
Soluble Endoglin Stimulates Inflammatory and Angiogenic Responses in Microglia That Are Associated with Endothelial Dysfunction

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
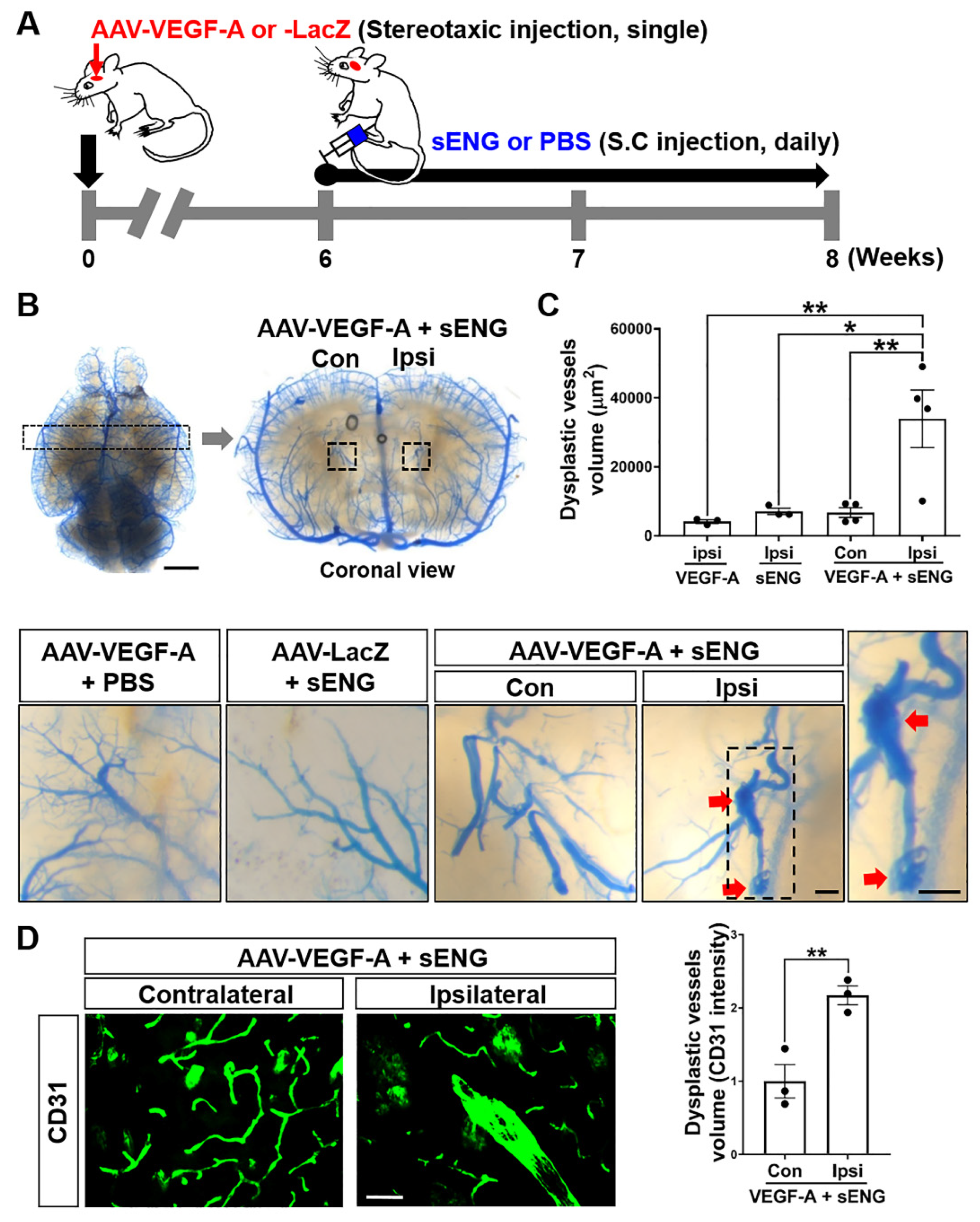
2.1. Soluble ENG Induced Vascular Dysplasia in the Mouse Brain with VEGF-A Overexpression
2.2. Microglia with Enhanced Inflammasome Marker Were Recruited around sENG/VEGF-A-Induced Dysplastic Vessels in Mice
2.3. Soluble ENG Induced Expression of VEGF-A and Inflammatory/Inflammasome Markers in Cultured Microglia
2.4. Soluble ENG-Stimulated Microglia Modulated Angiogenic Factors in Endothelial Cells
2.5. Soluble ENG-Stimulated Microglia Modulated Endothelial Cell Functions
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Treatment with AAV1-VEGF-A and sENG in Mice
4.3. Systemic Latex Vascular Casting and Analysis
4.4. Immunofluorescence Staining and Analysis
4.5. Cell Culture and sENG Treatment
4.6. EC Function Assays
4.7. Quantitative Real-Time PCR
| VEGF-A | (F) | CTCACCAAAGCCAGCACATA |
| (R) | AATGCTTTCTCCGCTCTGAA | |
| TGFβ | (F) | GCCCTTCCTGCTCCTCATG |
| (R) | CCGCACACAGCAGTTCTTCTC | |
| Notch-1 | (F) | CTGAGGCAAGGATTGGAGTC |
| (R) | GAATGGAGGTAGGTGCGAAG | |
| IL-6 | (F) | TGGTACTCCAGAAGACCAGAGG |
| (R) | AACGATGATGCACTTGCAGA | |
| TNF-α | (F) | ATGGCCTCCCTCTCATCAGT |
| (R) | TTTGCTACGACGTGGGCTAC | |
| MMP-9 | (F) | GCCGACTTTTGTGGTCTTCC |
| (R) | TACAAGTATGCCTCTGCCAGC | |
| NLRP3 | (F) | CTTCTAGCTTCTGCCGTGGTCTCT |
| (R) | CGAAGCAGCATTGATGGGACA | |
| Caspase-1 | (F) | GTACACGTCTTGCCCTCATTATCTG |
| (R) | TTTCACCTCTTTCACCATCTCCAG | |
| ASC | (F) | CTGAGCAGCTGCAAACGACTAAA |
| (R) | CTTCTGTGACCCTGGCAATGAGT | |
| IL-1β | (F) | CAACCAACAAGTGATATTCTCCATG |
| (R) | GATCCACACTCTCCAGCTGCA | |
| GAPDH | (F) | GGAGTCAACGGATTTGGTCG |
| (R) | GGAATCATATTGGAACATGTAAACC |
4.8. Western Blotting
4.9. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lawton, M.T.; Rutledge, W.C.; Kim, H.; Stapf, C.; Whitehead, K.J.; Li, D.Y.; Krings, T.; terBrugge, K.; Kondziolka, D.; Morgan, M.K.; et al. Brain arteriovenous malformations. Nat. Rev. Dis. Primers 2015, 1, 15008. [Google Scholar] [CrossRef]
