
Probing the Dynamics of Streptococcus pyogenes Cas9 Endonuclease Bound to the sgRNA Complex Using Hydrogen-Deuterium Exchange Mass Spectrometry

 , , and
, , and
Abstract
:1. Introduction
2. Results
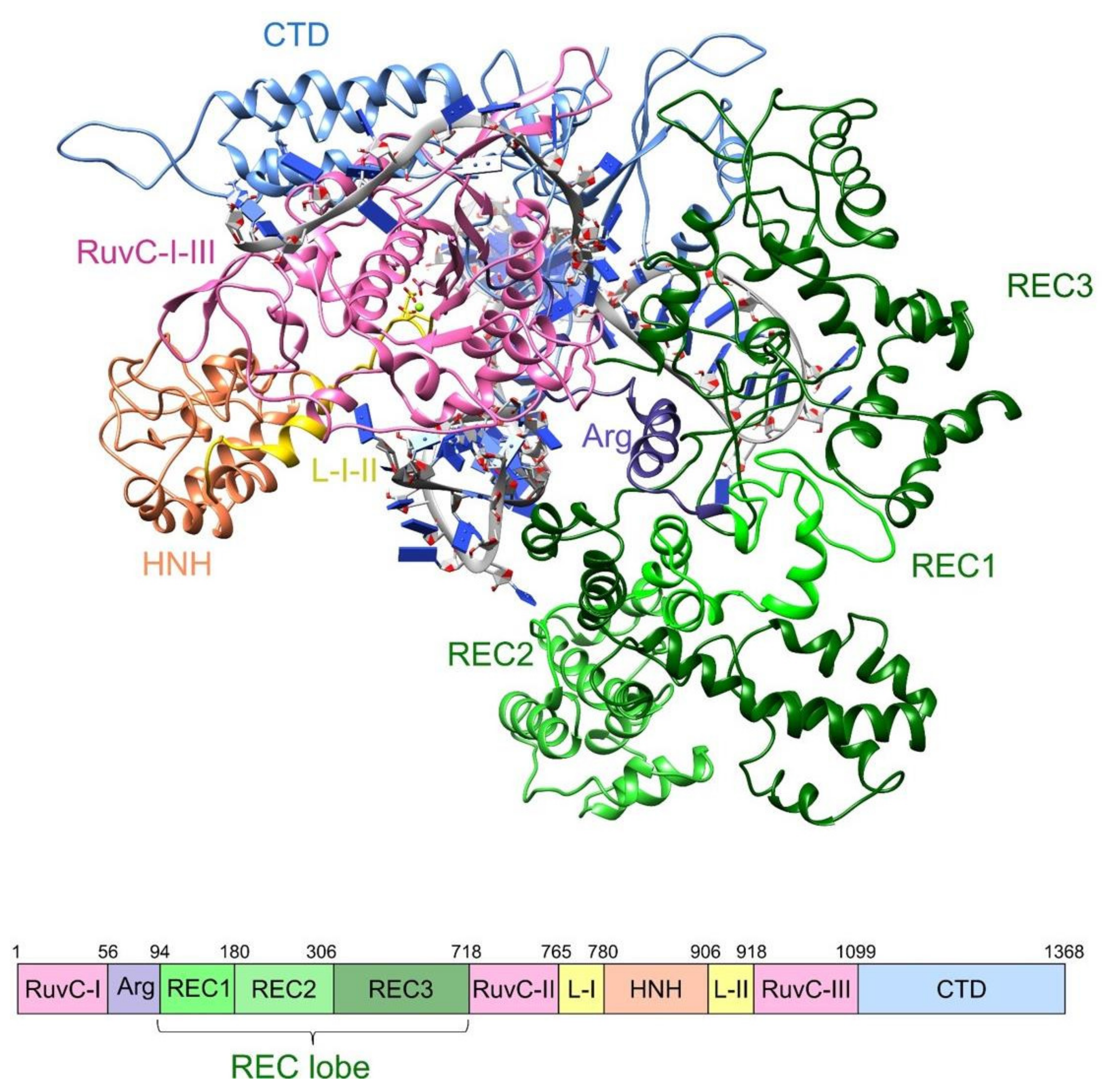
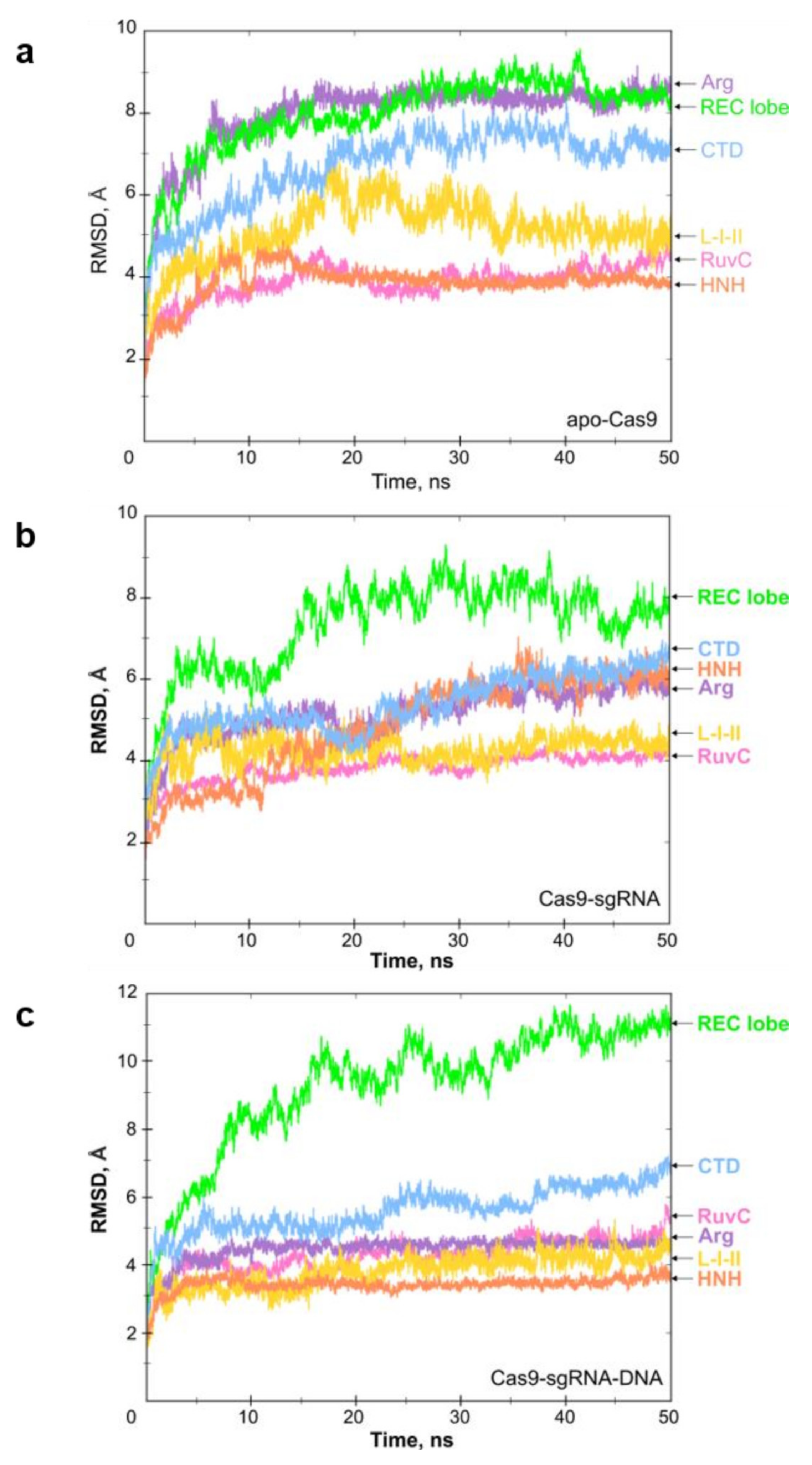
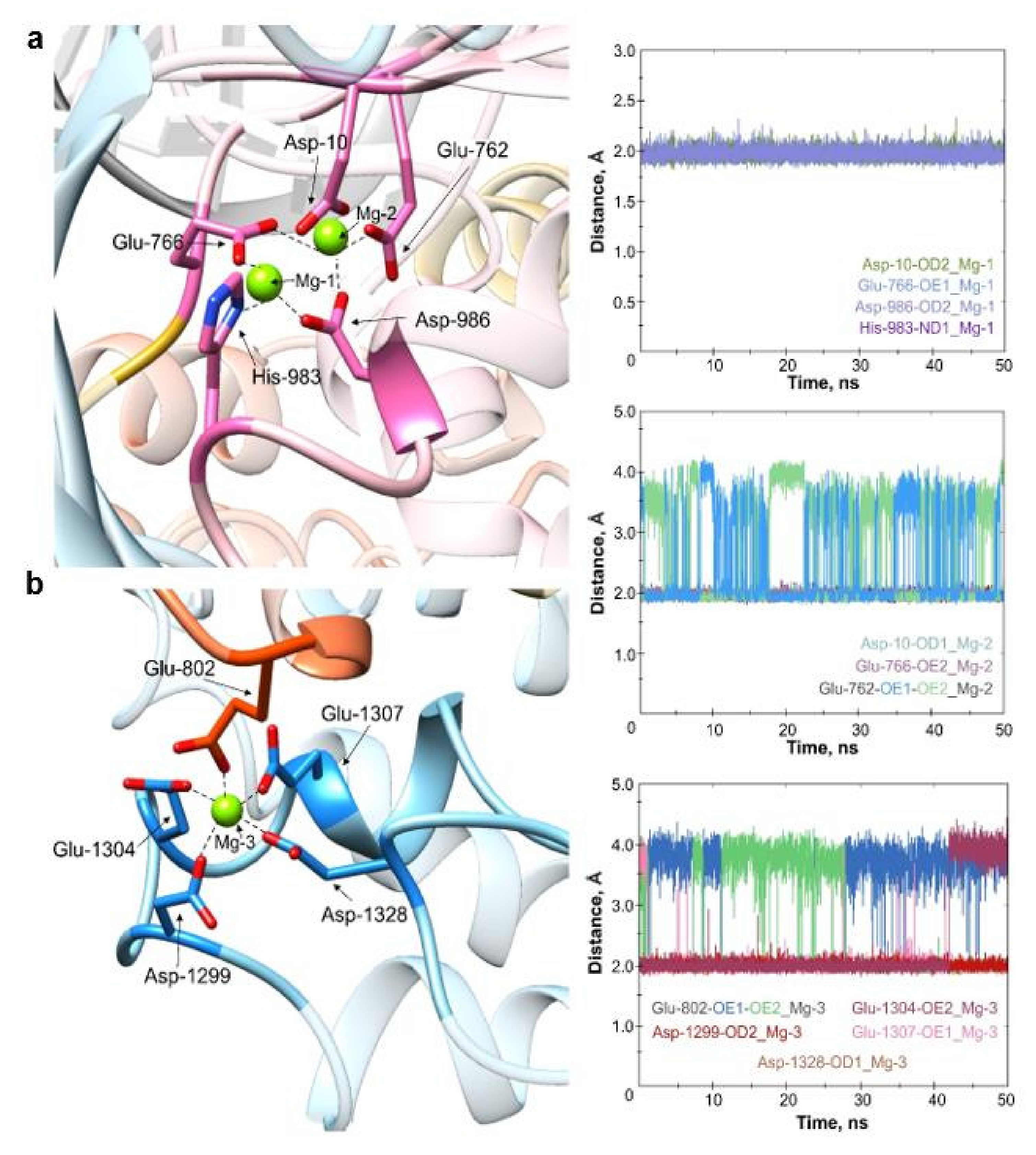
2.1. Molecular Dynamics
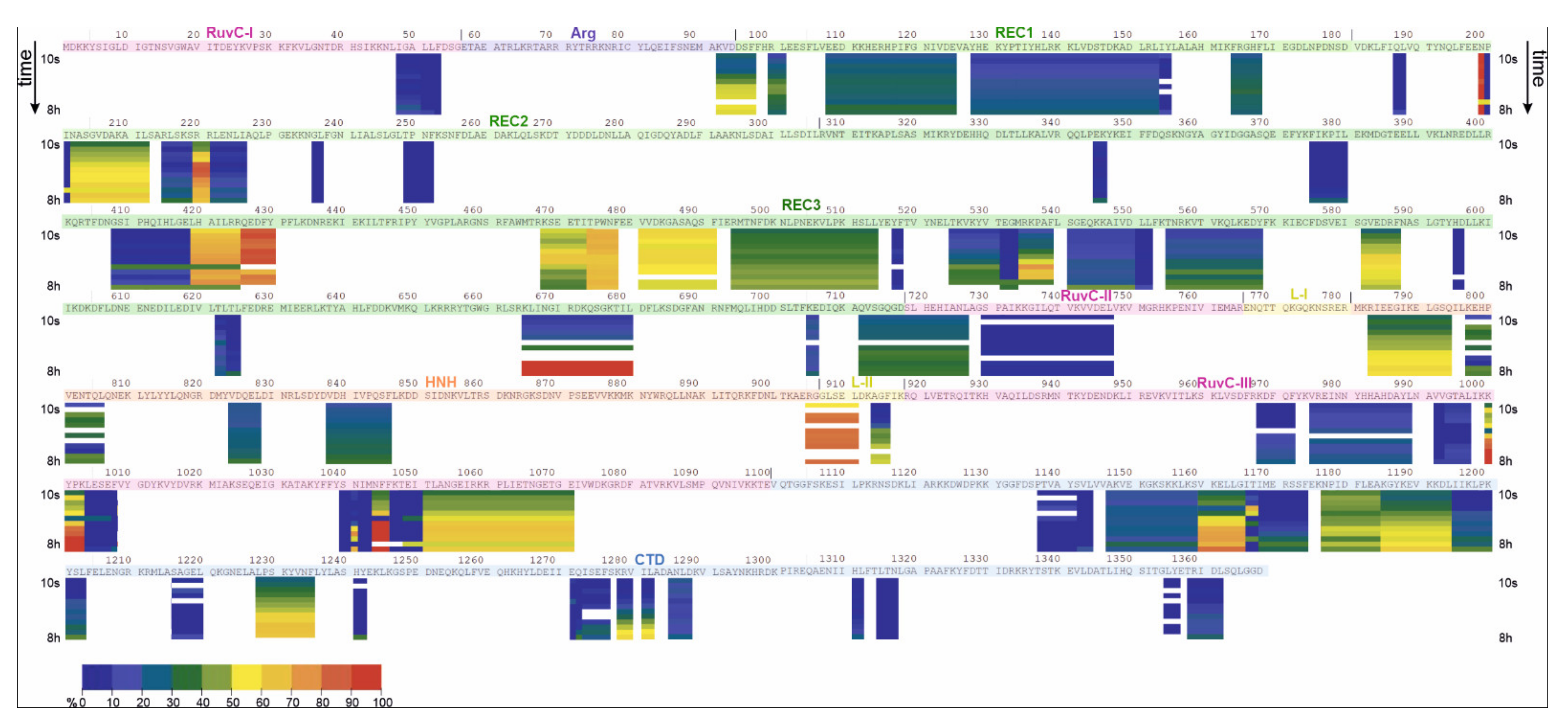
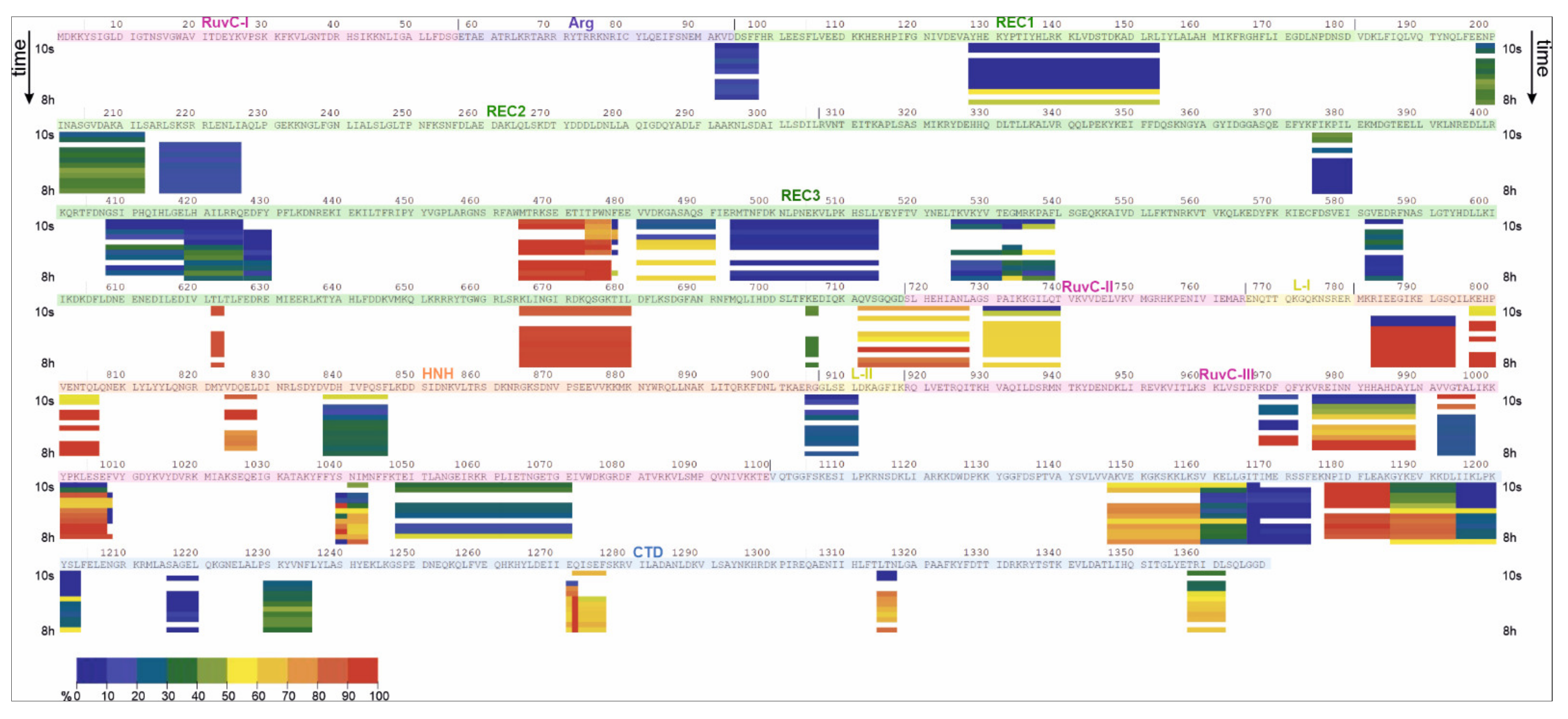
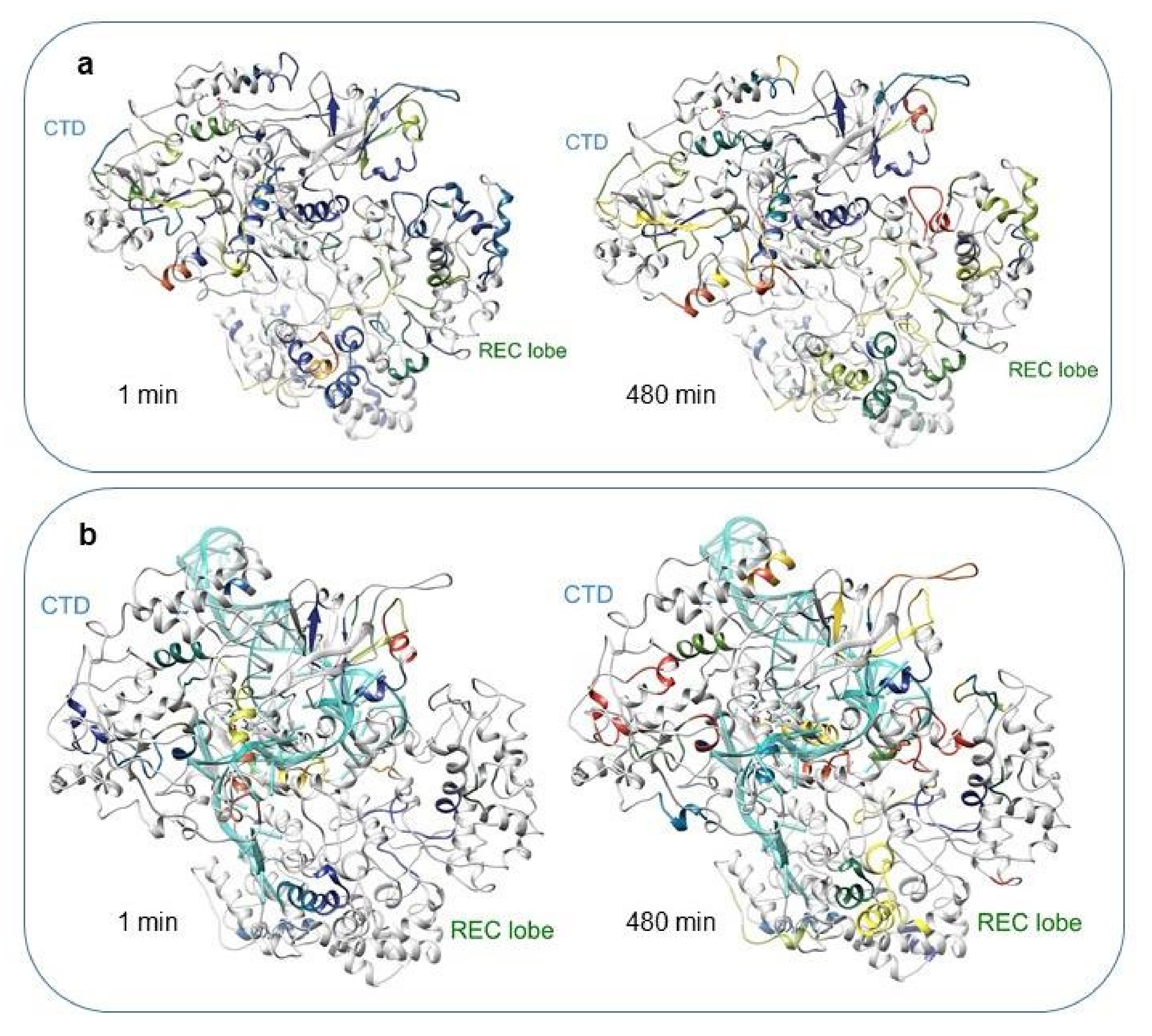
2.2. HDX-MS
3. Materials and Methods
3.1. Purification of the Cas9 Protein
3.2. Preparation of the Cas9–sgRNA Complex
3.3. Computer Simulation
3.4. HDX-MS
3.5. LC-MS Analysis
3.6. Data Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Terns, M.P.; Terns, R.M. CRISPR-based adaptive immune systems. Curr. Opin. Microbiol. 2011, 14, 321–327. [Google Scholar] [CrossRef] [Green Version]
- Bhaya, D.; Davison, M.; Barrangou, R. CRISPR-Cas systems in bacteria and archaea: Versatile small RNAs for adaptive defense and regulation. Annu. Rev. Genet. 2011, 45, 273–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saleh-Gohari, N.; Helleday, T. Conservative homologous recombination preferentially repairs DNA double-strand breaks in the S phase of the cell cycle in human cells. Nucleic Acids Res. 2004, 32, 3683–3688. [Google Scholar] [CrossRef]
- Doench, J.G.; Fusi, N.; Sullender, M.; Hegde, M.; Vaimberg, E.W.; Donovan, K.F.; Smith, I.; Tothova, Z.; Wilen, C.; Orchard, R.; et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol. 2016, 34, 184–191. [Google Scholar] [CrossRef] [Green Version]
- Bisaria, N.; Jarmoskaite, I.; Herschlag, D. Lessons from Enzyme Kinetics Reveal Specificity Principles for RNA-Guided Nucleases in RNA Interference and CRISPR-Based Genome Editing. Cell Syst. 2017, 4, 21–29. [Google Scholar] [CrossRef] [Green Version]
- Casini, A.; Olivieri, M.; Petris, G.; Montagna, C.; Reginato, G.; Maule, G.; Lorenzin, F.; Prandi, D.; Romanel, A.; Demichelis, F.; et al. A highly specific SpCas9 variant is identified by in vivo screening in yeast. Nat. Biotechnol. 2018, 36, 265–271. [Google Scholar] [CrossRef]
- Kleinstiver, B.P.; Pattanayak, V.; Prew, M.S.; Tsai, S.Q.; Nguyen, N.T.; Zheng, Z.; Joung, J.K. High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects. Nature 2016, 529, 490–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kocak, D.D.; Josephs, E.A.; Bhandarkar, V.; Adkar, S.S.; Kwon, J.B.; Gersbach, C.A. Increasing the specificity of CRISPR systems with engineered RNA secondary structures. Nat. Biotechnol. 2019, 37, 657–666. [Google Scholar] [CrossRef]
- Donohoue, P.D.; Pacesa, M.; Lau, E.; Vidal, B.; Irby, M.J.; Nyer, D.B.; Rotstein, T.; Banh, L.; Toh, M.S.; Gibson, J.; et al. Conformational control of Cas9 by CRISPR hybrid RNA-DNA guides mitigates off-target activity in T cells. Mol. Cell 2021, 81, 3637–3649.e5. [Google Scholar] [CrossRef] [PubMed]
- Heler, R.; Samai, P.; Modell, J.W.; Weiner, C.; Goldberg, G.W.; Bikard, D.; Marraffini, L.A. Cas9 specifies functional viral targets during CRISPR-Cas adaptation. Nature 2015, 519, 199–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jinek, M.; Jiang, F.; Taylor, D.W.; Sternberg, S.H.; Kaya, E.; Ma, E.; Anders, C.; Hauer, M.; Zhou, K.; Lin, S.; et al. Structures of Cas9 endonucleases reveal RNA-mediated conformational activation. Science 2014, 343, 1247997. [Google Scholar] [CrossRef] [Green Version]
- Biertumpfel, C.; Yang, W.; Suck, D. Crystal structure of T4 endonuclease VII resolving a Holliday junction. Nature 2007, 449, 616–620. [Google Scholar] [CrossRef]
- Gorecka, K.M.; Komorowska, W.; Nowotny, M. Crystal structure of RuvC resolvase in complex with Holliday junction substrate. Nucleic Acids Res. 2013, 41, 9945–9955. [Google Scholar] [CrossRef]
- Nishimasu, H.; Ran, F.A.; Hsu, P.D.; Konermann, S.; Shehata, S.I.; Dohmae, N.; Ishitani, R.; Zhang, F.; Nureki, O. Crystal structure of Cas9 in complex with guide RNA and target DNA. Cell 2014, 156, 935–949. [Google Scholar] [CrossRef] [Green Version]
- Huai, C.; Li, G.; Yao, R.; Zhang, Y.; Cao, M.; Kong, L.; Jia, C.; Yuan, H.; Chen, H.; Lu, D.; et al. Structural insights into DNA cleavage activation of CRISPR-Cas9 system. Nat. Commun. 2017, 8, 1375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, F.G.; Doudna, J.A. CRISPR-Cas9 Structures and Mechanisms. Annu. Rev. Biophys. 2017, 46, 505–529. [Google Scholar] [CrossRef] [Green Version]
- Palermo, G.; Ricci, C.G.; Fernando, A.; Basak, R.; Jinek, M.; Rivalta, I.; Batista, V.S.; McCammon, J.A. Protospacer Adjacent Motif-Induced Allostery Activates CRISPR-Cas9. J. Am. Chem. Soc. 2017, 139, 16028–16031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palermo, G.; Chen, J.S.; Ricci, C.G.; Rivalta, I.; Jinek, M.; Batista, V.S.; Doudna, J.A.; McCammon, J.A. Key role of the REC lobe during CRISPR-Cas9 activation by ‘sensing’, ‘regulating’, and ‘locking’ the catalytic HNH domain. Q. Rev. Biophys. 2018, 51, e9. [Google Scholar] [CrossRef] [Green Version]
- Nishimasu, H.; Cong, L.; Yan, W.X.; Ran, F.A.; Zetsche, B.; Li, Y.; Kurabayashi, A.; Ishitani, R.; Zhang, F.; Nureki, O. Crystal Structure of Staphylococcus aureus Cas9. Cell 2015, 162, 1113–1126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sternberg, S.H.; LaFrance, B.; Kaplan, M.; Doudna, J.A. Conformational control of DNA target cleavage by CRISPR-Cas9. Nature 2015, 527, 110–113. [Google Scholar] [CrossRef]
- Palermo, G.; Miao, Y.; Walker, R.C.; Jinek, M.; McCammon, J.A. Striking Plasticity of CRISPR-Cas9 and Key Role of Non-target DNA, as Revealed by Molecular Simulations. ACS Cent. Sci. 2016, 2, 756–763. [Google Scholar] [CrossRef]
- Singh, D.; Sternberg, S.H.; Fei, J.; Doudna, J.A.; Ha, T. Real-time observation of DNA recognition and rejection by the RNA-guided endonuclease Cas9. Nat. Commun. 2016, 7, 12778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osuka, S.; Isomura, K.; Kajimoto, S.; Komori, T.; Nishimasu, H.; Shima, T.; Nureki, O.; Uemura, S. Real-time observation of flexible domain movements in CRISPR-Cas9. EMBO J. 2018, 37, e96941. [Google Scholar] [CrossRef] [Green Version]
- Wales, T.E.; Engen, J.R. Hydrogen exchange mass spectrometry for the analysis of protein dynamics. Mass Spectrom. Rev. 2006, 25, 158–170. [Google Scholar] [CrossRef]
- Skinner, J.J.; Lim, W.K.; Bedard, S.; Black, B.E.; Englander, S.W. Protein dynamics viewed by hydrogen exchange. Protein Sci. 2012, 21, 996–1005. [Google Scholar] [CrossRef] [Green Version]
- Zhdanova, P.V.; Ishchenko, A.A.; Chernonosov, A.A.; Zharkov, D.O.; Koval, V.V. Dynamics and Conformational Changes in Human NEIL2 DNA Glycosylase Analyzed by Hydrogen/Deuterium Exchange Mass Spectrometry. J. Mol. Biol. 2021, 434, 167334. [Google Scholar] [CrossRef]
- van Erp, P.B.G.; Patterson, A.; Kant, R.; Berry, L.; Golden, S.M.; Forsman, B.L.; Carter, J.; Jackson, R.N.; Bothner, B.; Wiedenheft, B. Conformational Dynamics of DNA Binding and Cas3 Recruitment by the CRISPR RNA-Guided Cascade Complex. ACS Chem. Biol. 2018, 13, 481–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, F.; Zhou, K.; Ma, L.; Gressel, S.; Doudna, J.A. STRUCTURAL BIOLOGY. A Cas9-guide RNA complex preorganized for target DNA recognition. Science 2015, 348, 1477–1481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anders, C.; Jinek, M. In Vitro Enzymology of Cas9. Use of Crispr/Cas9, Zfns, and Talens in Generating Site-Specific Genome Alterations. Methods Enzymol. 2014, 546, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef]
- Wright, A.V.; Sternberg, S.H.; Taylor, D.W.; Staahl, B.T.; Bardales, J.A.; Kornfeld, J.E.; Doudna, J.A. Rational design of a split-Cas9 enzyme complex. Proc. Natl. Acad. Sci. USA 2015, 112, 2984–2989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madeira, F.; Park, Y.M.; Lee, J.; Buso, N.; Gur, T.; Madhusoodanan, N.; Basutkar, P.; Tivey, A.R.N.; Potter, S.C.; Finn, R.D.; et al. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 2019, 47, W636–W641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Šali, A.; Blundell, T.L. Comparative Protein Modelling by Satisfaction of Spatial Restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.B.K.; Ben-Shalom, I.Y.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.E.; Cruzeiro, I.V.W.D.; Darden, T.A.; Duke, R.E.; Giambasu, G.; Gilson, M.K.; et al. Amber 2020; University of California: San Francisco, CA, USA, 2020. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| apoCas9 (Free State) | Cas9-sgRNA (Bound State) | |||||||
|---|---|---|---|---|---|---|---|---|
| Domain | Low uptake (<30%) | Medium uptake (30–70%) | High uptake (70–90%) | Gradual uptake | Low uptake (<30%) | Medium uptake (30–70%) | High uptake (70–90%) | Gradual uptake |
| RuvC I | 48–53 | |||||||
| RuvC II | 730–747 | 719–727 | 730–740 | 719–727 | ||||
| RuvC III | 968–972 975–989 992–997 1004–1008 1039–1043 1047–1050 | 1051–1071 | 1000–1003, 1044–1046 | 1048–1071 | 992–997 | 975–989 | ||
| 968–972 | 999–1008 | |||||||
| 1038–1043 | ||||||||
| Arg | 92–97 | 92–97 | ||||||
| REC1 | 128–155 | 99–101 108–125 164–168 | 128–153 | |||||
| REC2 | 6 peptides between a.a. 187 and 418 | 202–212 | 220–221 | 216–226 | 198–212 | |||
| 220–221 | ||||||||
| REC3 | 346–347 375–380 408–418 4 peptides between a.a. 517 and 596 622–625 705–706 | 468–488 481–491 494–514 582–587 712–718 | 419–425 426–430 474–478 | 535–539 555–568, 582–587 665–680 | 375–380 | 532–539 | 465–477 | 481–491 |
| 408–418 | 705–706 | 622–623 | ||||||
| 419–430 | 665–680 | |||||||
| 494–514 | 712–718 | |||||||
| 525–531 | ||||||||
| 582–587 | ||||||||
| L-II | 905–911 | 914–916 | 905–911 | |||||
| HNH | 783–794 797–806 824–828 838–846 | 838–846 | 783–794 797–806 824–828 | |||||
| CTD | 1137–1144 1147–1159 1166–1174 1195–1203 1217–1220 1241–1242 1271–1276 1285–1288 1311–1312 1315–1317 1355–1356 1358–1362 | 1177–1184 | 1160–1165 1185–1194 1228–1235 | 1166–1174 1195–1203 1216–1220 | 1160–1165 1229–1235 | 1147–1159 | 1186–1194 | |
| 1177–1185 | 1315–1317 1358–1362 | |||||||
| 1271–1276 | ||||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhdanova, P.V.; Chernonosov, A.A.; Prokhorova, D.V.; Stepanov, G.A.; Kanazhevskaya, L.Y.; Koval, V.V. Probing the Dynamics of Streptococcus pyogenes Cas9 Endonuclease Bound to the sgRNA Complex Using Hydrogen-Deuterium Exchange Mass Spectrometry. Int. J. Mol. Sci. 2022, 23, 1129. https://doi.org/10.3390/ijms23031129
Zhdanova PV, Chernonosov AA, Prokhorova DV, Stepanov GA, Kanazhevskaya LY, Koval VV. Probing the Dynamics of Streptococcus pyogenes Cas9 Endonuclease Bound to the sgRNA Complex Using Hydrogen-Deuterium Exchange Mass Spectrometry. International Journal of Molecular Sciences. 2022; 23(3):1129. https://doi.org/10.3390/ijms23031129
Chicago/Turabian StyleZhdanova, Polina V., Alexander A. Chernonosov, Daria V. Prokhorova, Grigory A. Stepanov, Lyubov Yu. Kanazhevskaya, and Vladimir V. Koval. 2022. "Probing the Dynamics of Streptococcus pyogenes Cas9 Endonuclease Bound to the sgRNA Complex Using Hydrogen-Deuterium Exchange Mass Spectrometry" International Journal of Molecular Sciences 23, no. 3: 1129. https://doi.org/10.3390/ijms23031129
APA StyleZhdanova, P. V., Chernonosov, A. A., Prokhorova, D. V., Stepanov, G. A., Kanazhevskaya, L. Y., & Koval, V. V. (2022). Probing the Dynamics of Streptococcus pyogenes Cas9 Endonuclease Bound to the sgRNA Complex Using Hydrogen-Deuterium Exchange Mass Spectrometry. International Journal of Molecular Sciences, 23(3), 1129. https://doi.org/10.3390/ijms23031129