2.1. Age-Dependent Changes in the Density of Neuronal Progenitor Cells in the DG of OXYS and Wistar Rats; Effects of SkQ1
We showed that the ANP density is more than twofold lower in the DG of OXYS rats at 3 months of age (analysis of variance (ANOVA): F
1,12 = 7.4,
p < 0.02), indicating a lower intensity of neurogenesis at this age (
Figure 1A). At the same time, the density of quiescent neural progenitors (QNPs), which may develop into a neuronal or glial cell lineage, remained unchanged (
Figure 1B). Nonetheless, factorial ANOVA showed a significant main effect of the strain (genotype) on the QNP density in the DG: the parameter was higher in OXYS rats than in Wistar rats (F
1,28 = 7.9,
p < 0.009). The QNP density decreased with age in both rat strains (two-way ANOVA, main effect (age): F
1,28 = 82.9,
p < 0.0001) but was higher in 18-month-old OXYS rats than in 18-month-old Wistar rats (ANOVA: F
1,16 = 28.5,
p < 0.0001), thereby possibly reflecting the lower intensity of neurogenesis throughout the OXYS lifespan (
Figure 1B). The ANP density was also influenced by the genotype (two-way ANOVA: F
1,28 = 7.4,
p < 0.01) and was lower in OXYS rats than in Wistar rats; the parameter naturally decreased with age in both rat strains (two-way ANOVA, main effect (age): F
1,28 = 36.8,
p < 0.0001), indicating an age-related slowdown of hippocampal neurogenesis. The rates of density changes were different between the strains (
Figure 1A): from 3 to 18 months of age, the parameter decreased 10-fold in Wistar rats (ANOVA: F
1,13 = 21.9,
p < 0.0005) but only threefold in OXYS rats (ANOVA: F
1,15 = 17.0,
p < 0.001). This observation may support the hypothesis of the lower rate of neurogenesis in the DG of OXYS rats throughout their lifespan: the ANP density at 3 months of age was already lower in OXYS rats than in Wistar rats, and the age-related decrease was not as prominent in OXYS rats as in Wistar rats. Further research is needed to test this hypothesis and to clarify the possible connection between neurodegeneration and the rate of neurogenesis.
Previously, we have shown that, in OXYS rats, the development and progression of the AD-like pathology proceed concurrently with changes in the expression of hippocampal genes affecting all parameters of neurogenesis: cell proliferation, migration, incorporation into a synaptic network, and apoptosis [
32]. Undoubtedly, neurogenesis is a highly energy-consuming process that requires healthy mitochondria. Mitochondrial abnormalities, together with synaptic degeneration, are the earliest and most prominent features of vulnerable neurons in the brain of AD patients [
33,
34,
35]. For a decade, we have researched the role of mitochondria in the AD-affected brain using mitochondria-targeted antioxidant SkQ1 [
31,
32,
36]. We have found that prophylactic and therapeutic effects of this antioxidant in all cases are associated with an improvement of the mitochondrial apparatus. Here, we did not find significant main effects of treatment with SkQ1 on the density of QNPs (F
1,31 = 0.6,
p = 0.44) and ANPs (F
1,31 = 0.06,
p = 0.81) in the DG of rats from both strains (
Figure 1A,B). It should be noted that the results presented here and previously [
31,
32] were obtained in the same groups of animals for a more complete understanding of the anti-AD mechanisms of action of SkQ1 (all figures show only our new data). Thus, we can draw a conclusion that treatment with SkQ1 did not affect the density of neuronal progenitors but possibly facilitated the activation of the remaining undamaged neurons and synapses. This notion is supported by SkQ1-induced changes in the expression of OXYS hippocampal genes related to neuronal and synaptic processes [
31]. On the other hand, it is well known that proper neuronal and synaptic function is supported by glial cells, including astrocytes and microglia [
37,
38]; besides, these cells are reported to play a crucial part in AD pathogenesis [
39].
Figure 1C,D show representative immunohistochemical images of QNPs and ANPs in the DG of OXYS and Wistar rats.
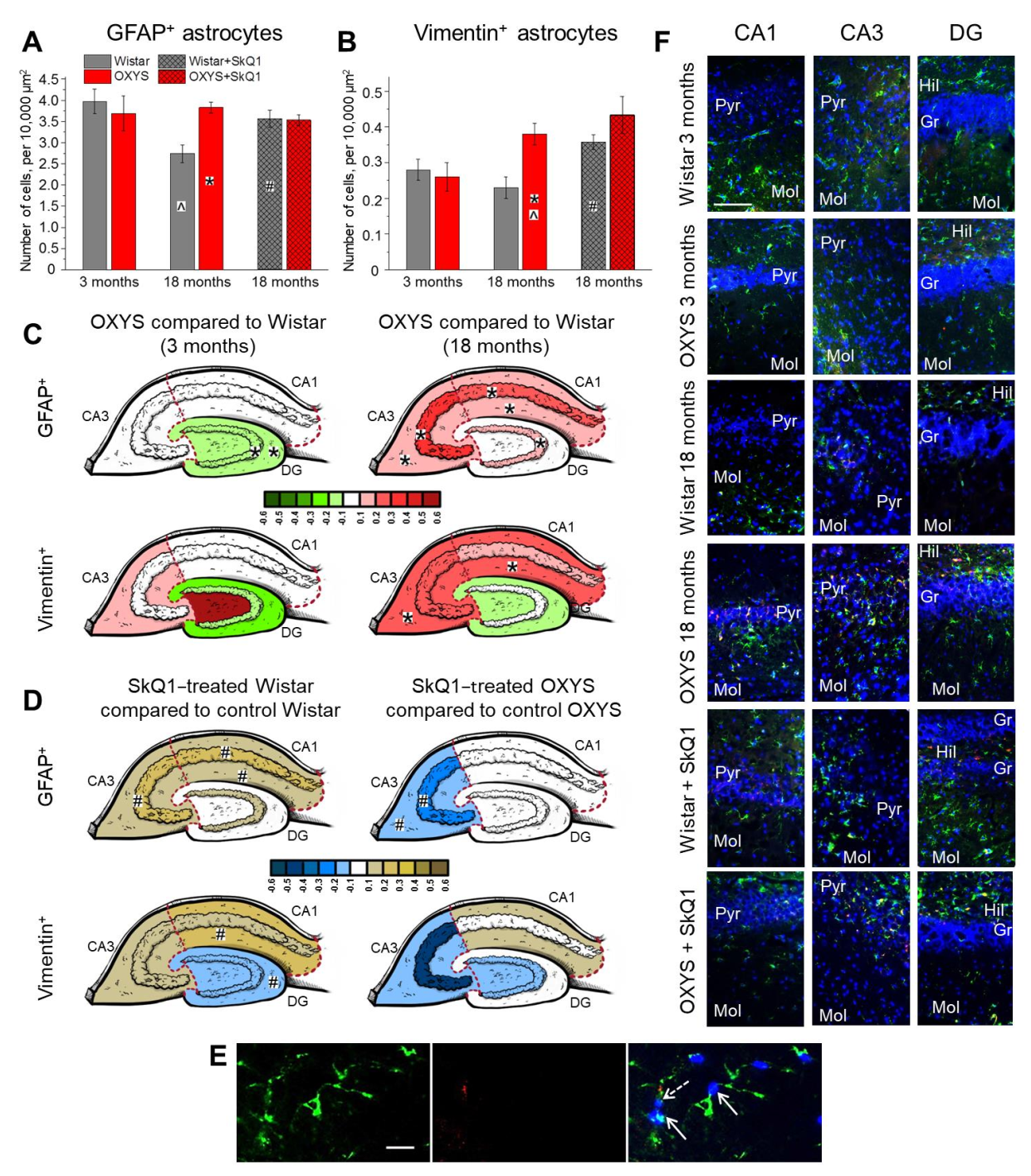
2.2. Age-Dependent Changes in the Density of Astrocytes in the Hippocampus of OXYS and Wistar Rats; Effects of SkQ1