- Solomon, R.A.; Connolly, E.S., Jr. Arteriovenous Malformations of the Brain. N. Engl. J. Med. 2017, 376, 1859–1866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bayrak-Toydemir, P.; McDonald, J.; Markewitz, B.; Lewin, S.; Miller, F.; Chou, L.S.; Gedge, F.; Tang, W.; Coon, H.; Mao, R. Genotype-phenotype correlation in hereditary hemorrhagic telangiectasia: Mutations and manifestations. Am. J. Med. Genet. A 2006, 140, 463–470. [Google Scholar] [CrossRef] [PubMed]
- Govani, F.S.; Shovlin, C.L. Hereditary haemorrhagic telangiectasia: A clinical and scientific review. Eur. J. Hum. Genet. 2009, 17, 860–871. [Google Scholar] [CrossRef] [Green Version]
- Sabba, C.; Pasculli, G.; Lenato, G.M.; Suppressa, P.; Lastella, P.; Memeo, M.; Dicuonzo, F.; Guant, G. Hereditary hemorrhagic telangiectasia: Clinical features in ENG and ALK1 mutation carriers. J. Thromb. Haemost. 2007, 5, 1149–1157. [Google Scholar] [CrossRef]
- Rossi, E.; Bernabeu, C.; SSmadja, D.M. Endoglin as an Adhesion Molecule in Mature and Progenitor Endothelial Cells: A Function Beyond TGF-beta. Front. Med. 2019, 6, 10. [Google Scholar] [CrossRef]
- Albinana, V.; Zafra, M.P.; Colau, J.; Zarrabeitia, R.; Recio-Poveda, L.; Olavarrieta, L.; Perez-Perez, J.; Botella, L.M. Mutation affecting the proximal promoter of Endoglin as the origin of hereditary hemorrhagic telangiectasia type 1. BMC Med. Genet. 2017, 18, 20. [Google Scholar] [CrossRef] [Green Version]
- Choi, E.J.; Chen, W.; Jun, K.; Arthur, H.M.; Young, W.L.; Su, H. Novel brain arteriovenous malformation mouse models for type 1 hereditary hemorrhagic telangiectasia. PLoS ONE 2014, 9, e88511. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Hao, Q.; Kim, H.; Su, H.; Letarte, M.; Karumanchi, S.A.; Lawton, M.T.; Barbaro, N.M.; Yang, G.Y.; Young, W.L. Soluble endoglin modulates aberrant cerebral vascular remodeling. Ann. Neurol. 2009, 66, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Mouchtouris, N.; Jabbour, P.M.; Starke, R.M.; Hasan, D.M.; Zanaty, M.; Theofanis, T.; Ding, D.; Tjoumakaris, S.I.; Dumont, A.S.; Ghobrial, G.M.; et al. Biology of cerebral arteriovenous malformations with a focus on inflammation. J. Cereb. Blood Flow. Metab. 2015, 35, 167–175. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.; Han, Z.; Degos, V.; Shen, F.; Choi, E.J.; Sun, Z.; Kang, S.; Wong, M.; Zhu, W.; Zhan, L.; et al. Persistent infiltration and pro-inflammatory differentiation of monocytes cause unresolved inflammation in brain arteriovenous malformation. Angiogenesis 2016, 19, 451–461. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Zhu, W.; Bollen, A.W.; Lawton, M.T.; Barbaro, N.M.; Dowd, C.F.; Hashimoto, T.; Yang, G.Y.; Young, W.L. Evidence of inflammatory cell involvement in brain arteriovenous malformations. Neurosurgery 2008, 62, 1340–134; discussion 1349–1350. [Google Scholar] [CrossRef] [Green Version]
- Germans, M.R.; Sun, W.; Sebok, M.; Keller, A.; Regli, L. Molecular Signature of Brain Arteriovenous Malformation Hemorrhage: A Systematic Review. World Neurosurg. 2022, 157, 143–151. [Google Scholar] [CrossRef]
- Varejckova, M.; Gallardo-Vara, E.; Vicen, M.; Vitverova, B.; Fikrova, P.; Dolezelova, E.; Rathouska, J.; Prasnicka, A.; Blazickova, K.; Micuda, S.; et al. Soluble endoglin modulates the pro-inflammatory mediators NF-kappaB and IL-6 in cultured human endothelial cells. Life Sci. 2017, 175, 52–60. [Google Scholar] [CrossRef]
- Guo, Y.; Tihan, T.; Kim, H.; Hess, C.; Lawton, M.T.; Young, W.L.; Zhao, Y.; Su, H. Distinctive distribution of lymphocytes in unruptured and previously untreated brain arteriovenous malformation. Neuroimmunol. Neuroinflamm. 2014, 1, 147–152. [Google Scholar] [CrossRef] [Green Version]
- Park, E.S.; Kim, S.; Huang, S.; Yoo, J.Y.; Korbelin, J.; Lee, T.J.; Kaur, B.; Dash, P.K.; Chen, P.R.; Kim, E. Selective Endothelial Hyperactivation of Oncogenic KRAS Induces Brain Arteriovenous Malformations in Mice. Ann. Neurol. 2021, 89, 926–941. [Google Scholar] [CrossRef] [PubMed]
- Eggen, B.J.; Raj, D.; Hanisch, U.K.; Boddeke, H.W. Microglial phenotype and adaptation. J. Neuroimmune Pharmacol. 2013, 8, 807–823. [Google Scholar] [CrossRef] [PubMed]
- Kettenmann, H.; Hanisch, U.K.; Noda, M.; Verkhratsky, A. Physiology of microglia. Physiol. Rev. 2011, 91, 461–553. [Google Scholar] [CrossRef] [PubMed]
- Kreutzberg, G.W. Microglia: A sensor for pathological events in the CNS. Trends Neurosci. 1996, 19, 312–318. [Google Scholar] [CrossRef]
- Shirasuna, K.; Karasawa, T.; Takahashi, M. