of mtDNA mutations. Cell 2010, 141, 280–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva Ramos, E.; Motori, E.; Bruser, C.; Kuhl, I.; Yeroslaviz, A.; Ruzzenente, B.; Kauppila, J.H.K.; Busch, J.D.; Hultenby, K.; Habermann, B.H.; et al. Mitochondrial fusion is required for regulation of mitochondrial DNA replication. PLoS Genet. 2019, 15, e1008085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tondera, D.; Grandemange, S.; Jourdain, A.; Karbowski, M.; Mattenberger, Y.; Herzig, S.; Da Cruz, S.; Clerc, P.; Raschke, I.; Merkwirth, C.; et al. SLP-2 is required for stress-induced mitochondrial hyperfusion. EMBO J. 2009, 28, 1589–1600. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Detmer, S.A.; Ewald, A.J.; Griffin, E.E.; Fraser, S.E.; Chan, D.C. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J. Cell Biol. 2003, 160, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Daste, F.; Sauvanet, C.; Bavdek, A.; Baye, J.; Pierre, F.; Le Borgne, R.; David, C.; Rojo, M.; Fuchs, P.; Tareste, D. The heptad repeat domain 1 of Mitofusin has membrane destabilization function in mitochondrial fusion. EMBO Rep. 2018, 19, e43637. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, X.; Bai, J.; Tian, X.; Zhao, X.; Liu, W.; Duan, X.; Shang, W.; Fan, H.Y.; Tong, C. Mitoguardin Regulates Mitochondrial Fusion through MitoPLD and Is Required for Neuronal Homeostasis. Mol. Cell 2016, 61, 111–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, S.Y.; Huang, P.; Jenkins, G.M.; Chan, D.C.; Schiller, J.; Frohman, M.A. A common lipid links Mfn-mediated mitochondrial fusion and SNARE-regulated exocytosis. Nat. Cell Biol. 2006, 8, 1255–1262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Head, B.; Griparic, L.; Amiri, M.; Gandre-Babbe, S.; van der Bliek, A.M. Inducible proteolytic inactivation of OPA1 mediated by the OMA1 protease in mammalian cells. J. Cell Biol. 2009, 187, 959–966. [Google Scholar] [CrossRef] [PubMed]
- Ge, Y.; Shi, X.; Boopathy, S.; McDonald, J.; Smith, A.W.; Chao, L.H. Two forms of Opa1 cooperate to complete fusion of the mitochondrial inner-membrane. eLife 2020, 9, e50973. [Google Scholar] [CrossRef] [PubMed]
- Cipolat, S.; Martins de Brito, O.; Dal Zilio, B.; Scorrano, L. OPA1 requires mitofusin 1 to promote mitochondrial fusion. Proc. Natl. Acad. Sci. USA 2004, 101, 15927–15932. [Google Scholar] [CrossRef] [Green Version]
- Ban, T.; Ishihara, T.; Kohno, H.; Saita, S.; Ichimura, A.; Maenaka, K.; Oka, T.; Mihara, K.; Ishihara, N. Molecular basis of selective mitochondrial fusion by heterotypic action between OPA1 and cardiolipin. Nat. Cell Biol. 2017, 19, 856–863. [Google Scholar] [CrossRef] [PubMed]
- Anand, R.; Wai, T.; Baker, M.J.; Kladt, N.; Schauss, A.C.; Rugarli, E.; Langer, T. The i-AAA protease YME1L and OMA1 cleave OPA1 to balance mitochondrial fusion and fission. J. Cell Biol. 2014, 204, 919–929. [Google Scholar] [CrossRef]
- Ehses, S.; Raschke, I.; Mancuso, G.; Bernacchia, A.; Geimer, S.; Tondera, D.; Martinou, J.C.; Westermann, B.; Rugarli, E.I.; Langer, T. Regulation of OPA1 processing and mitochondrial fusion by m-AAA protease isoenzymes and OMA1. J. Cell Biol. 2009, 187, 1023–1036. [Google Scholar] [CrossRef]
- Consolato, F.; Maltecca, F.; Tulli, S.; Sambri, I.; Casari, G. m-AAA and i-AAA complexes coordinate to regulate OMA1, the stress-activated supervisor of mitochondrial dynamics. J. Cell Sci. 2018, 131, jcs213546. [Google Scholar] [CrossRef] [Green Version]
- Nan, J.; Nan, C.; Ye, J.; Qian, L.; Geng, Y.; Xing, D.; Rahman, M.S.U.; Huang, M. EGCG protects cardiomyocytes against hypoxia-reperfusion injury through inhibition of OMA1 activation. J. Cell Sci. 2019, 132, jcs220871. [Google Scholar] [CrossRef] [Green Version]
- Merkwirth, C.; Dargazanli, S.; Tatsuta, T.; Geimer, S.; Lower, B.; Wunderlich, F.T.; von Kleist-Retzow, J.C.; Waisman, A.; Westermann, B.; Langer, T. Prohibitins control cell proliferation and apoptosis by regulating OPA1-dependent cristae morphogenesis in mitochondria. Genes Dev. 2008, 22, 476–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Sheu, S.-S.; Wang, W. Fission Promotes Respiration and ROS Production in Individual Mitochondria. Biophys. J. 2014, 106, 28a. [Google Scholar] [CrossRef] [Green Version]
- Smirnova, E.; Griparic, L.; Shurland, D.L.; van der Bliek, A.M. Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol. Biol. Cell 2001, 12, 2245–2256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smirnova, E.; Shurland, D.L.; Ryazantsev, S.N.; van der Bliek, A.M. A human dynamin-related protein controls the distribution of mitochondria. J. Cell Biol. 1998, 143, 351–358. [Google Scholar] [CrossRef] [Green Version]
- Cho, B.; Cho, H.M.; Jo, Y.; Kim, H.D.; Song, M.; Moon, C.; Kim, H.; Kim, K.; Sesaki, H.; Rhyu, I.J.; et al. Constriction of the mitochondrial inner compartment is a priming event for mitochondrial division. Nat. Commun. 2017, 8, 15754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Xu, S.; Roelofs, B.A.; Boyman, L.; Lederer, W.J.; Sesaki, H.; Karbowski, M. Transient assembly of F-actin on the outer mitochondrial membrane contributes to mitochondrial fission. J. Cell Biol. 2015, 208, 109–123. [Google Scholar] [CrossRef] [Green Version]
- Manor, U.; Bartholomew, S.; Golani, G.; Christenson, E.; Kozlov, M.; Higgs, H.; Spudich, J.; Lippincott-Schwartz, J. A mitochondria-anchored isoform of the actin-nucleating spire protein regulates mitochondrial division. eLife 2015, 4, e08828. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, R.; Ji, W.K.; Stan, R.V.; de Juan Sanz, J.; Ryan, T.A.; Higgs, H.N. INF2-mediated actin polymerization at the ER stimulates mitochondrial calcium uptake, inner membrane constriction, and division. J. Cell Biol. 2018, 217, 251–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korobova, F.; Ramabhadran, V.; Higgs, H.N. An actin-dependent step in mitochondrial fission mediated by the ER-associated formin INF2. Science 2013, 339, 464–467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, Y.C.; Ysselstein, D.; Krainc, D. Mitochondria-lysosome contacts regulate mitochondrial fission via RAB7 GTP hydrolysis. Nature 2018, 554, 382–386. [Google Scholar] [CrossRef] [PubMed]
- Loson, O.C.; Song, Z.; Chen, H.; Chan, D.C. Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol. Biol. Cell 2013, 24, 659–667. [Google Scholar] [CrossRef]
- Macdonald, P.; Stepanyants, N.; Singh, A.; Clinton, R.; Osellame, L.; Ryan, M.; Ramachandran, R. MiD51 and Mff Co-assemble in Cardiolipin-Enriched Membrane Microdomains to Cooperatively Regulate Drp1-Mediated Mitochondrial Fission. Biophys. J. 2018, 114, 603a. [Google Scholar] [CrossRef]
- Francy, C.A.; Clinton, R.W.; Frohlich, C.; Murphy, C.; Mears, J.A. Cryo-EM Studies of Drp1 Reveal Cardiolipin Interactions that Activate the Helical Oligomer. Sci. Rep. 2017, 7, 10744. [Google Scholar] [CrossRef]
- Nagashima, S.; Tabara, L.C.; Tilokani, L.; Paupe, V.; Anand, H.; Pogson, J.H.; Zunino, R.; McBride, H.M.; Prudent, J. Golgi-derived PI(4)P-containing vesicles drive late steps of mitochondrial division. Science 2020, 367, 1366–1371. [Google Scholar] [CrossRef]
- Lee, J.E.; Westrate, L.M.; Wu, H.; Page, C.; Voeltz, G.K. Multiple dynamin family members collaborate to drive mitochondrial division. Nature 2016, 540, 139–143. [Google Scholar] [CrossRef]
- Yu, R.; Jin, S.B.; Lendahl, U.; Nister, M.; Zhao, J. Human Fis1 regulates mitochondrial dynamics through inhibition of the fusion machinery. EMBO J. 2019, 38, e99748. [Google Scholar] [CrossRef]
- Murakawa, T.; Yamaguchi, O.; Hashimoto, A.; Hikoso, S.; Takeda, T.; Oka, T.; Yasui, H.; Ueda, H.; Akazawa, Y.; Nakayama, H.; et al. Bcl-2-like protein 13 is a mammalian Atg32 homologue that mediates mitophagy and mitochondrial fragmentation. Nat. Commun. 2015, 6, 7527. [Google Scholar] [CrossRef] [Green Version]
- Gustafsson, A.B.; Gottlieb, R.A. Heart mitochondria: Gates of life and death. Cardiovasc. Res. 2008, 77, 334–343. [Google Scholar] [CrossRef] [PubMed]
- Papanicolaou, K.N.; Ngoh, G.A.; Dabkowski, E.R.; O’Connell, K.A.; Ribeiro, R.F., Jr.; Stanley, W.C.; Walsh, K. Cardiomyocyte deletion of mitofusin-1 leads to mitochondrial fragmentation and improves tolerance to ROS-induced mitochondrial dysfunction and cell death. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H167–H179. [Google Scholar] [CrossRef] [Green Version]
- Kalkhoran, S.B.; Hall, A.R.; White, I.J.; Cooper, J.; Fan, Q.; Ong, S.B.; Hernandez-Resendiz, S.; Cabrera-Fuentes, H.; Chinda, K.; Chakraborty, B.; et al. Assessing the effects of mitofusin 2 deficiency in the adult heart using 3D electron tomography. Physiol. Rep. 2017, 5, e13437. [Google Scholar] [CrossRef] [PubMed]
- Papanicolaou, K.N.; Khairallah, R.J.; Ngoh, G.A.; Chikando, A.; Luptak, I.; O’Shea, K.M.; Riley, D.D.; Lugus, J.J.; Colucci, W.S.; Lederer, W.J.; et al. Mitofusin-2 maintains mitochondrial structure and contributes to stress-induced permeability transition in cardiac myocytes. Mol. Cell Biol. 2011, 31, 1309–1328. [Google Scholar] [CrossRef] [Green Version]
- Dorn, G.W., 2nd; Song, M.; Walsh, K. Functional implications of mitofusin 2-mediated mitochondrial-SR tethering. J. Mol. Cell. Cardiol. 2015, 78, 123–128. [Google Scholar] [CrossRef] [Green Version]
- Seidlmayer, L.K.; Mages, C.; Berbner, A.; Eder-Negrin, P.; Arias-Loza, P.A.; Kaspar, M.; Song, M.; Dorn, G.W., II; Kohlhaas, M.; Frantz, S.; et al. Mitofusin 2 Is Essential for IP3-Mediated SR/Mitochondria Metabolic Feedback in Ventricular Myocytes. Front. Physiol. 2019, 10, 733. [Google Scholar] [CrossRef] [Green Version]
- Hall, A.R.; Burke, N.; Dongworth, R.K.; Kalkhoran, S.B.; Dyson, A.; Vicencio, J.M.; Dorn, G.W., II; Yellon, D.M.; Hausenloy, D.J. Hearts deficient in both Mfn1 and Mfn2 are protected against acute myocardial infarction. Cell Death Dis. 2016, 7, e2238. [Google Scholar] [CrossRef] [PubMed]
- Burke, N.; Hall, A.R.; Hausenloy, D.J. OPA1 in Cardiovascular Health and Disease. Curr. Drug Targets 2015, 16, 912–920. [Google Scholar] [CrossRef] [Green Version]
- Piquereau, J.; Caffin, F.; Novotova, M.; Prola, A.; Garnier, A.; Mateo, P.; Fortin, D.; Huynh, L.H.; Nicolas, V.; Alavi, M.V.; et al. Down-regulation of OPA1 alters mouse mitochondrial morphology, PTP function, and cardiac adaptation to pressure overload. Cardiovasc. Res. 2012, 94, 408–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Page, S.; Niro, M.; Fauconnier, J.; Cellier, L.; Tamareille, S.; Gharib, A.; Chevrollier, A.; Loufrani, L.; Grenier, C.; Kamel, R.; et al. Increase in Cardiac Ischemia-Reperfusion Injuries in Opa1+/− Mouse Model. PLoS ONE 2016, 11, e0164066. [Google Scholar] [CrossRef] [Green Version]
- Bouche, L.; Kamel, R.; Tamareille, S.; Garcia, G.; Villedieu, C.; Pillot, B.; Gueguen, N.; Chehaitly, A.; Chao de la Barca, J.M.; Beaumont, J.; et al. DRP1 haploinsufficiency attenuates cardiac ischemia/reperfusion injuries. PLoS ONE 2021, 16, e0248554. [Google Scholar] [CrossRef]
- Ikeda, Y.; Shirakabe, A.; Maejima, Y.; Zhai, P.; Sciarretta, S.; Toli, J.; Nomura, M.; Mihara, K.; Egashira, K.; Ohishi, M.; et al. Endogenous Drp1 mediates mitochondrial autophagy and protects the heart against energy stress. Circ. Res. 2015, 116, 264–278. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, T.; Ban-Ishihara, R.; Maeda, M.; Matsunaga, Y.; Ichimura, A.; Kyogoku, S.; Aoki, H.; Katada, S.; Nakada, K.; Nomura, M.; et al. Dynamics of mitochondrial DNA nucleoids regulated by mitochondrial fission is essential for maintenance of homogeneously active mitochondria during neonatal heart development. Mol. Cell Biol. 2015, 35, 211–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Satohisa, S.; Zhang, H.H.; Feng, L.; Yang, Y.Y.; Huang, L.; Chen, D.B. Endogenous NO upon estradiol-17beta stimulation and NO donor differentially regulate mitochondrial S-nitrosylation in endothelial cells. Endocrinology 2014, 155, 3005–3016. [Google Scholar] [CrossRef] [Green Version]
- Junior, R.F.R.; Rodrigues, P.L.; Morra, E.A.; Ronconi, K.S.; Do Val Lima, P.R.; Porto, M.L.; Simoes, M.R.; Vassallo, D.V.; Figueiredo, S.G.; Stefanon, I. Estrogen regulates spatially distinct cardiac mitochondrial subpopulations. Mitochondrion 2017, 35, 87–96. [Google Scholar] [CrossRef]
- Martin, O.J.; Lai, L.; Soundarapandian, M.M.; Leone, T.C.; Zorzano, A.; Keller, M.P.; Attie, A.D.; Muoio, D.M.; Kelly, D.P. A role for peroxisome proliferator-activated receptor gamma coactivator-1 in the control of mitochondrial dynamics during postnatal cardiac growth. Circ. Res. 2014, 114, 626–636. [Google Scholar] [CrossRef] [Green Version]
- Dworatzek, E.; Mahmoodzadeh, S.; Schubert, C.; Westphal, C.; Leber, J.; Kusch, A.; Kararigas, G.; Fliegner, D.; Moulin, M.; Ventura-Clapier, R.; et al. Sex differences in exercise-induced physiological myocardial hypertrophy are modulated by oestrogen receptor beta. Cardiovasc. Res. 2014, 102, 418–428. [Google Scholar] [CrossRef] [Green Version]
- Du, M.; Shan, J.; Feng, A.; Schmull, S.; Gu, J.; Xue, S. Oestrogen Receptor beta Activation Protects Against Myocardial Infarction via Notch1 Signalling. Cardiovasc. Drugs Ther. 2020, 34, 165–178. [Google Scholar] [CrossRef] [PubMed]