Here, for the first time, we estimated the density of astrocytes in the whole hippocampus of Wistar and OXYS rats at different stages of the progression of AD signs (at 3 and 18 months of age); we also investigated the effects of SkQ1 treatment from 12 to 18 months of age on the astrocyte density.
Factorial ANOVA revealed that the density of GFAP
+ astrocytes was affected by age (main effect (age): F
1,46 = 4.9,
p < 0.03) and was not affected by the genotype (main effect (genotype): F
1,46 = 2.7,
p = 0.11), but there was an interaction between these factors (interaction effect: F
1,46 = 7.8,
p < 0.008). Indeed, we observed that, in Wistar rats, the astrocyte density decreased from 3 to 18 months of age (ANOVA: F
1,22 = 12.3,
p < 0.002), whereas only a slight increase in this parameter occurred in OXYS rats (
Figure 2A). Pairwise comparisons showed that the density of astrocytes was significantly higher in OXYS rats than in Wistar rats at 18 months of age (Tukey’s test:
p < 0.005).
An aging-related decrease in astrocyte density in CA1 and CA3 hippocampal areas in rodents has been documented elsewhere [
40,
41]. Our analysis of the astrocyte distribution in hippocampal regions of OXYS and Wistar rats revealed that the astrocyte density in the DG and CA1 area was influenced by age (two-way ANOVA, main effect (age): F
1,
46 = 4.4,
p < 0.04 for DG; F
1,46 = 12.6,
p < 0.0009 for CA1) and by an interaction between the genotype and age (two-way ANOVA, interaction effect: F
1,46 = 13.7,
p < 0.0006 for the DG; F
1,46 = 10.1,
p < 0.003 for CA1), just as in the whole hippocampus (
Figure 2C, upper panel;
Table S1). This was not the case for the CA3 region, where the density of astrocytes was higher in OXYS rats than in Wistar rats (two-way ANOVA, main effect (genotype): F
1,44 = 6.0,
p < 0.02) and was affected by age (two-way ANOVA, main effect (age): F
1,44 = 6.9,
p < 0.01). Regarding the DG, at 3 months of age, the density of astrocytes was lower in OXYS rats than in Wistar rats (
Figure 2C; Tukey’s test:
p < 0.05), mainly because of a decreased astrocyte density in granular and molecular layers (ANOVA: F
1,16 = 5.7,
p < 0.03 for the granular layer; F
1,16 = 7.2,
p < 0.02 for the molecular layer); however, at 18 months of age, the astrocyte density in the DG was higher in OXYS rats than Wistar rats (Tukey’s test:
p < 0.004). Our data in some ways are consistent with the findings of Olabarria and colleagues [
42]: those authors failed to detect changes in astroglial density in the hippocampus of 3xTG mice; however, they postulated a morphological atrophy of astrocytes already at an early stage of the familial-AD-like pathology—namely, in the DG—prior to Aβ plaque aggregation. We can hypothesize that the decline of astroglial density observed here contributes to the abnormally low intensity of neurogenesis seen at 3 months of age in the DG of OXYS rats.
In the CA1 region, the density of astrocytes decreased with age only in Wistar rats (Tukey’s test:
p < 0.0008) because of a decrease in the pyramidal (Tukey’s test:
p < 0.02) and molecular (Tukey’s test:
p < 0.0005) layers, resulting in a higher astrocyte density at 18 months of age in OXYS rats than in Wistar rats (
Figure 2C; Tukey’s test:
p < 0.002). As for the CA3 area (
Figure 2C), at 18 months of age, the density of astrocytes was higher in OXYS rats compared to Wistar rats, mostly because of a more than twofold higher astrocyte density in the pyramidal layer (Tukey’s test:
p < 0.0002) and an increase in this parameter in the molecular layer (Tukey’s test:
p < 0.003) of the CA3 region. Thus, in Wistar rats, the density of GFAP
+ astrocytes naturally decreased with age in CA1 and CA3 regions; this was not the case in OXYS rats, where the astrocyte density in the DG even increased by 18 months of age. The unchanged astrocyte density in the hippocampus of OXYS rats at the advanced age, as compared to young OXYS rats, may be a consequence of a slower death of astrocytes or their re-entry into the cell cycle. Taking into account the increased cell death in the hippocampus of OXYS rats between 12 and 18 months of age [
25,
27], we can theorize that the latter scenario—namely cell cycle re-entry—takes place in OXYS rats; this hypothesis needs to be tested in future studies.
Treatment with SkQ1 caused an increase in the GFAP
+ astrocyte density in Wistar rats (ANOVA: F
1,21 = 6.6,
p < 0.02) and did not affect it in OXYS rats (
Figure 2A). The regional analysis showed that, in CA1, the astrocyte density was influenced by the genotype (two-way ANOVA, main effect (genotype): F
1,45 = 10.4,
p < 0.003) and treatment (two-way ANOVA, main effect (treatment): F
1,45 = 4.1,
p < 0.05). Additionally, we observed an interaction between the genotype and treatment, and this interaction affected the astrocyte density in the CA3 region of the hippocampus (two-way ANOVA, interaction effect: F
1,45 = 8.6,
p < 0.005;
Figure 2D, upper panel;
Table S1). Indeed, the density of astrocytes in the CA3 region was significantly higher in OXYS rats than in Wistar rats (ANOVA: F
1,30 = 23.6,
p < 0.0001), and SkQ1 consumption decreased this parameter (ANOVA: F
1,24 = 7.4,
p < 0.02). At the same time, SkQ1 treatment led to an increase in the astrocyte density in the CA3 hippocampal area of Wistar rats (ANOVA: F
1,21 = 7.1,
p < 0.02). SkQ1 did not influence the astrocyte density in the DG of both rat strains. As a result of the crucial role of astrocytes in neurogenesis, this absence of an SkQ1 influence on their density may result in the absence of SkQ1 effects on neurogenesis, as discussed above.
Next, we counted vimentin-containing astrocytes. Vimentin is known to be expressed in astrocyte progenitors [
43] and reactive astrocytes [
44]. Although in a developing brain and in the DG, vimentin-positive astrocytes are thought to be astrocyte progenitors [
43], in other hippocampal areas of the aged brain, these cells should be regarded as reactive [
45]. Firstly, we found that, in the hippocampus, the density of vimentin-containing astrocytes is only much lower than the density of GFAP
+ astrocytes (the
t test for dependent samples:
p < 0.0001). Then, we revealed that the density of vimentin
+ astrocytes was not affected by age (two-way ANOVA: F
1,46 = 1.3,
p = 0.26) or the genotype (two-way ANOVA: F
1,46 = 3.2,
p = 0.08); however, there was an interaction between these factors (two-way ANOVA, interaction effect: F
1,46 = 5.7,
p < 0.02). Pairwise comparisons indicated that, at 18 months of age, the density of vimentin-containing astrocytes was more than 1.5-fold higher in OXYS rats relative to Wistar rats (Tukey’s test:
p < 0.008;
Figure 2B).
The regional analysis revealed that, in the CA1 area, the density of vimentin
+ astrocytes increased with age (two-way ANOVA, main effect (age): F
1,46 = 11.8,
p < 0.001), and there was an interaction between the age and genotype (two-way ANOVA, interaction effect: F
1,46 = 4.6,
p < 0.04). In the CA3 region, the parameter also increased with age (two-way ANOVA, main effect (age): F
1,44 = 13.4,
p < 0.0007) and was higher in OXYS rats than in Wistar rats (two-way ANOVA, main effect (genotype): F
1,44 = 5.9,
p < 0.02). Pairwise comparisons showed that the density of vimentin-containing astrocytes in both CA1 and CA3 areas increased more than twofold from 3 to 18 months only in OXYS rats (Tukey’s test:
p < 0.006 for both regions), becoming higher relative to Wistar rats (Tukey’s test:
p < 0.02 for the CA1 region;
p < 0.003 for the CA3 region). In both regions, the observed increase in this parameter was a consequence of a higher vimentin
+ astrocyte density in the molecular layer (
Figure 2C, lower panel;
Table S2).
Therefore, here, we revealed that, at the early stage of the spontaneously developing AD-like pathology, astrocyte deficiency rather than reactivity takes place in the hippocampus; by contrast, the progressive stage of this pathology is characterized by a higher density of GFAP
+ astrocytes and vimentin
+ astrocytes. Studies on the APPswePS1dE9 mouse model of AD have revealed that the cortical expression of GFAP and vimentin is increased during this pathology; however, other components of the intermediate-filament network, such as synemin and nestin, are not upregulated [
46].
Concerning the effects of SkQ1, the density of vimentin
+ astrocytes in the hippocampus increased only in Wistar rats after SkQ1 treatment (ANOVA: F
1,21 = 6.0,
p < 0.03;
Figure 2B). Again, the CA1 area was the most affected region (
Figure 2D, lower panel;
Table S2): the density of vimentin-containing astrocytes was higher in OXYS rats than in Wistar rats (two-way ANOVA, main effect (genotype): F
1,45 = 6.3,
p < 0.02) and increased after SkQ1 supplementation (two-way ANOVA, main effect (treatment): F
1,45 = 8.6,
p < 0.005). In the CA3 region, the SkQ1 treatment exerted opposite effects on the density of vimentin
+ astrocytes (
Figure 2D, lower panel;
Table S1): treatment with SkQ1 decreased this parameter in OXYS rats (ANOVA: F
1,24 = 5.1,
p < 0.04) but increased it in Wistar rats (ANOVA: F
1,21 = 10.1,
p < 0.005). Representative immunohistochemical images of GFAP
+ and vimentin
+ astrocytes in the hippocampus of OXYS and Wistar rats are provided in
Figure 2E,F.
Taken together, the results suggested that, at the progressive stage of the AD-like pathology, OXYS rats manifest the signs of astrogliosis; the most affected region of the hippocampus is CA3. Treatment with SkQ1 restored the astrocyte density in the CA3 region of OXYS rats (almost to the level seen in the normal control strain, Wistar rats), thus probably alleviating astrogliosis. On the other hand, the astrocyte density increased after SkQ1 treatment in the hippocampus of Wistar rats.
2.3. Genes Differentially Expressed between OXYS and Wistar Rats in Astrocytes
We analyzed the hippocampal transcriptome using lists of cell-type specific genes created on the basis of data from single-cell high-throughput RNA sequencing (single-cell RNA-Seq) [
47].
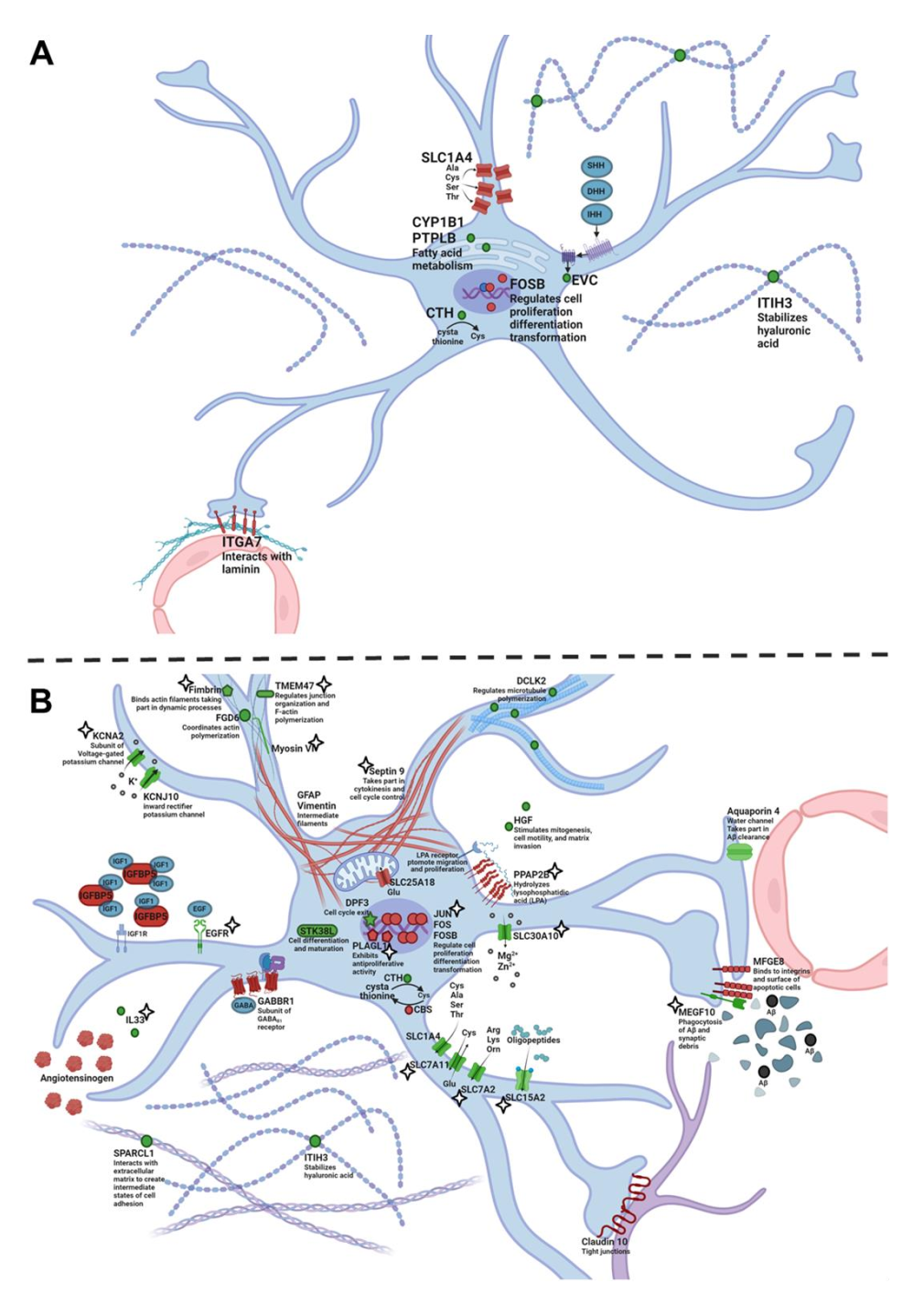
According to the RNA-Seq data, there were 10 differentially expressed genes (DEGs) specific for astrocytes in the hippocampus of OXYS rats compared to Wistar rats at 5 months of age (
Table S3). Among them, four genes had a higher expression (
Fosb,
Itga7,
Slc1a4, and
Tom1l1), and six genes had a lower expression in OXYS rats (
Cth,
Cyp1b1,
Evc,
Gem,
Itih3, and
Ptplb). It is important to point out that four genes (
Evc,
Gem,
Itga7, and
Slc1a4) are known to be associated with the cell membrane; the proteins encoded by these genes take part in actin cytoskeleton organization and interactions with the extracellular matrix. Of note,
Itih3 codes for a component of the extracellular matrix. Proteins encoded by genes
Cth,
Cyp1b1, and
Ptplb are enzymes that participate in the metabolism of amino acids and fatty acids. We can conclude that interactions with the extracellular matrix and some metabolic functions may become abnormal in the astrocytes of OXYS rats during the manifestation of AD signs (
Figure 3A).
During the progression of the AD-like pathology from 5 to 18 months of age, we found 218 DEGs specific for astrocytes in OXYS rats and 167 DEGs in Wistar rats at 18 months of age as compared to an age of 5 months (
Table S3); among them, 136 DEGs were common between the two strains and had the same pattern of altered expression. Forty-eight DEGs found only in OXYS rats decreased their expression, whereas 34 DEGs increased their expression from 5 to 18 months of age. We revealed that, among the 82 time-dependent DEGs seen only in OXYS rats, 36 genes encode membrane proteins, and products of 12 genes are associated with the extracellular space. We noticed that the expression of genes
Rb1 and
Dpf3 decreased, whereas the expression of
Sept9 increased with age. Due to the fact that the products of
Rb1 and
Dpf3 are negative regulators of the cell cycle—whereas the protein encoded by
Sept9 is involved in cytokinesis and cell cycle control—we can theorize that the cell cycle control in astrocytes weakened during the progression of AD signs in OXYS rats (
Figure 3B). It is widely accepted that, during AD, reactive astrocytes change their intermediate-filament network but do not proliferate [
48]. On the other hand, the weakness of the cell cycle control may be caused by Aβ, as demonstrated in neurons [
49], and drives aberrant cell cycle re-entry and DNA duplication without cytokinesis rather than proper cell division. Furthermore, the observed increase in the vimentin
+ cell density, together with an elevated
Gfap expression and
Kcnj10 underexpression, may point to the development of reactive astrogliosis from 5 to 18 months of age in OXYS rats; this is because such expression alterations are among potential markers of reactive astrocytes [
50]. Additionally, we documented the downregulation of
Megf10 and
Aqp4 in astrocytes by 18 months of age. These genes code for proteins crucial for Aβ phagocytosis and Aβ removal throughout the BBB in the blood stream and lymphatic vessels. Due to the fact that Aβ toxicity is considered to be the major damaging factor during AD, the impairment of Aβ clearance, along with its overproduction, may be a cause of AD [
51]. The key role in Aβ clearance is played by astrocytes, through which, Aβ is transferred to the blood stream and lymphatic drainage vessels [
52]. Thus, the downregulation of
Aqp4 and
Megf10 in hippocampal astrocytes by 18 months of age may result in an altered Aβ clearance and may contribute to the Aβ pathology observed in OXYS rats [
25].
At 18 months of age, there were 45 DEGs specific for astrocytes in OXYS rats relative to Wistar rats (
Table S3); among them, 18 genes had a higher expression, and 27 genes had a lower expression in OXYS rats. Among the DEGs, five genes (
Agt,
Cbs,
Gfap,
Sept9, and
Slc25a34) turned out to be upregulated because of their age-related increase only in OXYS rats, and the
Chsy1 gene had a higher expression because of the absence of its age-related decrease (this decrease is characteristic for Wistar rats). Besides, the expression of seven genes (
Alpk1,
Igsf11,
Megf10,
Pdgfrb,
Rgs20,
Slc30a10, and
Tmc7) was lower as a consequence of an age-related decrease only in OXYS rats; the expression of
Dclk2 was lower in OXYS rats because of the absence of its age-related upregulation seen in Wistar rats. Moreover, there were seven astrocyte-specific DEGs common between 5 and 18 months of age in OXYS rats: five of them were found to be downregulated (
Cth,
Cyp1b1,
Evc,
Gem, and
Itih3), and two genes were upregulated (
Fosb and
Tom1l1). Similar to the age of 5 months, at 18 months of age, nearly half of the DEGs were found to encode membrane proteins (20 of 45 genes), and 10 genes proved to encode proteins localized in the extracellular space. Furthermore, the expression of genes coding for cytoskeletal proteins in astroglia (
Gfap,
Myo6,
Pls1, and
Sept9) was found to change in OXYS rats relative to Wistar rats. Taken together, these results may point to an altered structure of astrocytes and their interaction with extracellular molecules (
Figure 3B). A higher expression of genes
Fox,
Foxb, and
Sept9 and a lower expression of
Megf10 and
Zfp217 in OXYS rats relative to Wistar rats again indicate the dysregulation of cell cycle re-entry. Astrocytes are known to induce the formation of excitatory synapses by secreting some protein factors, one of which being SPARCL1 [
53]. Additionally, the MEGF10 protein serves as a phagocytic receptor mediating synaptic phagocytosis [
54]. OXYS rats featured a lower expression of
Sparcl1 and
Megf10 in the hippocampus at 18 months of age (as compared to Wistar rats). This aberration may contribute to altered synaptic plasticity—which means a decreased synaptic formation and pruning—at the progressive stage of the AD-like pathology, as demonstrated earlier [
55]. Additionally, the
Rgs20 gene (encoding a regulator of G
q and G
i protein signaling) is downregulated in OXYS rats relative to Wistar rats. The activation of the G
q protein induces an increase in intracellular Ca
2+ concentration [
56]; thus, the underexpression of its regulator may cause a decrease in intracellular Ca
2+ concentration. The observed change in gene expression may be regarded as a compensatory reaction designed to reduce the higher Ca
2+ concentration in the astrocytes. In this regard, it is known that the intracellular Ca
2+ level is elevated in reactive astrocytes located near amyloid plaques [
57]. This assumption needs to be verified.
Insofar as astrocyte senescence is induced by Aβ and oxidative stress [
58], the influence of SkQ1 supplementation on the astrocyte gene expression profile is to be expected. Indeed, previously, we have shown that major effects of SkQ1 on astrocytes are related to the regulation of processes such as biosynthesis, cell proliferation, transcription, apoptosis, tube development, and blood vessel development [
31]. Here, we revealed that SkQ1 treatment prevented the age-associated (from 5 to 18 months of age) upregulation of four genes specific for astrocytes (
Hhipl1,
Jun,
Paqr7, and
Sept9) and downregulation of seven genes (
Tmem47,
Alpk1,
Megf10,
Pdgfrb,
Rgs20,
Slc30a10, and
Tmc7) in OXYS rats. This means that we failed to detect differences in the expression of these genes between SkQ1-treated OXYS rats at 18 months of age and OXYS rats at 5 months of age (
Table S3). Moreover, supplementation with SkQ1 normalized the expression of five upregulated genes (
Chsy1,
Hes1,
Plagl1,
Ppap2b, and
Tpbg) and 12 downregulated genes (
Egfr,
Elovl5,
Evc,
Gem,
Il33,
Kcna2,
Myo6,
Pls1,
Scara3,
Slc15a2,
Slc7a11, and
Slc7a2) in OXYS rats at 18 months of age. This means that we could not find any differences in the expression of these genes at 18 months of age between SkQ1-treated OXYS rats and untreated Wistar rats (healthy control rats). The majority (21 of 28) of genes whose expression was restored by SkQ1—which means the prevention of both age-related differences (from 5 to 18 months of age in OXYS rats) and interstrain differences (OXYS vs. Wistar rats at 18 months of age)—code for proteins from the extracellular space or plasma membrane. These data indicate that SkQ1 improved the astrocyte structure and interaction with extracellular stimuli (
Figure 3B). In addition, SkQ1 restored the expression of almost all genes associated with the cytoskeleton (
Figure 3B), with the exception of
Gfap, in line with the absence of SkQ1’s influence on the GFAP
+ cell density in OXYS rats. It is important to point out that SkQ1 treatment prevented abnormal
Megf10 underexpression, thereby possibly improving Aβ phagocytosis and the pruning of dysfunctional synapses [
32].
2.4. Age-Dependent Changes in the Density of Microglia in the Hippocampus of OXYS and Wistar Rats; Effects of SkQ1
We measured the density of resting (Iba1
+) and activated (Iba1
+CD68
+) microglia, as well as the total microglial density, in several hippocampal regions of OXYS and Wistar rats at 3 and 18 months of age. We found that the total microglial density in the whole hippocampus was affected only by the genotype (two-way ANOVA, main effect [genotype]: F
1,59 = 6.3,
p < 0.02) and was higher in OXYS rats than in Wistar rats. In the meantime, the density of resting microglia in the whole hippocampus was influenced by neither the genotype (two-way ANOVA: F
1,63 = 1.6,
p = 0.21) nor age (two-way ANOVA: F
1,63 = 1.7,
p = 0.19). By contrast, the density of activated microglia was affected by the genotype (two-way ANOVA, main effect (genotype): F
1,61 = 4.3,
p < 0.05) and was higher in OXYS rats than in Wistar rats (
Figure 4A). Thus, we may conclude that the increased total microglial density in OXYS rats was due to a higher density of activated microglia.
As for the regional analysis (
Figure 4B;
Tables S4 and S5), we noticed a significant main effect of the hippocampal region (multifactorial ANOVA: F
1,59 = 8.0,
p < 0.007): in the CA3 area, the microglial density was the highest among all tested regions in both rat strains. In the DG, the total microglial density significantly increased from 3 to 18 months of age (two-way ANOVA, main effect (age): F
1,27 = 15.9,
p < 0.0005), largely because of a higher resting-microglia density (two-way ANOVA, main effect (age): F
1,27 = 57.1,
p < 0.0001). Neither the genotype nor age affected the total microglial density in the CA3 region (two-way ANOVA: F
1,27 = 0.4,
p = 0.55 for effects of the genotype; F
1,27 = 1.3,
p = 0.26 for effects of age). Nonetheless, there was an interaction between these factors in the CA1 area (two-way ANOVA, interaction effect: F
1,28 = 4.7,
p < 0.04). Indeed, this parameter rose with age only in OXYS rats (ANOVA: F
1,17 = 13.5,
p < 0.002) via an increase in the resting-microglia density (ANOVA: F
1,17 = 107.1,
p < 0.0001), whereas, in Wistar rats, these parameters remained unchanged.
It is widely accepted that activated microglia secrete factors that induce the transformation of astrocytes to the reactive state; reactive astrocytes, in turn, intensify the acquisition of the activated phenotype by microglia [
50]. Next, we analyzed a possible correlation between the densities of vimentin
+ astrocytes (considered “reactive”) and CD68
+ microglia (regarded as “activated”) in hippocampal regions of OXYS and Wistar rats at an advanced age (meaning 18 months;
Figure S1). As expected, we detected a positive correlation between the parameters in the CA1 area of Wistar rats (correlation: r = 0.84,
p < 0.04). Meanwhile, a strong negative correlation between the reactive-astrocyte density and activated-microglia density was detectable in the CA3 region of OXYS rats (correlation: r = −0.96,
p < 0.002). This result may indicate the prevalence of only one type of cell with a proinflammatory phenotype in the CA3 area during the progressive stage of the AD-like pathology in OXYS rats, likely owing to the recruitment of cells of the same type. This observation requires further investigation.
Thus, here, we found an increased density of CD68
+ activated microglia during both the manifestation and progression of the AD-like pathology. These data suggest that microglial changes occur before Aβ accumulation does, and progress during the disease-like pathology. Earlier, we detected the signs of destructive changes in hippocampal microglial cells, as well as in the astrocytes of 18-month-old OXYS rats: a low electron density of the hyaloplasm, enlarged mitochondria with partial or full disintegration of cristae, numerous vacuoles, membrane complexes, and lipofuscin inclusions [
55]. The alterations in the structure of microglial cells reflect the loss of their regulatory functions related to migration, the clearance of cellular waste, and ultimately, neuronal survival.
Next, we assayed the effects of SkQ1 treatment on microglial density. Factorial ANOVA detected only insignificant effects of the genotype and treatment on the total microglial density (main effect (genotype): F
1,28 = 3.3,
p = 0.079; main effect (treatment): F
1,28 = 3.9,
p = 0.059, respectively). Even though the total microglial density remained unchanged, SkQ1 supplementation affected the resting/activated microglia ratio (
Figure 4A;
Tables S4 and S5). In particular, SkQ1 treatment raised the resting-microglia density (two-way ANOVA, main effect (treatment): F
1,28 = 13.1,
p < 0.001) and lowered that of activated microglia (two-way ANOVA, main effect (treatment): F
1,28 = 8.8,
p < 0.006), thereby exerting an anti-inflammatory effect. Pairwise comparisons revealed that SkQ1 increased the density of resting microglia in the whole hippocampus of both rat strains (Tukey’s test:
p < 0.02 for Wistar rats;
p < 0.002 for OXYS rats) and reduced the activated-microglia density only in OXYS rats (Tukey’s test:
p < 0.04).
Supplementation with SkQ1 eliminated the correlations between the reactive-astrocyte density and activated-microglia density (see above) in hippocampal regions of both rat strains (
Figure S1).
After that, we examined the density of pyknotic nuclei and their percentage phagocyted by microglia (
Table S6;
Figure S2). We revealed that neither the genotype nor treatment affected the percentage of pyknotic nuclei phagocyted by microglia. On the contrary, treatment with SkQ1 lowered the total density of pyknotic nuclei in the hippocampus (two-way ANOVA, main effect [treatment]: F
1,31 = 7.5,
p < 0.01). Thus, SkQ1 treatment had an anti-inflammatory effect in the hippocampus of rats from both strains, as evidenced by a decreased activated-microglia density and increased resting-microglia density, partly because of SkQ1’s contribution to the decrease in the pyknotic-nuclei density.
2.5. Genes Differentially Expressed between OXYS and Wistar Rats in Microglia
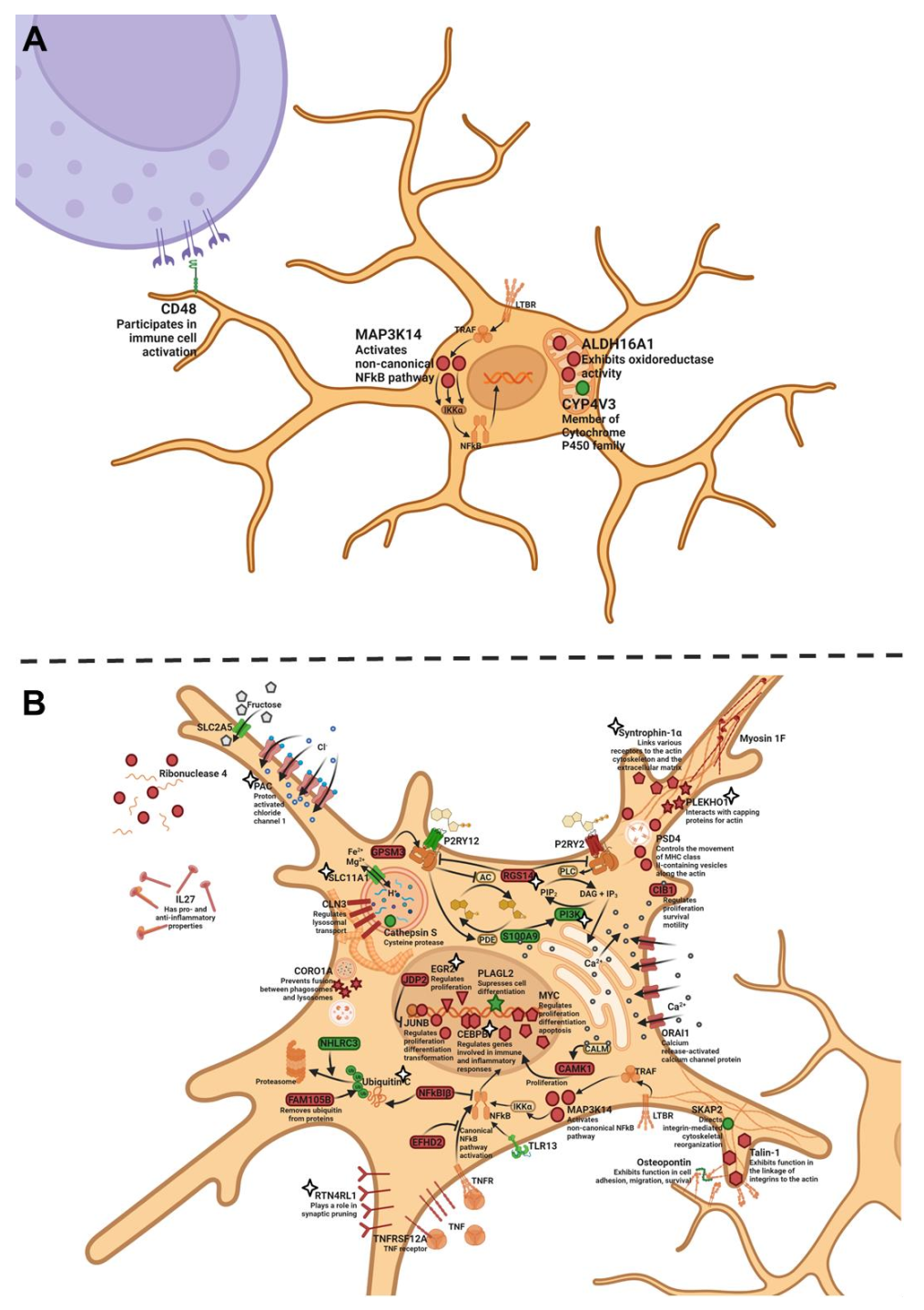
Next, we analyzed the expression of genes specific to microglia in the hippocampus during the development and progression of AD signs in OXYS rats and evaluated the possibility of SkQ1’s influence on the expression of these genes. At 5 months of age, there were four DEGs specific to microglia in the hippocampus of OXYS rats compared to Wistar rats. The expression of
Map3k14 and
Aldh16a1 was higher, and the expression of
Cd48 and
Cyp4v3 was lower in OXYS rats. Given that the
Map3k14 gene encodes a kinase of NF-κB, its overexpression may enhance the activation of the proinflammatory TNF pathway; at the same time,
Cd48 underexpression may mean a weaker activation of microglia by extracellular stimuli. Therefore, the decrease in the
Cd48 mRNA level may be considered to be a response that compensates for the increased activation of the TNF pathway (
Figure 5A).
As for age-related gene expression changes—from 5 to 18 months of age—we found 217 DEGs specific for microglia in Wistar rats and 185 DEGs in OXYS rats (
Table S7). Among the time-dependent DEGs in Wistar rats, 14 genes decreased their expression, whereas 203 genes increased their expression between these ages. In OXYS rats, 26 genes diminished and 159 genes increased their expression. From 5 to 18 months of age, 138 DEGs were common between OXYS and Wistar rats. In both rat strains, we observed an activation of the following pathways: glycan degradation (
HexA,
Hexb, and
Man2b1) and glycosaminoglycan degradation (
Glans,
Hexa,
Hexb, and
Naglu; FDR < 0.01); lysosome formation and function (
Cd68,
Ctsa,
Ctsb,
Ctsd,
Ctsf,
Ctss,
Ctsz,
Glans,
Hexa, and
Hexb); and apoptosis (
Ctsb,
Ctsd,
Ctsf,
Ctss,
Ctszb,
Ctszm,
Gadd46b,
Mcl1, and
Nfkb1). There were 47 genes that changed their expression from 5 to 18 months of age only in OXYS rats; among them, 12 genes decreased expression, and 35 genes were upregulated. Among these genes, 21 are associated with the cell membrane, and six genes with the cytoskeleton. Furthermore, an analysis of Gene Ontology categories (biological processes) revealed that 19 genes are related to cell communication.
At the progressive stage of neurodegeneration—meaning at 18 months of age—we found 33 strain-dependent DEGs specific to microglia in the hippocampus of OXYS rats relative to Wistar rats (
Table S7). Among them, 14 genes had a higher expression, and 19 genes had a lower expression in OXYS rats. Just as at the age of 5 months, OXYS rats at 18 months had a higher mRNA level of
Map3k14 and diminished mRNA levels of
Cd48 and
Cyp4v3 compared with Wistar rats. Aside from
Map3k14, OXYS rats manifested an overexpression of
Cebp and
Junb; products of all these genes take part in the triggering of TNF signaling. Meanwhile, the expression of
Pik3cg,
Spp1, and
Cdk6 was lower in OXYS rats than in Wistar rats; proteins encoded by these genes are components of mTOR signaling. In addition, the expression of
Ubc was lower in OXYS rats; this gene codes for a polyubiquitin precursor. The mRNA level of
Nhlrc3—another gene whose product is involved in ubiquitination—was lower in OXYS rats than in Wistar rats. The ubiquitin proteasome system in microglia is crucial for Aβ degradation [
59]. The upregulation of
Plekho1 and downregulation of
Skap2 mRNA expression are suggestive of greater actin stabilization, which, in turn, may worsen cellular and process motility, which are key features for the microglial response to AD [
60]. The level of
Ctss mRNA was significantly lower in OXYS rats than in Wistar rats; this gene encodes the cathepsin S protein, which belongs to the peptidase C1 family and may participate in the degradation of antigenic proteins for presentation to MHC class II molecules. To sum up, we found signs of an activation of proinflammatory signaling cascades and an altered interaction of microglia with the extracellular space during the progression of AD signs in OXYS rats (
Figure 5B).
As for the effects of SkQ1 treatment, previously, we reported that the following pathways are influenced: a response to an organic substance, the regulation of cell death, GTPase regulator activity, and ion homeostasis [
31]. Here, we demonstrated that treatment with SkQ1 prevented the age-associated increase (from 5 to 18 months of age) in the expression of seven genes (
Plekho1,
Arhgap22,
Dok3,
Fam110a,
Rogdi,
Rtn4rl1, and
Snta1) and downregulation of four genes (
Hk2,
Pik3cg,
Slc11a1, and
Ubc) specific for microglia in OXYS rats: this means that we could not find differences in the expression of these genes between SkQ1-treated 18-month-old OXYS rats and 5-month-old OXYS rats (
Table S7). Furthermore, the expression of
Dusp1 increased with age (from 5 to 18 months of age) in SkQ1-treated OXYS rats, just as in Wistar rats. Additionally, SkQ1 abrogated the upregulation of five genes (
Cebpb,
Egr2,
Rgs14,
Tmem206, and
Tor3a) in OXYS rats: this means that we failed to detect any differences in the expression of these genes at 18 months of age between SkQ1-treated OXYS rats and untreated Wistar rats (healthy control strain). Taken together, our results mean that the treatment with SkQ1 partly reversed the pathological alterations of mTOR signaling, of the ubiquitin proteasome system, and of actin cytoskeleton reorganization (
Figure 5B).
Summarizing the findings, we can conclude that the development of the AD-like pathology in OXYS rats is accompanied by such signs as a lower intensity of hippocampal neurogenesis accompanied by an insufficient astrocyte density in the neurogenic niche. Another conclusion is that the progressive stage of the AD-like pathology in OXYS rats is characterized by astrogliosis and microglial activation in the hippocampus. According to our results, both astrocytes and microglia are more vulnerable to AD-associated neurodegeneration in the CA3 area than in other hippocampal areas. Possible functional consequences of the changes in glia density and altered gene expression profiles are being investigated further. The treatment with mitochondria-targeted antioxidant SkQ1 from 12 to 18 months of age decreased the signs of astrogliosis in the hippocampus of OXYS rats, but increased these signs in Wistar rats. Additionally, SkQ1 treatment had an anti-inflammatory effect in the hippocampus of rats from both strains; this phenomenon was mediated by the observed shift of the resting-/activated-microglia density ratio toward the predominance of resting cells. The presented effects of SkQ1 may underline its ability to prevent neuronal loss and synaptic damage, to enhance a neurotrophic supply, and to decrease Aβ protein levels and tau hyperphosphorylation in the hippocampus [
32].


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




