
Antibody Conjugated PLGA Nanocarriers and Superparmagnetic Nanoparticles for Targeted Delivery of Oxaliplatin to Cells from Colorectal Carcinoma


 , , and
, , and
Abstract
:
1. Introduction
2. Results
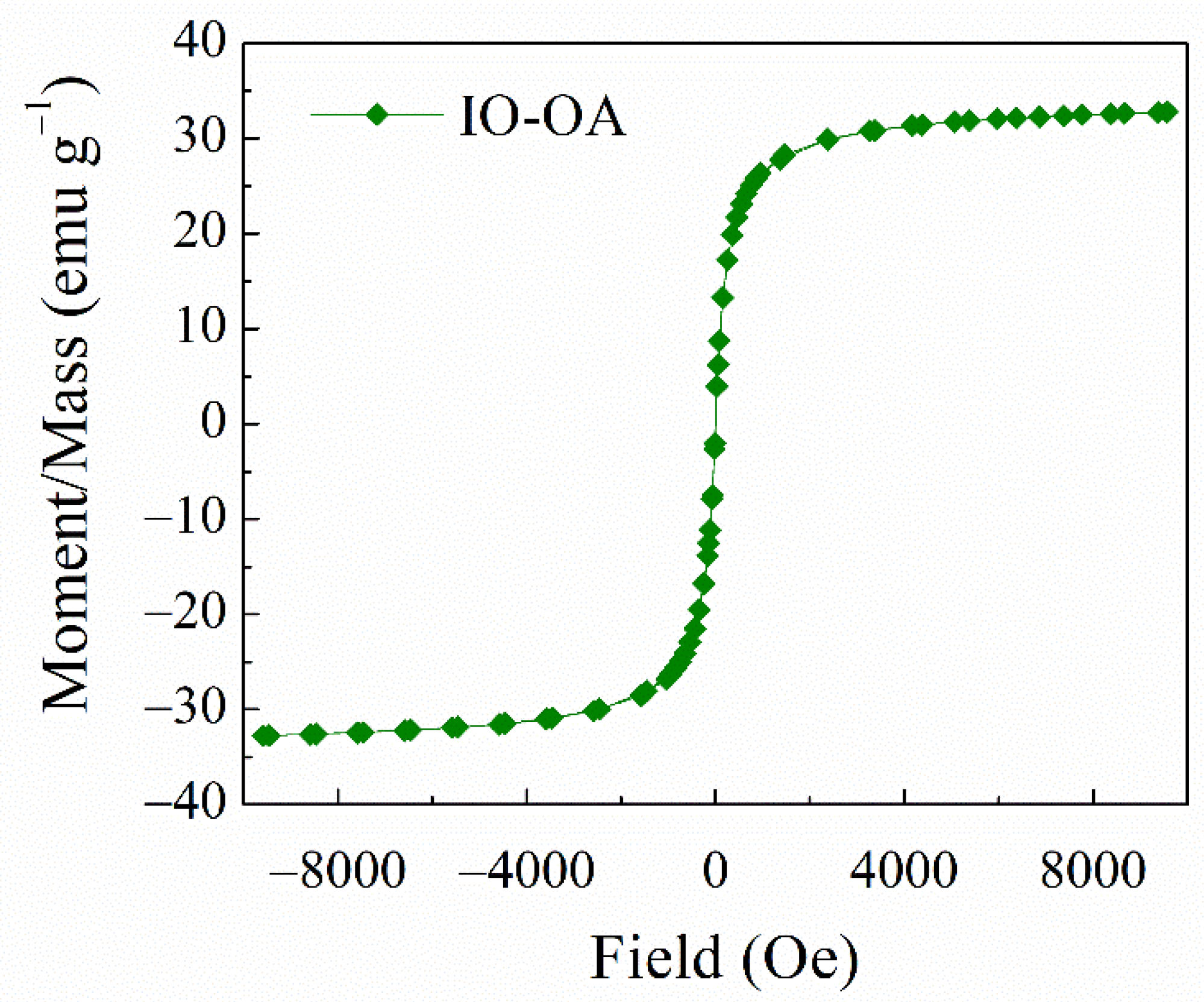
2.1. Physicochemical Characteristics of the Iron Oxide Nanoparticles
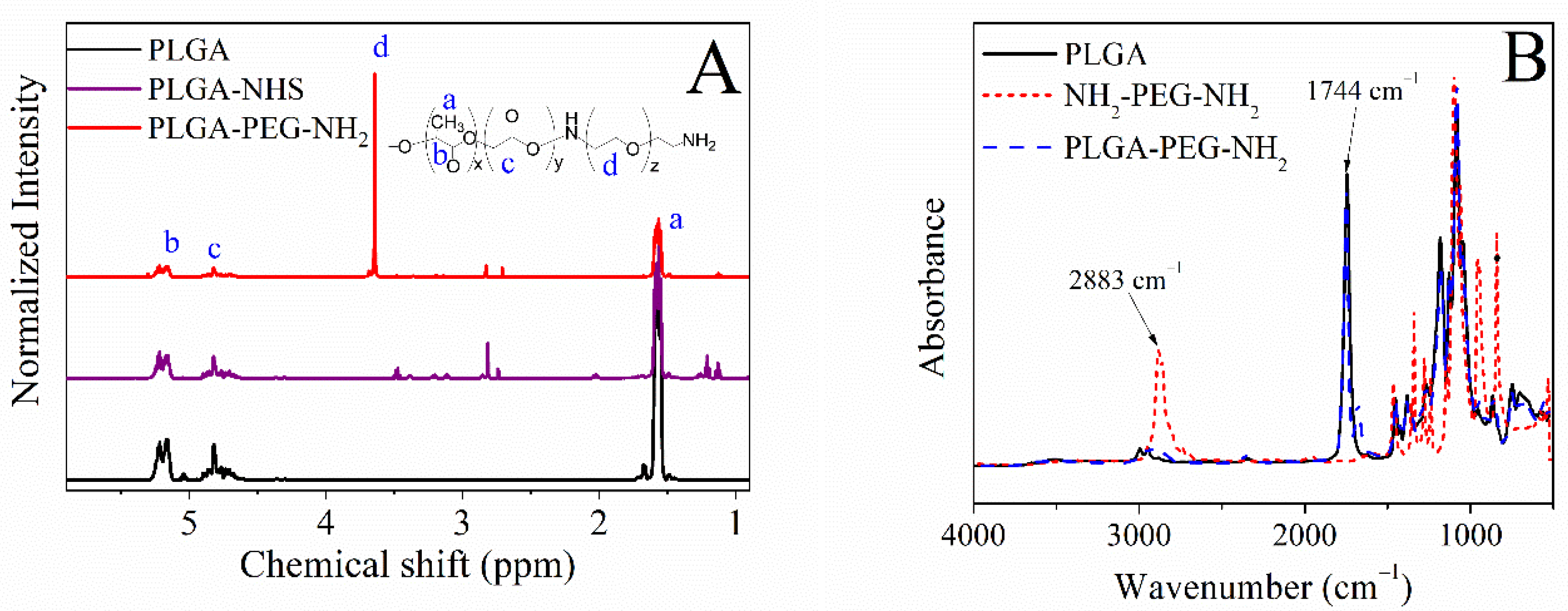
2.2. Chemical Structure of the Diblock Copolymer PLGA-PEG-NH2
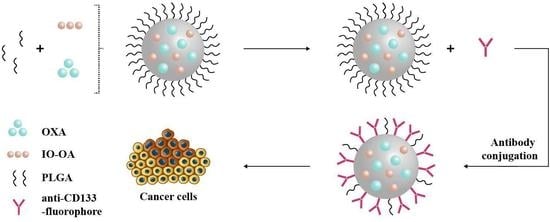
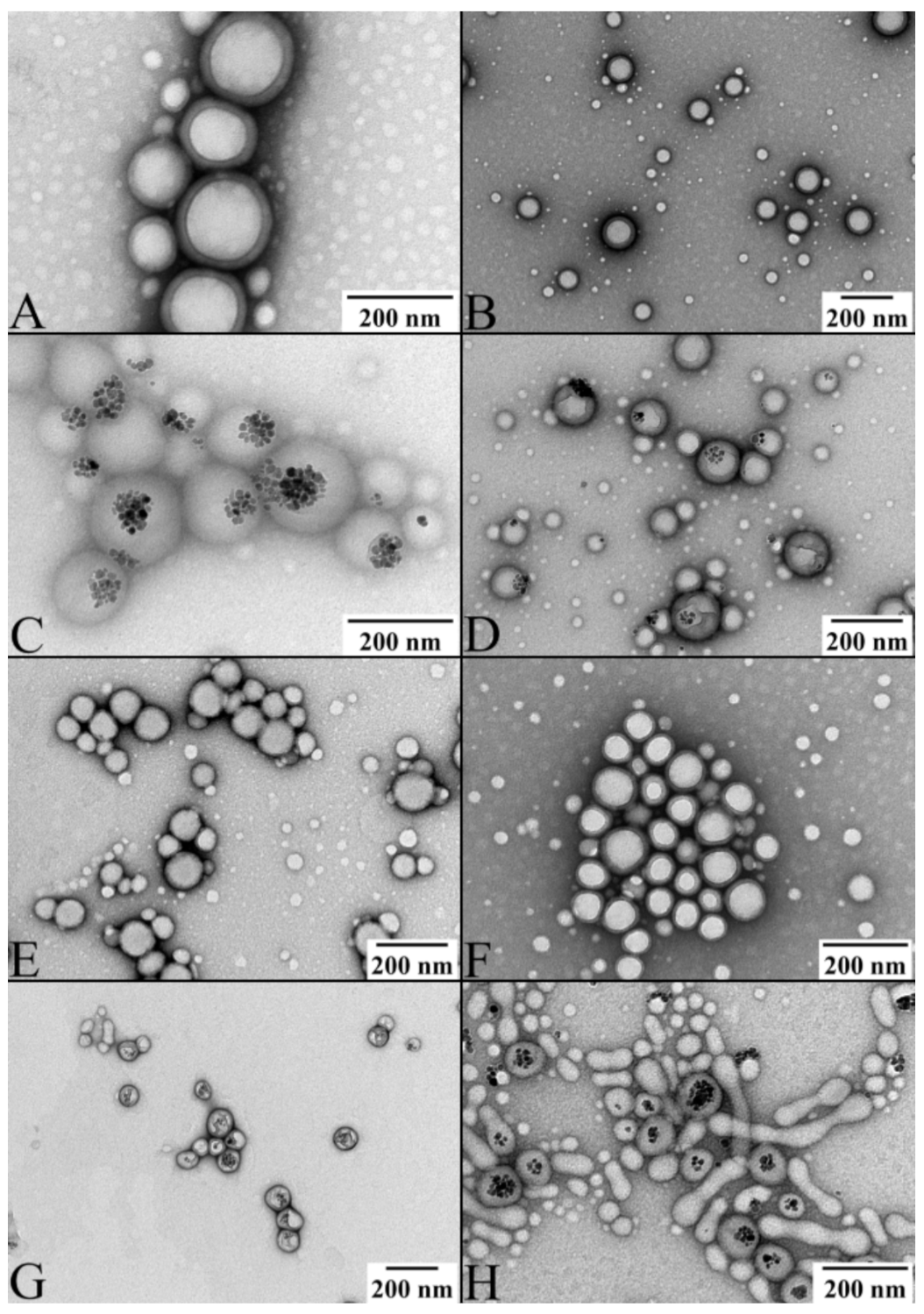
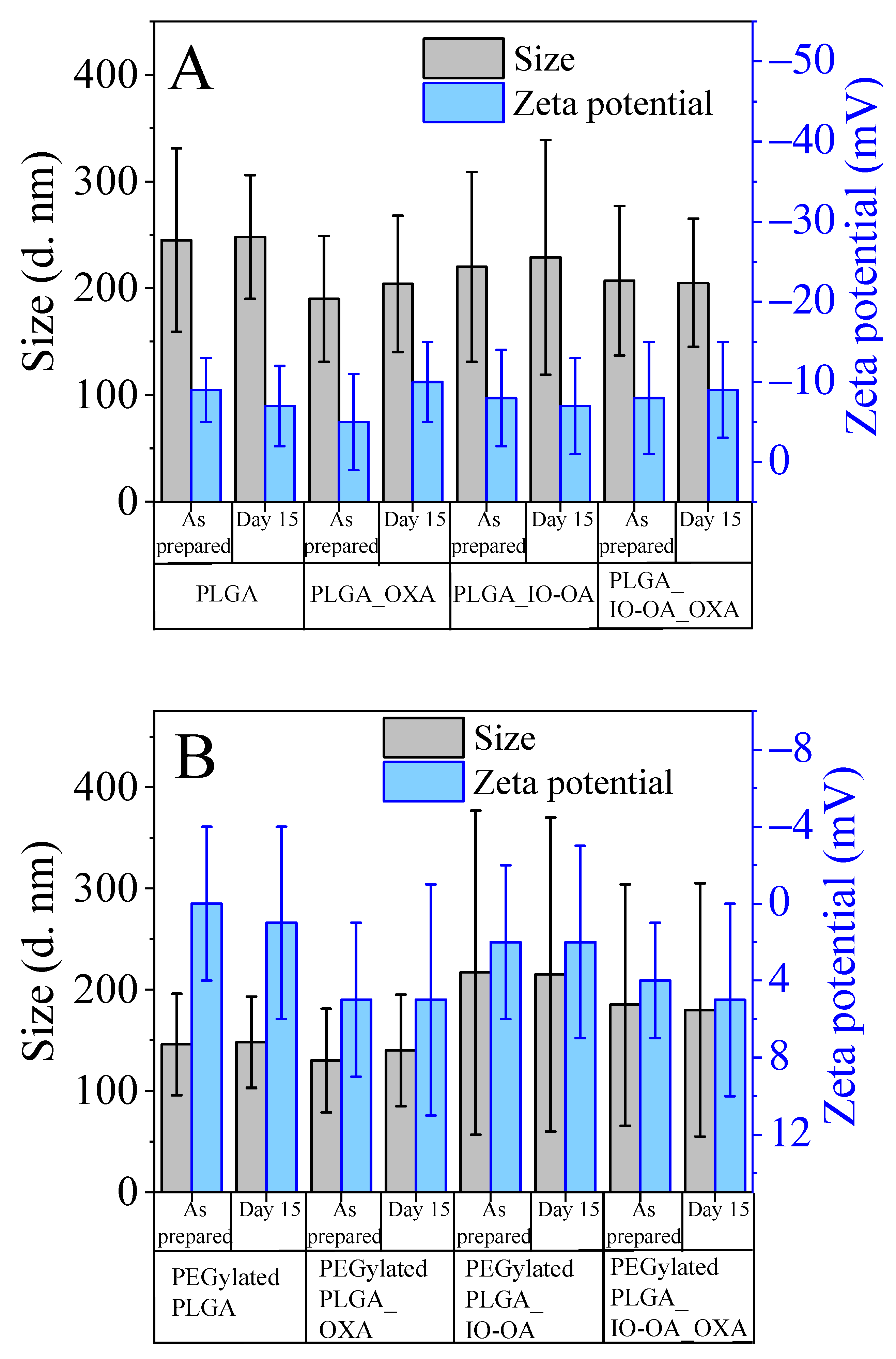
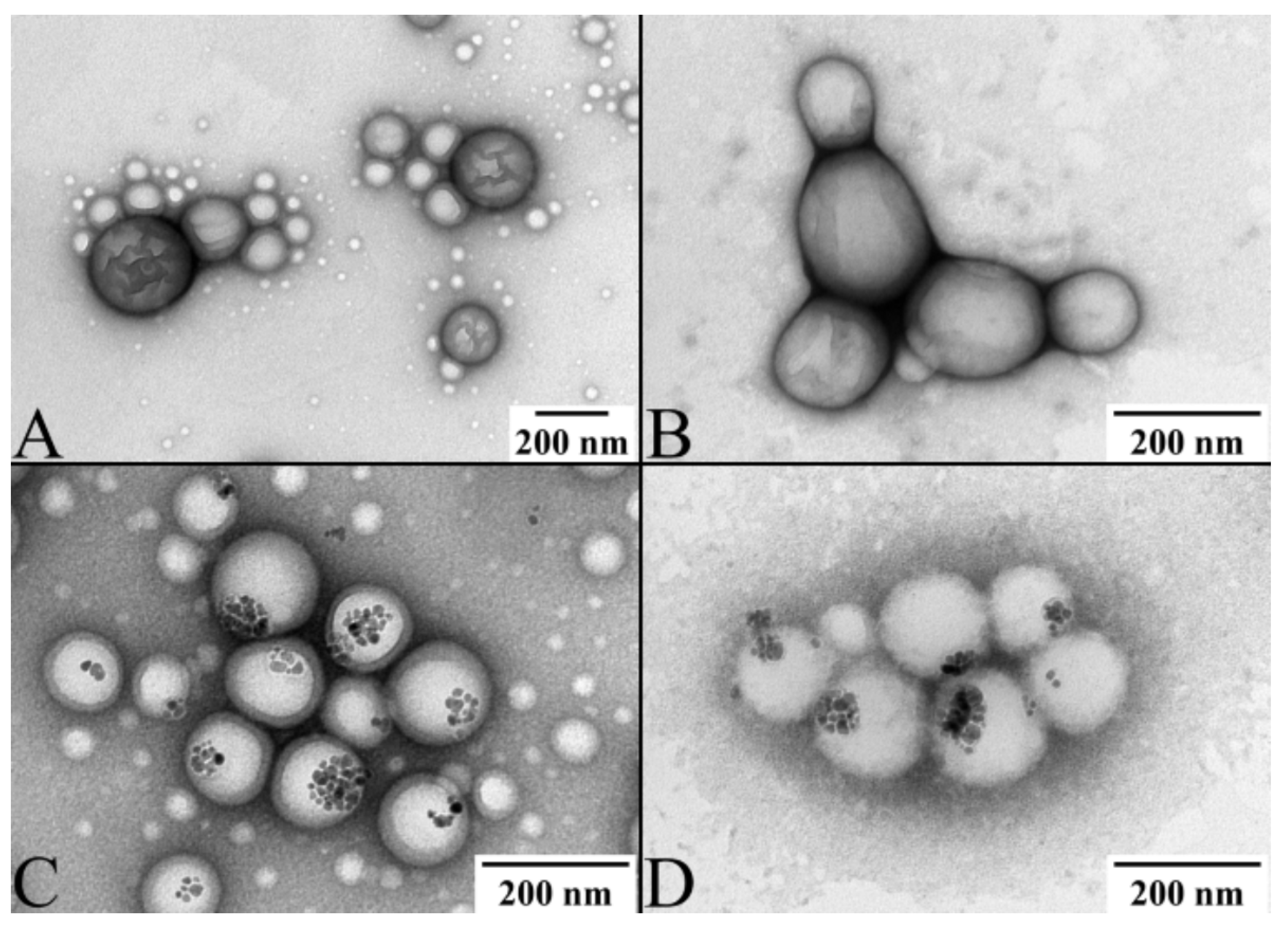
2.3. Multicomponent Drug Delivery Systems: Mean Size, Morphology, and Surface Properties
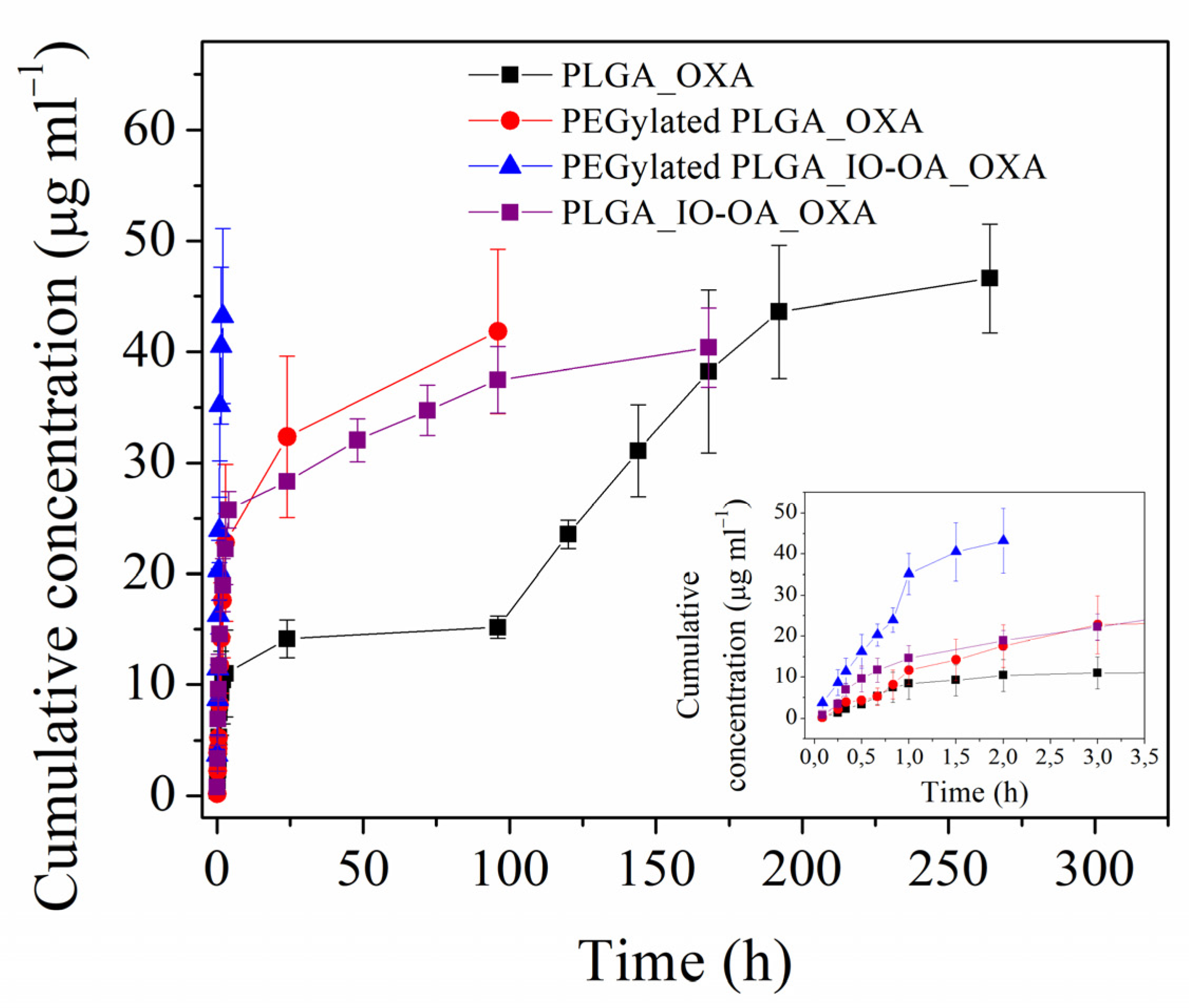
2.4. Drug Loading and Drug Release Kinetics
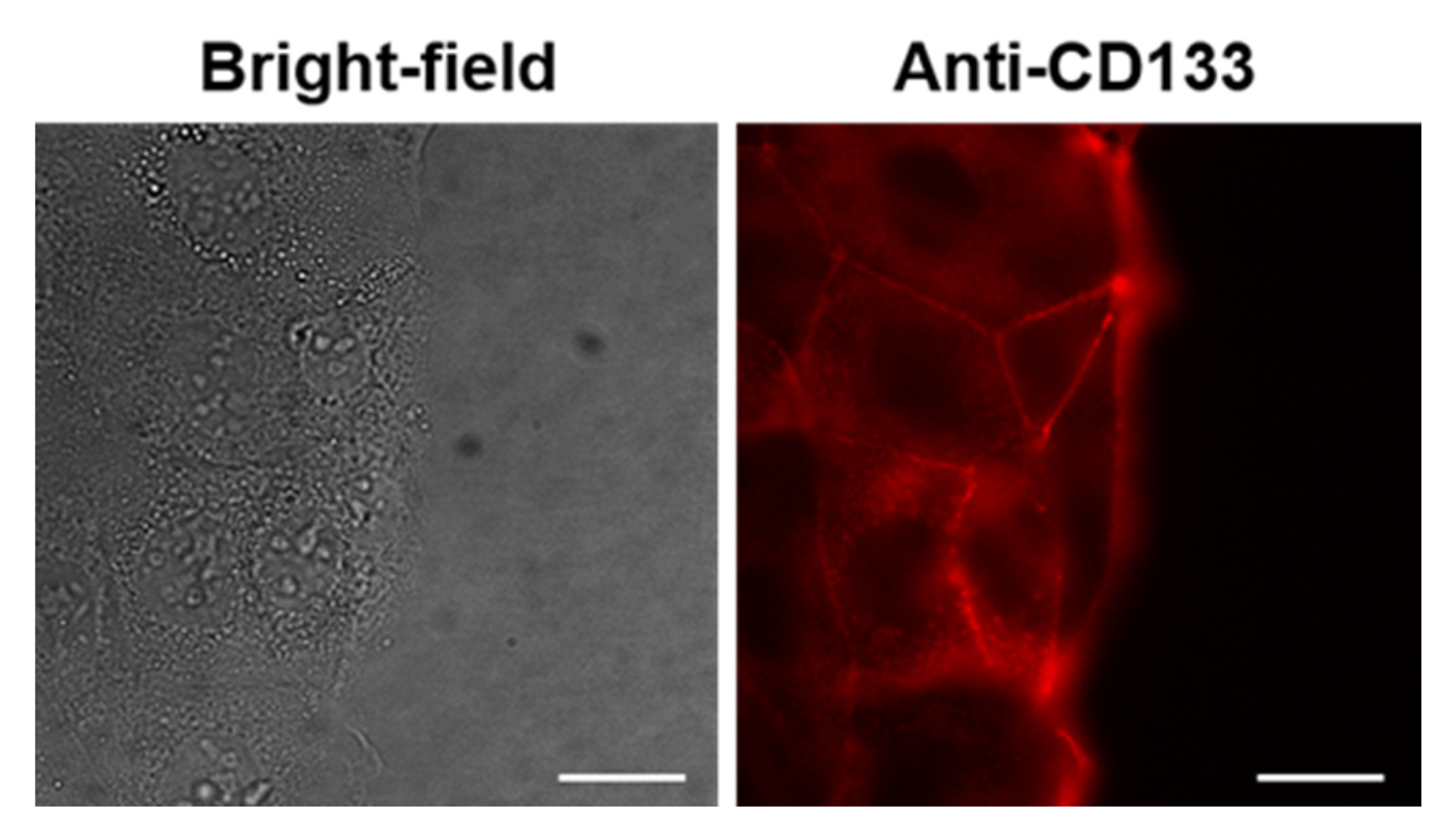
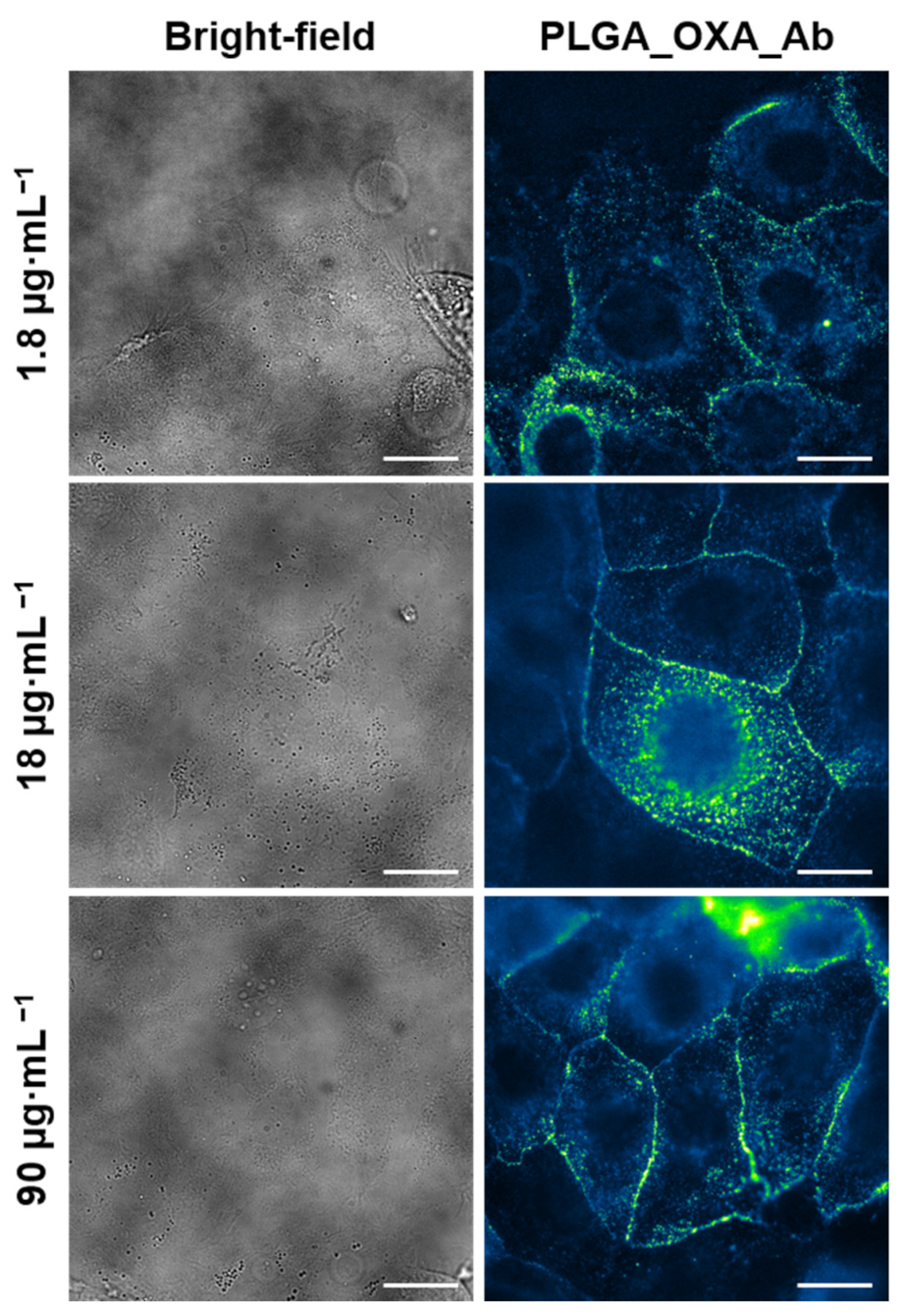
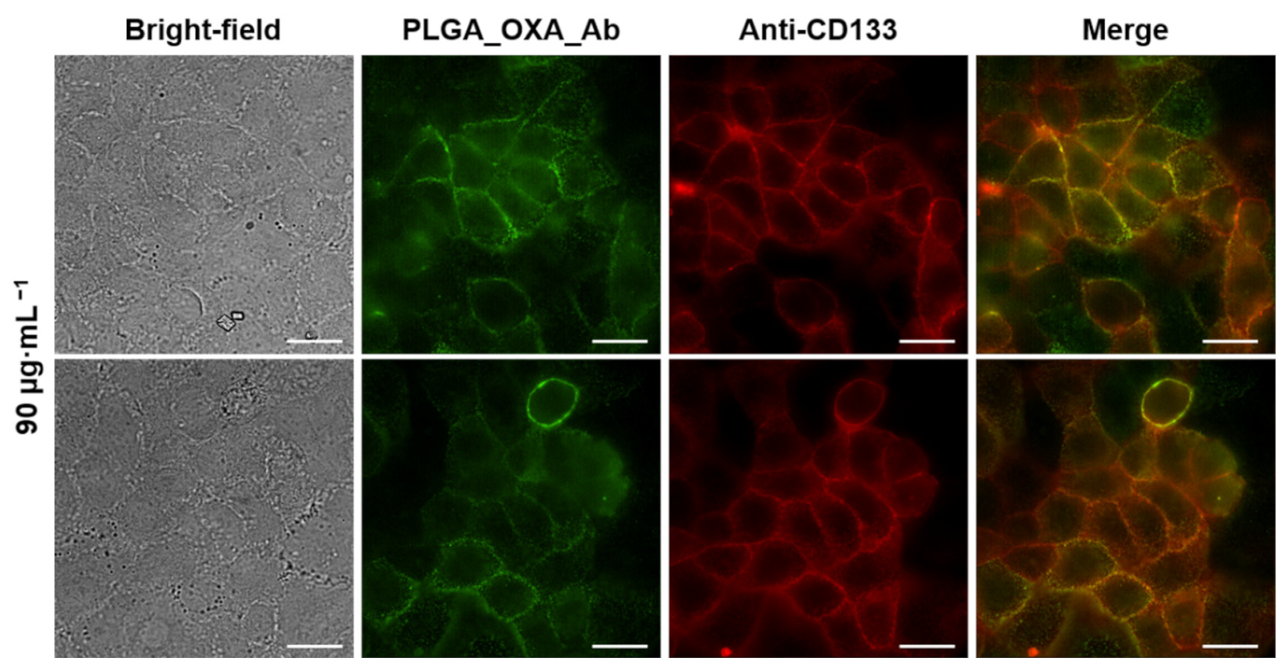
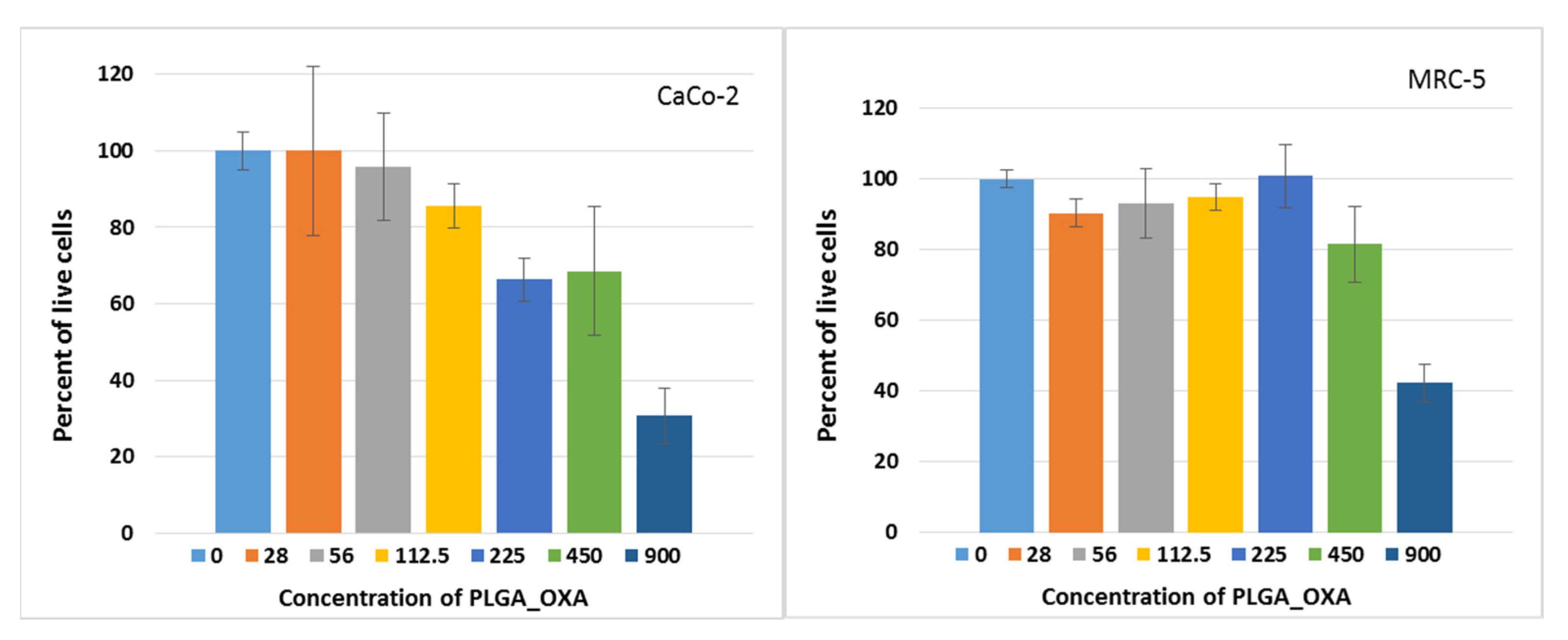
2.5. In Vitro Antitumor Activity of PLGA_IO-OA_OXA_Ab
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Synthesis of Iron Oxide Nanoparticles
4.3. Synthesis of PLGA-PEG-NH2 Copolymer
4.4. Preparation of the Multicomponent Drug Delivery Systems
4.5. Antibody Conjugation
4.6. Drug Loading and Dissolution Test
4.7. In Vitro Studies in Human Cells
4.7.1. Sample Fixation
4.7.2. Fluorescence Microscopy
4.7.3. Metabolic Activity Assay
4.8. Characterization
Dynamic Light Scattering
Zetasizer Potential
Transmission Electron Microscopy
Attenuated Total Reflection Fourier-Transformed Infrared Spectroscopy
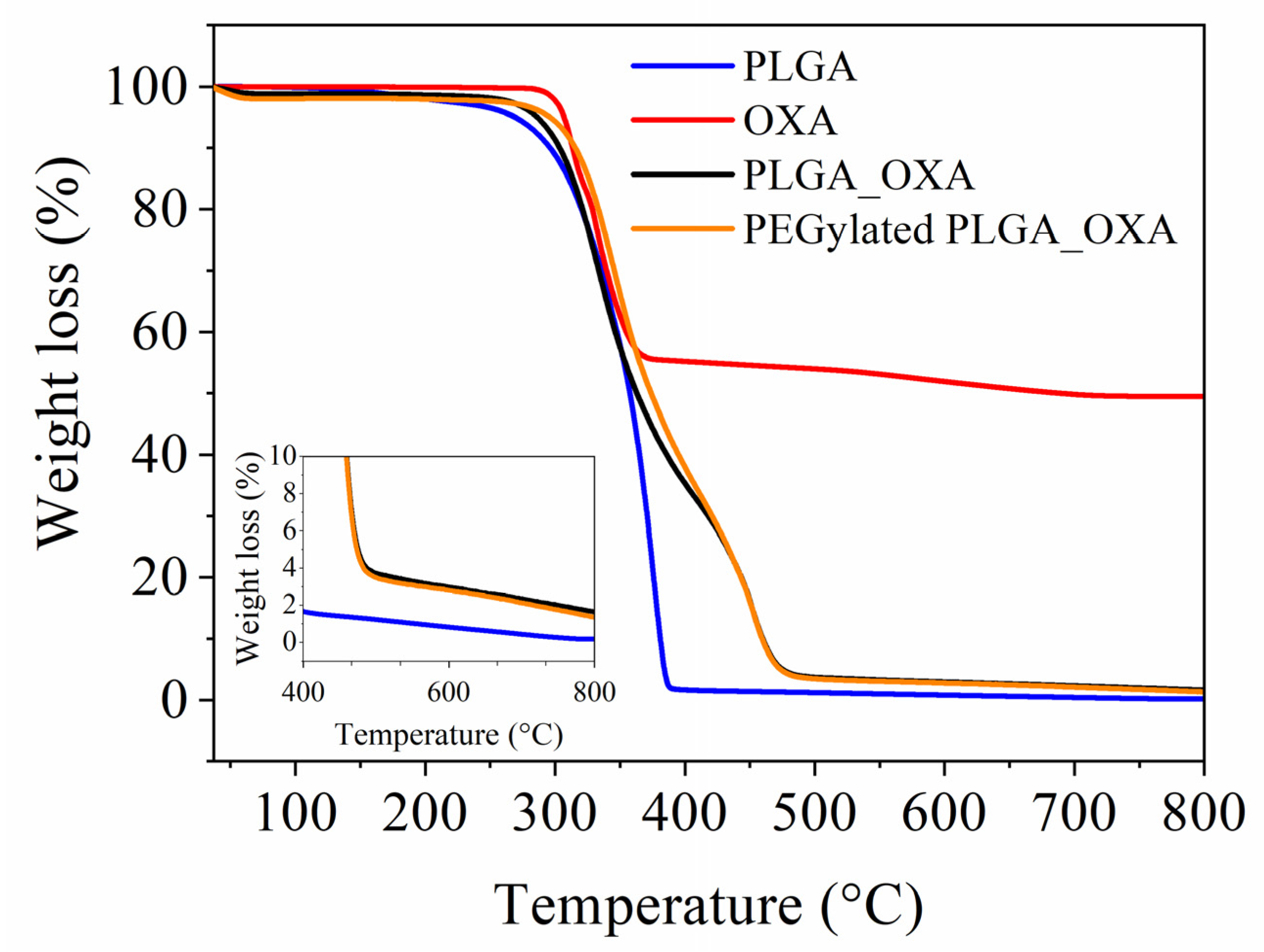
Thermogravimetric Analysis
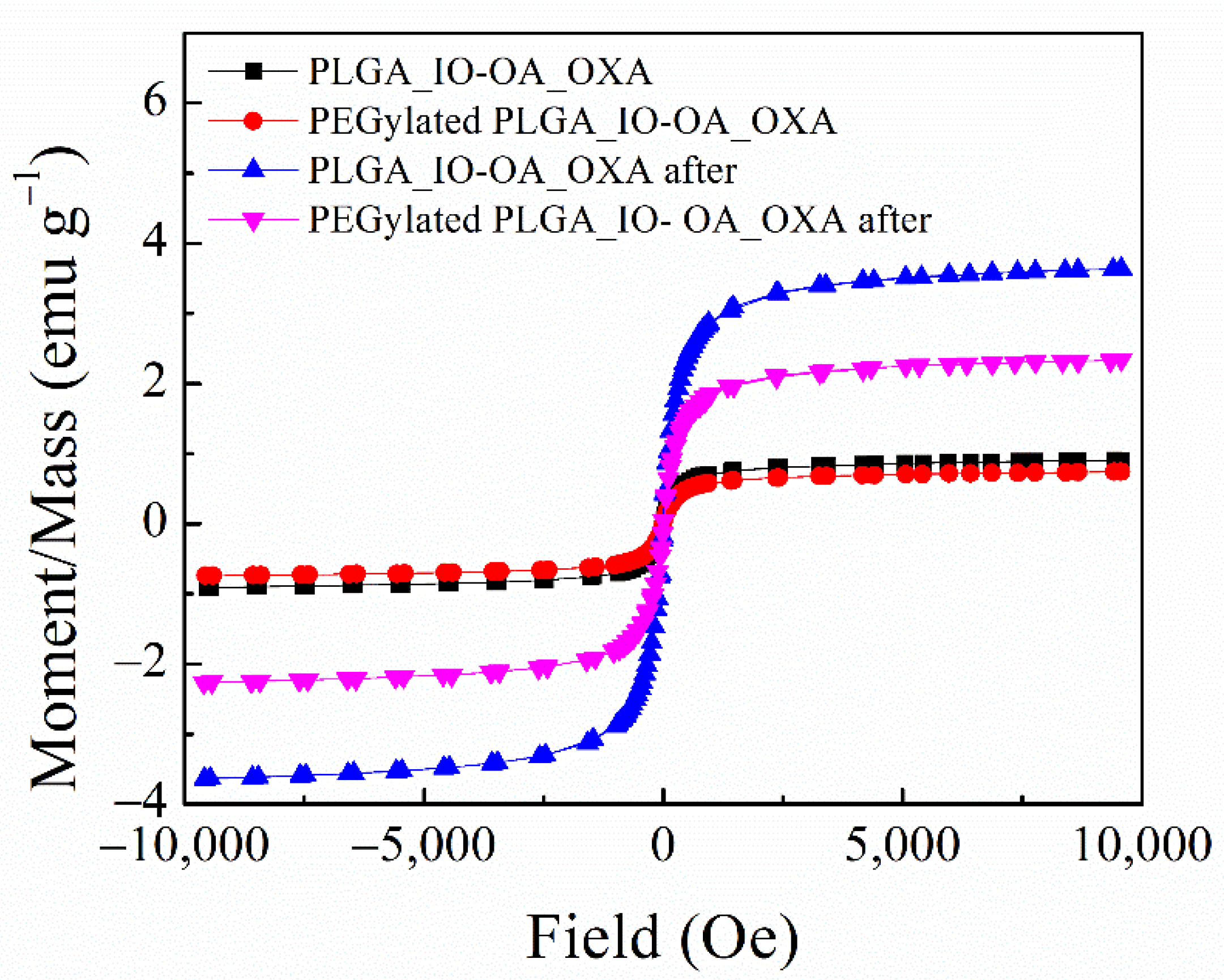
Vibrating Sample Magnetometer
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statemen
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 1H-NMR | Proton Nuclear Magnetic Resonance |
| 5-FU | 5-Fluorouracil |
| Ab | Antibody |
| anti-CD133-TMP4 | Monoclonal antibody CD133-TMP4 conjugated to Alexa Fluor 488 |
| CaCo-2 | Human cells derived from colorectal carcinoma |
| CSC | Cancer stem cells |
| DCC | N,N′-dicyclohexylcarbodiimide |
| DCM | Dichloromethane |
| DLS | Dymanic Light Scattering |
| DMEM | Dulbecco’s Modified Eagles’ medium |
| DNA | Deoxyribonucleic acid |
| EDC | N-(3-dimethyl aminoproyl)-N′-ethylcarbodiimide hydrochloride |
| EMEM | Eagle’s Minimum Essential Medium |
| FTIR | Fourier-transform Infrared spectroscopy |
| IO | Iron Oxide nanoparticles |
| IO-OA | Oleic acid-coated iron oxide nanoparticles |
| IPN | Induced peripheral neurotoxicity |
| MRC-5 | Noncancerous human primary fibroblasts |
| MRI | Magnetic Resonance Imaging |
| Ms | Saturation magnetization |
| NH2-PEG-NH2 | Poly(ethylene glycol diamine) |
| NHS | N-hydroxysuccinimide |
| OA | Oleic acid |
| OXA | Oxaliplatin |
| PBS | Phosphate Buffer Saline |
| PDI | Polydispersity index |
| PEGylated PLGA | PEGylated poly(lactide-co-glycolide) nanoparticles |
| PEGylated PLGA_IO-OA | Oleic acid-coated iron oxide-loaded PEGylated poly(lactide-co-glycolide) nanoparticles |
| PEGylated PLGA_IO-OA_OXA | Oleic acid-coated iron oxide and oxaliplatin-loaded PEGylated poly(lactide-co-glycolide) multicomponent delivery system |
| PEGylated PLGA_OXA | Oxaliplatin-loaded PEGylated poly(lactide-co-glycolide) nanoparticles |
| PLGA | Poly(lactide-co-glycolide) |
| PLGA_IO-OA | Oleic acid-coated iron oxide-loaded poly(lactide-co-glycolide) nanoparticles |
| PLGA_IO-OA_OXA | Oleic acid-coated iron oxide and oxaliplatin-loaded poly(lactide-co-glycolide) multicomponent delivery system |
| PLGA_IO-OA_OXA_Ab | anti-CD133 antibody conjugated oleic acid-coated iron oxide and oxaliplatin-loaded poly(lactide-co-glycolide) multicomponent delivery system |
| PLGA_OXA | Oxaliplatin-loaded poly(lactide-co-glycolide) nanoparticles |
| PLGA_OXA_Ab | anti-CD133 antibody conjugated oxaliplatin-loaded poly(lactide-co-glycolide) nanoparticles |
| PLGA-PEG-NH2 | Poly(lactide-co-glycolide)-poly(ethylene glycol) bearing amino end group |
| PVA | Polyvinyl alcohol |
| RNA | Ribonucleic acid |
| SD | Standard Deviation |
| SEC | Size Exlcusion Chromatography |
| TEM | Transmission Electron Microscopy |
| TGA | Thermogravimetric Analysis |
| THF | Tetrahydrofuran |
| VSM | Vibrating Sample Magnetometer |
| XRD | X-ray Diffraction |
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Ferlay, J.; Ervik, M.; Lam, F.; Colombet, M.; Mery, L.; Piñeros, M.; Znaor, A.; Soerjomataram, I.; Bray, F. Global Cancer Observatory: Cancer Today; International Agency for Research on Cancer: Lyon, France, 2020. [Google Scholar]
- Bishehsari, F.; Mahdavinia, M.; Vacca, M.; Malekzadeh, R.; Mariani-Costantini, R. Epidemiological transition of colorectal cancer in developing countries: Environmental factors, molecular pathways, and opportunities for prevention. World J. Gastroenterol. 2014, 20, 6055–6072. [Google Scholar] [CrossRef]
- Arnold, M.; Abnet, C.C.; Neale, R.E.; Vignat, J.; Giovannucci, E.L.; McGlynn, K.A.; Bray, F. Global Burden of 5 Major Types of Gastrointestinal Cancer. Gastroenterology 2020, 159, 335–349. [Google Scholar] [CrossRef]
- Heidelberger, C.; Chaudhuri, N.K.; Danneberg, P.; Mooren, D.; Griesbach, L.; Duschinsky, R.; Schnitzer, R.J.; Pleven, E.; Scheiner, J. Fluorinated pyrimidines, a new class of tumour-inhibitory compounds. Nature 1957, 179, 663–666. [Google Scholar] [CrossRef] [PubMed]
- Danenberg, P.V. Thymidylate synthetase—A target enzyme in cancer chemotherapy. Biochim. Biophys. Acta 1977, 473, 73–92. [Google Scholar] [CrossRef]
- Poon, M.A.; O’Connell, M.J.; Moertel, C.G.; Wieand, H.S.; Cullinan, S.A.; Everson, L.K.; Krook, J.E.; Mailliard, J.A.; Laurie, J.A.; Tschetter, L.K.; et al. Biochemical modulation of fluorouracil: Evidence of significant improvement of survival and quality of life in patients with advanced colorectal carcinoma. J. Clin. Oncol. 1989, 7, 1407–1418. [Google Scholar] [CrossRef] [PubMed]
- Mini, E.; Trave, F.; Rustum, Y.M.; Bertino, J.R. Enhancement of the antitumor effects of 5-fluorouracil by folinic acid. Pharmacol. Ther. 1990, 47, 1–19. [Google Scholar] [CrossRef]
- Saltz, L.B.; Cox, J.V.; Blanke, C.; Rosen, L.S.; Fehrenbacher, L.; Moore, M.J.; Maroun, J.A.; Ackland, S.P.; Locker, P.K.; Pirotta, N.; et al. Irinotecan plus fluorouracil and leucovorin for metastatic colorectal cancer. Irinotecan Study Group. New Engl. J. Med. 2000, 343, 905–914. [Google Scholar] [CrossRef]
- Goldberg, R.M.; Sargent, D.J.; Morton, R.F.; Fuchs, C.S.; Ramanathan, R.K.; Williamson, S.K.; Findlay, B.P.; Pitot, H.C.; Alberts, S.R. A randomized controlled trial of fluorouracil plus leucovorin, irinotecan, and oxaliplatin combinations in patients with previously untreated metastatic colorectal cancer. J. Clin. Oncol. 2004, 22, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.M.; Cardona, K.; Russell, M.C. Historical perspective: Two decades of progress in treating metastatic colorectal cancer. J. Surg. Oncol. 2019, 119, 549–563. [Google Scholar] [CrossRef]
- Gustavsson, B.; Carlsson, G.; Machover, D.; Petrelli, N.; Roth, A.; Schmoll, H.J.; Tveit, K.M.; Gibson, F. A review of the evolution of systemic chemotherapy in the management of colorectal cancer. Clin. Colorectal Cancer 2015, 14, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Lievre, A.; Bachet, J.B.; Le Corre, D.; Boige, V.; Landi, B.; Emile, J.F.; Cote, J.F.; Tomasic, G.; Penna, C.; Ducreux, M.; et al. KRAS mutation status is predictive of response to cetuximab therapy in colorectal cancer. Cancer Res. 2006, 66, 3992–3995. [Google Scholar] [CrossRef] [Green Version]
- Matos, A.I.; Carreira, B.; Peres, C.; Moura, L.I.F.; Conniot, J.; Fourniols, T.; Scomparin, A.; Martinez-Barriocanal, A.; Arango, D.; Conde, J.P.; et al. Nanotechnology is an important strategy for combinational innovative chemo-immunotherapies against colorectal cancer. J. Control. Release 2019, 307, 108–138. [Google Scholar] [CrossRef]
- Yassin, A.E.; Anwer, M.K.; Mowafy, H.A.; El-Bagory, I.M.; Bayomi, M.A.; Alsarra, I.A. Optimization of 5-flurouracil solid-lipid nanoparticles: A preliminary study to treat colon cancer. Int. J. Med. Sci. 2010, 7, 398–408. [Google Scholar] [CrossRef] [Green Version]
- Mattos, A.C.; Altmeyer, C.; Tominaga, T.T.; Khalil, N.M.; Mainardes, R.M. Polymeric nanoparticles for oral delivery of 5-fluorouracil: Formulation optimization, cytotoxicity assay and pre-clinical pharmacokinetics study. Eur. J. Pharm. Sci. 2016, 84, 83–91. [Google Scholar] [CrossRef]
- Feng, S.T.; Li, J.; Luo, Y.; Yin, T.; Cai, H.; Wang, Y.; Dong, Z.; Shuai, X.; Li, Z.P. pH-sensitive nanomicelles for controlled and efficient drug delivery to human colorectal carcinoma LoVo cells. PLoS ONE 2014, 9, e100732. [Google Scholar] [CrossRef]
- Hosseinzadeh, H.; Atyabi, F.; Varnamkhasti, B.S.; Hosseinzadeh, R.; Ostad, S.N.; Ghahremani, M.H.; Dinarvand, R. SN38 conjugated hyaluronic acid gold nanoparticles as a novel system against metastatic colon cancer cells. Int. J. Pharm. 2017, 526, 339–352. [Google Scholar] [CrossRef] [PubMed]
- Kuo, C.Y.; Liu, T.Y.; Chan, T.Y.; Tsai, S.C.; Hardiansyah, A.; Huang, L.Y.; Yang, M.C.; Lu, R.H.; Jiang, J.K.; Yang, C.Y.; et al. Magnetically triggered nanovehicles for controlled drug release as a colorectal cancer therapy. Colloids Surf. B: Biointerfaces 2016, 140, 567–573. [Google Scholar] [CrossRef] [PubMed]
- Di Francesco, A.M.; Ruggiero, A.; Riccardi, R. Cellular and molecular aspects of drugs of the future: Oxaliplatin. Cell. Mol. Life Sci. 2002, 59, 1914–1927. [Google Scholar] [CrossRef]
- Kim, G.P.; Erlichman, C. Oxaliplatin in the treatment of colorectal cancer. Expert Opin. Drug Metab. Toxicol. 2007, 3, 281–294. [Google Scholar] [CrossRef] [PubMed]
- Pietrangeli, A.; Leandri, M.; Terzoli, E.; Jandolo, B.; Garufi, C. Persistence of high-dose oxaliplatin-induced neuropathy at long-term follow-up. Eur. Neurol. 2006, 56, 13–16. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, A.V.; Goldstein, D.; Friedlander, M.; Kiernan, M.C. Oxaliplatin-induced neurotoxicity and the development of neuropathy. Muscle Nerve 2005, 32, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Beijers, A.J.; Mols, F.; Vreugdenhil, G. A systematic review on chronic oxaliplatin-induced peripheral neuropathy and the relation with oxaliplatin administration. Support. Care Cancer 2014, 22, 1999–2007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makadia, H.K.; Siegel, S.J. Poly Lactic-co-Glycolic Acid (PLGA) as Biodegradable Controlled Drug Delivery Carrier. Polymers 2011, 3, 1377–1397. [Google Scholar] [CrossRef]
- Sinha, V.R.; Bansal, K.; Kaushik, R.; Kumria, R.; Trehan, A. Poly-ϵ-caprolactone microspheres and nanospheres: An overview. Int. J. Pharm. 2004, 278, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Llorens, E.; Pérez-Madrigal, M.M.; Armelin, E.; del Valle, L.J.; Puiggalí, J.; Alemán, C. Hybrid nanofibers from biodegradable polylactide and polythiophene for scaffolds. RSC Adv. 2014, 4, 15245–15255. [Google Scholar] [CrossRef]
- Kapoor, D.N.; Bhatia, A.; Kaur, R.; Sharma, R.; Kaur, G.; Dhawan, S. PLGA: A unique polymer for drug delivery. Ther. Deliv. 2015, 6, 41–58. [Google Scholar] [CrossRef]
- D’Avila Carvalho Erbetta, C. Synthesis and Characterization of Poly(D,L-Lactide-co-Glycolide) Copolymer. J. Biomater. Nanobiotechnology 2012, 3, 208–225. [Google Scholar] [CrossRef]
- Hermanson, G.T. Chapter 18—PEGylation and Synthetic Polymer Modification. In Bioconjugate Techniques, 3rd ed.; Hermanson, G.T., Ed.; Academic Press: Boston, MA, USA, 2013; pp. 787–838. [Google Scholar]
- Kocbek, P.; Obermajer, N.; Cegnar, M.; Kos, J.; Kristl, J. Targeting cancer cells using PLGA nanoparticles surface modified with monoclonal antibody. J. Control. Release 2007, 120, 18–26. [Google Scholar] [CrossRef]
- Vij, N.; Min, T.; Marasigan, R.; Belcher, C.N.; Mazur, S.; Ding, H.; Yong, K.T.; Roy, I. Development of PEGylated PLGA nanoparticle for controlled and sustained drug delivery in cystic fibrosis. J. Nanobiotechnology 2010, 8, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zumaya, A.L.V.; Ulbrich, P.; Vilčáková, J.; Dendisová, M.; Fulem, M.; Šoóš, M.; Hassouna, F. Comparison between two multicomponent drug delivery systems based on PEGylated-poly (l-lactide-co-glycolide) and superparamagnetic nanoparticles: Nanoparticulate versus nanocluster systems. J. Drug Deliv. Sci. Technol. 2021, 64, 102643. [Google Scholar] [CrossRef]
- Zumaya, A.L.V.; Martynek, D.; Bautkinová, T.; Šoóš, M.; Ulbrich, P.; Raquez, J.-M.; Dendisová, M.; Merna, J.; Vilčáková, J.; Kopecký, D.; et al. Self-assembly of poly(L-lactide-co-glycolide) and magnetic nanoparticles into nanoclusters for controlled drug delivery. Eur. Polym. J. 2020, 133, 109795. [Google Scholar] [CrossRef]
- Jia, Y.; Yuan, M.; Yuan, H.; Huang, X.; Sui, X.; Cui, X.; Tang, F.; Peng, J.; Chen, J.; Lu, S.; et al. Co-encapsulation of magnetic Fe3O4 nanoparticles and doxorubicin into biodegradable PLGA nanocarriers for intratumoral drug delivery. Int. J. Nanomed. 2012, 7, 1697–1708. [Google Scholar] [CrossRef] [Green Version]
- Laurent, S.; Dutz, S.; Hafeli, U.O.; Mahmoudi, M. Magnetic fluid hyperthermia: Focus on superparamagnetic iron oxide nanoparticles. Adv. Colloid Interface Sci. 2011, 166, 8–23. [Google Scholar] [CrossRef] [PubMed]
- Soares, P.I.; Alves, A.M.; Pereira, L.C.; Coutinho, J.T.; Ferreira, I.M.; Novo, C.M.; Borges, J.P. Effects of surfactants on the magnetic properties of iron oxide colloids. J. Colloid Interface Sci. 2014, 419, 46–51. [Google Scholar] [CrossRef]
- Soares, P.I.P.; Laia, C.A.T.; Carvalho, A.; Pereira, L.C.J.; Coutinho, J.T.; Ferreira, I.M.M.; Novo, C.M.M.; Borges, J.P. Iron oxide nanoparticles stabilized with a bilayer of oleic acid for magnetic hyperthermia and MRI applications. Appl. Surf. Sci. 2016, 383, 240–247. [Google Scholar] [CrossRef]
- Patel, J.; Amrutiya, J.; Bhatt, P.; Javia, A.; Jain, M.; Misra, A. Targeted delivery of monoclonal antibody conjugated docetaxel loaded PLGA nanoparticles into EGFR overexpressed lung tumour cells. J. Microencapsul. 2018, 35, 204–217. [Google Scholar] [CrossRef]
- Singh, S.; Kumar, N.K.; Dwiwedi, P.; Charan, J.; Kaur, R.; Sidhu, P.; Chugh, V.K. Monoclonal Antibodies: A Review. Curr. Clin. Pharmacol. 2018, 13, 85–99. [Google Scholar] [CrossRef] [PubMed]
- Belov, L.; Zhou, J.; Christopherson, R.I. Cell surface markers in colorectal cancer prognosis. Int. J. Mol. Sci. 2010, 12, 78–113. [Google Scholar] [CrossRef] [Green Version]
- Moreno, D.; de Ilarduya, C.T.; Bandres, E.; Bunuales, M.; Azcona, M.; Garcia-Foncillas, J.; Garrido, M.J. Characterization of cisplatin cytotoxicity delivered from PLGA-systems. Eur. J. Pharm. Biopharm. 2008, 68, 503–512. [Google Scholar] [CrossRef]
- Carter, T.; Mulholland, P.; Chester, K. Antibody-targeted nanoparticles for cancer treatment. Immunotherapy 2016, 8, 941–958. [Google Scholar] [CrossRef]
- Veiseh, O.; Gunn, J.W.; Zhang, M. Design and fabrication of magnetic nanoparticles for targeted drug delivery and imaging. Adv. Drug Del. Rev. 2010, 62, 284–304. [Google Scholar] [CrossRef] [Green Version]
- Cheng, J.; Teply, B.A.; Sherifi, I.; Sung, J.; Luther, G.; Gu, F.X.; Levy-Nissenbaum, E.; Radovic-Moreno, A.F.; Langer, R.; Farokhzad, O.C. Formulation of functionalized PLGA-PEG nanoparticles for in vivo targeted drug delivery. Biomaterials 2007, 28, 869–876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.P.; Pei, Y.Y.; Zhang, X.Y.; Gu, Z.H.; Zhou, Z.H.; Yuan, W.F.; Zhou, J.J.; Zhu, J.H.; Gao, X.J. PEGylated PLGA nanoparticles as protein carriers: Synthesis, preparation and biodistribution in rats. J. Control. Release 2001, 71, 203–211. [Google Scholar] [CrossRef]
- Betancourt, T.; Byrne, J.D.; Sunaryo, N.; Crowder, S.W.; Kadapakkam, M.; Patel, S.; Casciato, S.; Brannon-Peppas, L. PEGylation strategies for active targeting of PLA/PLGA nanoparticles. J. Biomed. Mater. Res. A 2009, 91, 263–276. [Google Scholar] [CrossRef] [PubMed]
- Ngaboni Okassa, L.; Marchais, H.; Douziech-Eyrolles, L.; Cohen-Jonathan, S.; Souce, M.; Dubois, P.; Chourpa, I. Development and characterization of sub-micron poly(D,L-lactide-co-glycolide) particles loaded with magnetite/maghemite nanoparticles. Int. J. Pharm. 2005, 302, 187–196. [Google Scholar] [CrossRef]
- Saez, V.; Cerruti, R.; Ramón, J.A.; Santos, E.R.F.; Silva, D.Z.; Pinto, J.C.; Souza, F.G. Quantification of Oxaliplatin Encapsulated into PLGA Microspheres by TGA. Macromol. Symp. 2016, 368, 116–121. [Google Scholar] [CrossRef]
- Yoo, J.; Won, Y.Y. Phenomenology of the Initial Burst Release of Drugs from PLGA Microparticles. ACS Biomater. Sci. Eng. 2020, 6, 6053–6062. [Google Scholar] [CrossRef]
- Avgoustakis, K. PLGA–mPEG nanoparticles of cisplatin: In vitro nanoparticle degradation, in vitro drug release and in vivo drug residence in blood properties. J. Control. Release 2002, 79, 123–135. [Google Scholar] [CrossRef]
- Uhlen, M.; Zhang, C.; Lee, S.; Sjöstedt, E.; Fagerberg, L.; Bidkhori, G.; Benfeitas, R.; Arif, M.; Liu, Z.; Edfors, F.; et al. A pathology atlas of the human cancer transcriptome. Science 2017, 357, eaan2507. [Google Scholar] [CrossRef] [Green Version]
- Grothey, A.; Goldberg, R.M. A review of oxaliplatin and its clinical use in colorectal cancer. Expert Opin. Pharmacother. 2004, 5, 2159–2170. [Google Scholar] [CrossRef]
- Stewart, D.J. Mechanisms of resistance to cisplatin and carboplatin. Crit. Rev. Oncol. Hematol. 2007, 63, 12–31. [Google Scholar] [CrossRef] [PubMed]
- Rixe, O.; Ortuzar, W.; Alvarez, M.; Parker, R.; Reed, E.; Paull, K.; Fojo, T. Oxaliplatin, tetraplatin, cisplatin, and carboplatin: Spectrum of activity in drug-resistant cell lines and in the cell lines of the national cancer institute’s anticancer drug screen panel. Biochem. Pharmacol. 1996, 52, 1855–1865. [Google Scholar] [CrossRef]
- Raymond, E.; Faivre, S.; Woynarowski, J.M.; Chaney, S.G. Oxaliplatin: Mechanism of action and antineoplastic activity. Semin. Oncol. 1998, 25, 4–12. [Google Scholar] [PubMed]
- Graham, J.; Mushin, M.; Kirkpatrick, P. Oxaliplatin. Nat. Rev. Drug Discov. 2004, 3, 11–12. [Google Scholar] [CrossRef]
- de Gramont, A.; Figer, A.; Seymour, M.; Homerin, M.; Hmissi, A.; Cassidy, J.; Boni, C.; Cortes-Funes, H.; Cervantes, A.; Freyer, G.; et al. Leucovorin and fluorouracil with or without oxaliplatin as first-line treatment in advanced colorectal cancer. J. Clin. Oncol. 2000, 18, 2938–2947. [Google Scholar] [CrossRef]
- Tummala, S.; Gowthamarajan, K.; Satish Kumar, M.N.; Wadhwani, A. Oxaliplatin immuno hybrid nanoparticles for active targeting: An approach for enhanced apoptotic activity and drug delivery to colorectal tumors. Drug Deliv. 2016, 23, 1773–1787. [Google Scholar] [CrossRef] [Green Version]
- Handali, S.; Ramezani, Z.; Moghimipour, E.; Rezaei, M.; Dorkoosh, F.A. A novel method for the simultaneous determination of 5-fluorouracil and oxaliplatin in new biodegradable PHBV/PLGA nanoparticles. J. Iran. Chem. Soc. 2018, 16, 609–615. [Google Scholar] [CrossRef]
- Handali, S.; Moghimipour, E.; Rezaei, M.; Saremy, S.; Dorkoosh, F.A. Co-delivery of 5-fluorouracil and oxaliplatin in novel poly(3-hydroxybutyrate-co-3-hydroxyvalerate acid)/poly(lactic-co-glycolic acid) nanoparticles for colon cancer therapy. Int. J. Biol. Macromol. 2019, 124, 1299–1311. [Google Scholar] [CrossRef]
- Rezvantalab, S.; Drude, N.I.; Moraveji, M.K.; Guvener, N.; Koons, E.K.; Shi, Y.; Lammers, T.; Kiessling, F. PLGA-Based Nanoparticles in Cancer Treatment. Front. Pharmacol. 2018, 9, 1260. [Google Scholar] [CrossRef] [Green Version]
- Mahmoudi, M.; Sant, S.; Wang, B.; Laurent, S.; Sen, T. Superparamagnetic iron oxide nanoparticles (SPIONs): Development, surface modification and applications in chemotherapy. Adv. Drug Del. Rev. 2011, 63, 24–46. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Alvarez, F.; Caro, C.; Garcia-Garcia, G.; Garcia-Martin, M.L.; Arias, J.L. Engineering of stealth (maghemite/PLGA)/chitosan (core/shell)/shell nanocomposites with potential applications for combined MRI and hyperthermia against cancer. J. Mater. Chem. B 2021, 9, 4963–4980. [Google Scholar] [CrossRef] [PubMed]
- Eynali, S.; Khoei, S.; Khoee, S.; Esmaelbeygi, E. Evaluation of the cytotoxic effects of hyperthermia and 5-fluorouracil-loaded magnetic nanoparticles on human colon cancer cell line HT-29. Int. J. Hyperth. 2017, 33, 327–335. [Google Scholar] [CrossRef] [PubMed]
- Costa Lima, S.A.; Gaspar, A.; Reis, S.; Duraes, L. Multifunctional nanospheres for co-delivery of methotrexate and mild hyperthermia to colon cancer cells. Mater. Sci. Eng. C 2017, 75, 1420–1426. [Google Scholar] [CrossRef] [PubMed]
- Mishra, P.; Nayak, B.; Dey, R.K. PEGylation in anti-cancer therapy: An overview. Asian J. Pharm. Health Sci. 2016, 11, 337–348. [Google Scholar] [CrossRef] [Green Version]
- Babos, G.; Biro, E.; Meiczinger, M.; Feczko, T. Dual Drug Delivery of Sorafenib and Doxorubicin from PLGA and PEG-PLGA Polymeric Nanoparticles. Polymers 2018, 10, 895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, W.T.; Ryu, C.J. Cancer stem cell surface markers on normal stem cells. BMB Rep. 2017, 50, 285–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Name | Mean Diameter (nm) | PDI * | Zeta Potential (mV) |
|---|---|---|---|
| IO-OA | 20 ± 4 | 0.13 ± 0.07 | −36 ± 4 |
| PLGA | 245 ± 86 | 0.05 ± 0.03 | −9 ± 4 |
| PLGA_IO-OA | 220 ± 89 | 0.08 ± 0.02 | −8 ± 6 |
| PLGA_OXA | 190 ± 59 | 0.06 ± 0.003 | −5 ± 6 |
| PLGA_OXA_Ab | 285 ± 74 | 0.191 ± 0.026 | −3 ± 4 |
| PLGA_IO-OA_OXA | 207 ± 70 | 0.08 ± 0.006 | −8 ± 7 |
| PLGA_IO-OA_OXA_Ab | 223 ± 70 | 0.136 ± 0.026 | −4 ± 5 |
| PEGylated PLGA | 146 ± 50 | 0.1 ± 0.01 | 0 ± 4 |
| PEGylated PLGA_OXA | 130 ± 51 | 0.2 ± 0.009 | 5 ± 4 |
| PEGylated PLGA_IO-OA | 217 ± 160 | 0.2 ± 0.014 | 2 ± 4 |
| PEGylated PLGA_IO-OA_OXA | 185 ± 119 | 0.2 ± 0.004 | 4 ± 3 |
| IC50 ± SD [µg·mL−1] of the Nanoparticles | ||
|---|---|---|
| Cell line | PLGA_OXA | PLGA_IO_OXA |
| CaCo-2 | 255 ± 11 | 560 ± 62 |
| MRC-5 | 525 ± 12 | 325 ± 31 |
| IC50 ± SD [µg·mL−1] of the Oxaliplatin | |||
|---|---|---|---|
| Cell line | PLGA_OXA | PLGA_IO_OXA | OXA |
| CaCo-2 | 5.66 ± 0.24 | 12.44 ± 1.38 | 8.35 ± 0.48 |
| MRC-5 | 11.66 ± 0.27 | 7.22 ± 0.69 | 14.30 ± 1.26 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zumaya, A.L.V.; Rimpelová, S.; Štějdířová, M.; Ulbrich, P.; Vilčáková, J.; Hassouna, F. Antibody Conjugated PLGA Nanocarriers and Superparmagnetic Nanoparticles for Targeted Delivery of Oxaliplatin to Cells from Colorectal Carcinoma. Int. J. Mol. Sci. 2022, 23, 1200. https://doi.org/10.3390/ijms23031200
Zumaya ALV, Rimpelová S, Štějdířová M, Ulbrich P, Vilčáková J, Hassouna F. Antibody Conjugated PLGA Nanocarriers and Superparmagnetic Nanoparticles for Targeted Delivery of Oxaliplatin to Cells from Colorectal Carcinoma. International Journal of Molecular Sciences. 2022; 23(3):1200. https://doi.org/10.3390/ijms23031200
Chicago/Turabian StyleZumaya, Alma Lucia Villela, Silvie Rimpelová, Markéta Štějdířová, Pavel Ulbrich, Jarmila Vilčáková, and Fatima Hassouna. 2022. "Antibody Conjugated PLGA Nanocarriers and Superparmagnetic Nanoparticles for Targeted Delivery of Oxaliplatin to Cells from Colorectal Carcinoma" International Journal of Molecular Sciences 23, no. 3: 1200. https://doi.org/10.3390/ijms23031200
APA StyleZumaya, A. L. V., Rimpelová, S., Štějdířová, M., Ulbrich, P., Vilčáková, J., & Hassouna, F. (2022). Antibody Conjugated PLGA Nanocarriers and Superparmagnetic Nanoparticles for Targeted Delivery of Oxaliplatin to Cells from Colorectal Carcinoma. International Journal of Molecular Sciences, 23(3), 1200. https://doi.org/10.3390/ijms23031200