- Ong, S.B.; Hall, A.R.; Dongworth, R.K.; Kalkhoran, S.; Pyakurel, A.; Scorrano, L.; Hausenloy, D.J. Akt protects the heart against ischaemia-reperfusion injury by modulating mitochondrial morphology. Thromb. Haemost. 2015, 113, 513–521. [Google Scholar] [CrossRef]
- Lagranha, C.J.; Deschamps, A.; Aponte, A.; Steenbergen, C.; Murphy, E. Sex differences in the phosphorylation of mitochondrial proteins result in reduced production of reactive oxygen species and cardioprotection in females. Circ. Res. 2010, 106, 1681–1691. [Google Scholar] [CrossRef] [Green Version]
- Paris, O.; Ferraro, L.; Grober, O.M.; Ravo, M.; De Filippo, M.R.; Giurato, G.; Nassa, G.; Tarallo, R.; Cantarella, C.; Rizzo, F.; et al. Direct regulation of microRNA biogenesis and expression by estrogen receptor beta in hormone-responsive breast cancer. Oncogene 2012, 31, 4196–4206. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Liu, C.Y.; Zhang, X.J.; Feng, C.; Zhou, L.Y.; Zhao, Y.; Li, P.F. miR-361-regulated prohibitin inhibits mitochondrial fission and apoptosis and protects heart from ischemia injury. Cell Death Differ. 2015, 22, 1058–1068. [Google Scholar] [CrossRef] [Green Version]
- Amput, P.; Palee, S.; Arunsak, B.; Pratchayasakul, W.; Thonusin, C.; Kerdphoo, S.; Jaiwongkam, T.; Chattipakorn, S.C.; Chattipakorn, N. PCSK9 inhibitor and atorvastatin reduce cardiac impairment in ovariectomized prediabetic rats via improved mitochondrial function and Ca(2+) regulation. J. Cell. Mol. Med. 2020, 24, 9189–9203. [Google Scholar] [CrossRef]
- Souvenir, R.A.; Renata, A.; Campbell, R.; Hinton, A.O.; Rondina, M.T.; Abel, E.D. SUN-572 Estrogen Synergistically Interacts with Optic Atrophy Protein 1 to Promote Thrombosis. J. Endocr. Soc. 2020, 4, SUN-572. [Google Scholar] [CrossRef]
- Devine, M.J.; Kittler, J.T. Mitochondria at the neuronal presynapse in health and disease. Nat. Rev. Neurosci. 2018, 19, 63–80. [Google Scholar] [CrossRef]
- Hollenbeck, P.J.; Saxton, W.M. The axonal transport of mitochondria. J. Cell Sci. 2005, 118, 5411–5419. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Okamoto, K.; Hayashi, Y.; Sheng, M. The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell 2004, 119, 873–887. [Google Scholar] [CrossRef] [Green Version]
- Bevan, R.J.; Williams, P.A.; Waters, C.T.; Thirgood, R.; Mui, A.; Seto, S.; Good, M.; Morgan, J.E.; Votruba, M.; Erchova, I. OPA1 deficiency accelerates hippocampal synaptic remodelling and age-related deficits in learning and memory. Brain Commun. 2020, 2, fcaa101. [Google Scholar] [CrossRef] [PubMed]
- Smith, T.G.; Seto, S.; Ganne, P.; Votruba, M. A randomized, placebo-controlled trial of the benzoquinone idebenone in a mouse model of OPA1-related dominant optic atrophy reveals a limited therapeutic effect on retinal ganglion cell dendropathy and visual function. Neuroscience 2016, 319, 92–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trevisan, T.; Pendin, D.; Montagna, A.; Bova, S.; Ghelli, A.M.; Daga, A. Manipulation of Mitochondria Dynamics Reveals Separate Roles for Form and Function in Mitochondria Distribution. Cell Rep. 2018, 23, 1742–1753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishihara, N.; Nomura, M.; Jofuku, A.; Kato, H.; Suzuki, S.O.; Masuda, K.; Otera, H.; Nakanishi, Y.; Nonaka, I.; Goto, Y.; et al. Mitochondrial fission factor Drp1 is essential for embryonic development and synapse formation in mice. Nat. Cell Biol. 2009, 11, 958–966. [Google Scholar] [CrossRef]
- Verstreken, P.; Ly, C.V.; Venken, K.J.; Koh, T.W.; Zhou, Y.; Bellen, H.J. Synaptic mitochondria are critical for mobilization of reserve pool vesicles at Drosophila neuromuscular junctions. Neuron 2005, 47, 365–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kageyama, Y.; Zhang, Z.; Roda, R.; Fukaya, M.; Wakabayashi, J.; Wakabayashi, N.; Kensler, T.W.; Reddy, P.H.; Iijima, M.; Sesaki, H. Mitochondrial division ensures the survival of postmitotic neurons by suppressing oxidative damage. J. Cell Biol. 2012, 197, 535–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; McCaffery, J.M.; Chan, D.C. Mitochondrial fusion protects against neurodegeneration in the cerebellum. Cell 2007, 130, 548–562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, S.; Nandy, P.; Austria, Q.; Siedlak, S.L.; Torres, S.; Fujioka, H.; Wang, W.; Zhu, X. Mfn2 Ablation in the Adult Mouse Hippocampus and Cortex Causes Neuronal Death. Cells 2020, 9, 116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Carmona, S.; Muhammad, A.; Bell, S.; Landeros, J.; Vazquez, M.; Ho, R.; Franco, A.; Lu, B.; Dorn, G.W., 2nd; et al. Restoring mitofusin balance prevents axonal degeneration in a Charcot-Marie-Tooth type 2A model. J. Clin. Investig. 2019, 129, 1756–1771. [Google Scholar] [CrossRef]
- Lee, S.; Sterky, F.H.; Mourier, A.; Terzioglu, M.; Cullheim, S.; Olson, L.; Larsson, N.G. Mitofusin 2 is necessary for striatal axonal projections of midbrain dopamine neurons. Hum. Mol. Genet. 2012, 21, 4827–4835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hara, Y.; Yuk, F.; Puri, R.; Janssen, W.G.; Rapp, P.R.; Morrison, J.H. Presynaptic mitochondrial morphology in monkey prefrontal cortex correlates with working memory and is improved with estrogen treatment. Proc. Natl. Acad. Sci. USA 2014, 111, 486–491. [Google Scholar] [CrossRef] [Green Version]
- Arnold, S.; de Araujo, G.W.; Beyer, C. Gender-specific regulation of mitochondrial fusion and fission gene transcription and viability of cortical astrocytes by steroid hormones. J. Mol. Endocrinol. 2008, 41, 289–300. [Google Scholar] [CrossRef]
- Sung, J.H.; Cho, E.H.; Min, W.; Kim, M.J.; Kim, M.O.; Jung, E.J.; Koh, P.O. Identification of proteins regulated by estradiol in focal cerebral ischemic injury--a proteomics approach. Neurosci. Lett. 2010, 477, 66–71. [Google Scholar] [CrossRef]
- Demarest, T.G.; Waite, E.L.; Kristian, T.; Puche, A.C.; Waddell, J.; McKenna, M.C.; Fiskum, G. Sex-dependent mitophagy and neuronal death following rat neonatal hypoxia-ischemia. Neuroscience 2016, 335, 103–113. [Google Scholar] [CrossRef] [Green Version]
- Namjoo, Z.; Moradi, F.; Aryanpour, R.; Piryaei, A.; Joghataei, M.T.; Abbasi, Y.; Hosseini, A.; Hassanzadeh, S.; Taklimie, F.R.; Beyer, C.; et al. Combined effects of rat Schwann cells and 17beta-estradiol in a spinal cord injury model. Metab. Brain Dis. 2018, 33, 1229–1242. [Google Scholar] [CrossRef] [PubMed]
- Kumari, S.; Mehta, S.L.; Milledge, G.Z.; Huang, X.; Li, H.; Li, P.A. Ubisol-Q10 Prevents Glutamate-Induced Cell Death by Blocking Mitochondrial Fragmentation and Permeability Transition Pore Opening. Int. J. Biol. Sci. 2016, 12, 688–700. [Google Scholar] [CrossRef] [Green Version]
- Koh, P.O. 17Beta-estradiol prevents the glutamate-induced decrease of Akt and its downstream targets in HT22 cells. J. Vet. Med. Sci. 2007, 69, 285–288. [Google Scholar] [CrossRef] [PubMed]
- Giordano, C.; Montopoli, M.; Perli, E.; Orlandi, M.; Fantin, M.; Ross-Cisneros, F.N.; Caparrotta, L.; Martinuzzi, A.; Ragazzi, E.; Ghelli, A.; et al. Oestrogens ameliorate mitochondrial dysfunction in Leber’s hereditary optic neuropathy. Brain 2011, 134, 220–234. [Google Scholar] [CrossRef]
- Sarkar, S.; Jun, S.; Simpkins, J.W. Estrogen amelioration of Abeta-induced defects in mitochondria is mediated by mitochondrial signaling pathway involving ERbeta, AKAP and Drp1. Brain Res. 2015, 1616, 101–111. [Google Scholar] [CrossRef] [Green Version]
- Prat, A.; Behrendt, M.; Marcinkiewicz, E.; Boridy, S.; Sairam, R.M.; Seidah, N.G.; Maysinger, D. A novel mouse model of Alzheimer’s disease with chronic estrogen deficiency leads to glial cell activation and hypertrophy. J. Aging Res. 2011, 2011, 251517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, J.; Irwin, R.; Chen, S.; Hamilton, R.; Cadenas, E.; Brinton, R.D. Ovarian hormone loss induces bioenergetic deficits and mitochondrial beta-amyloid. Neurobiol. Aging 2012, 33, 1507–1521. [Google Scholar] [CrossRef] [Green Version]
- Duarte, A.I.; Candeias, E.; Alves, I.N.; Mena, D.; Silva, D.F.; Machado, N.J.; Campos, E.J.; Santos, M.S.; Oliveira, C.R.; Moreira, P.I. Liraglutide Protects Against Brain Amyloid-beta1-42 Accumulation in Female Mice with Early Alzheimer’s Disease-Like Pathology by Partially Rescuing Oxidative/Nitrosative Stress and Inflammation. Int. J. Mol. Sci. 2020, 21, 1746. [Google Scholar] [CrossRef] [Green Version]
- Tahsili-Fahadan, P.; Geocadin, R.G. Heart-Brain Axis: Effects of Neurologic Injury on Cardiovascular Function. Circ. Res. 2017, 120, 559–572. [Google Scholar] [CrossRef]
- Riching, A.S.; Major, J.L.; Londono, P.; Bagchi, R.A. The Brain-Heart Axis: Alzheimer’s, Diabetes, and Hypertension. ACS Pharmacol. Transl. Sci. 2020, 3, 21–28. [Google Scholar] [CrossRef]
- Kerro, A.; Woods, T.; Chang, J.J. Neurogenic stunned myocardium in subarachnoid hemorrhage. J. Crit. Care 2017, 38, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Ashby, E.L.; Miners, J.S.; Kehoe, P.G.; Love, S. Effects of Hypertension and Anti-Hypertensive Treatment on Amyloid-beta (Abeta) Plaque Load and Abeta-Synthesizing and Abeta-Degrading Enzymes in Frontal Cortex. J. Alzheimers Dis. 2016, 50, 1191–1203. [Google Scholar] [CrossRef] [Green Version]
- Sundboll, J.; Horvath-Puho, E.; Adelborg, K.; Schmidt, M.; Pedersen, L.; Botker, H.E.; Henderson, V.W.; Sorensen, H.T. Higher Risk of Vascular Dementia in Myocardial Infarction Survivors. Circulation 2018, 137, 567–577. [Google Scholar] [CrossRef] [PubMed]
- Arboix, A. Cardiovascular risk factors for acute stroke: Risk profiles in the different subtypes of ischemic stroke. World J. Clin. Cases 2015, 3, 418–429. [Google Scholar] [CrossRef]
- Mrozek, S.; Gobin, J.; Constantin, J.M.; Fourcade, O.; Geeraerts, T. Crosstalk between brain, lung and heart in critical care. Anaesth. Crit. Care Pain Med. 2020, 39, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Xu, D.; Sabat, G.; Valdivia, H.; Xu, W.; Shi, N.Q. Disrupting KATP channels diminishes the estrogen-mediated protection in female mutant mice during ischemia-reperfusion. Clin. Proteom. 2014, 11, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minta, W.; Palee, S.; Mantor, D.; Sutham, W.; Jaiwongkam, T.; Kerdphoo, S.; Pratchayasakul, W.; Kumfu, S.; Chattipakorn, S.C.; Chattipakorn, N. Estrogen deprivation aggravates cardiometabolic dysfunction in obese-insulin resistant rats through the impairment of cardiac mitochondrial dynamics. Exp. Gerontol. 2018, 103, 107–114. [Google Scholar] [CrossRef]
- Carnevale, D.; Mascio, G.; D’Andrea, I.; Fardella, V.; Bell, R.D.; Branchi, I.; Pallante, F.; Zlokovic, B.; Yan, S.S.; Lembo, G. Hypertension induces brain beta-amyloid accumulation, cognitive impairment, and memory deterioration through activation of receptor for advanced glycation end products in brain vasculature. Hypertension 2012, 60, 188–197. [Google Scholar] [CrossRef] [Green Version]
- Ribaric, S. The Rationale for Insulin Therapy in Alzheimer’s Disease. Molecules 2016, 21, 689. [Google Scholar] [CrossRef] [Green Version]
- Previtali, M.; Repetto, A.; Panigada, S.; Camporotondo, R.; Tavazzi, L. Left ventricular apical ballooning syndrome: Prevalence, clinical characteristics and pathogenetic mechanisms in a European population. Int. J. Cardiol. 2009, 134, 91–96. [Google Scholar] [CrossRef] [PubMed]
- Templin, C.; Ghadri, J.R.; Diekmann, J.; Napp, L.C.; Bataiosu, D.R.; Jaguszewski, M.; Cammann, V.L.; Sarcon, A.; Geyer, V.; Neumann, C.A.; et al. Clinical Features and Outcomes of Takotsubo (Stress) Cardiomyopathy. N. Engl. J. Med. 2015, 373, 929–938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Battrawy, I.; Zhao, Z.; Lan, H.; Schunemann, J.D.; Sattler, K.; Buljubasic, F.; Patocskai, B.; Li, X.; Yucel, G.; Lang, S.; et al. Estradiol protection against toxic effects of catecholamine on electrical properties in human-induced pluripotent stem cell derived cardiomyocytes. Int. J. Cardiol. 2018, 254, 195–202. [Google Scholar] [CrossRef]
- Xu, S.; Wang, P.; Zhang, H.; Gong, G.; Gutierrez Cortes, N.; Zhu, W.; Yoon, Y.; Tian, R.; Wang, W. CaMKII induces permeability transition through Drp1 phosphorylation during chronic beta-AR stimulation. Nat. Commun. 2016, 7, 13189. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beikoghli Kalkhoran, S.; Kararigas, G. Oestrogenic Regulation of Mitochondrial Dynamics. Int. J. Mol. Sci. 2022, 23, 1118. https://doi.org/10.3390/ijms23031118
Beikoghli Kalkhoran S, Kararigas G. Oestrogenic Regulation of Mitochondrial Dynamics. International Journal of Molecular Sciences. 2022; 23(3):1118. https://doi.org/10.3390/ijms23031118
Chicago/Turabian StyleBeikoghli Kalkhoran, Siavash, and Georgios Kararigas. 2022. "Oestrogenic Regulation of Mitochondrial Dynamics" International Journal of Molecular Sciences 23, no. 3: 1118. https://doi.org/10.3390/ijms23031118
APA StyleBeikoghli Kalkhoran, S., & Kararigas, G. (2022). Oestrogenic Regulation of Mitochondrial Dynamics. International Journal of Molecular Sciences, 23(3), 1118. https://doi.org/10.3390/ijms23031118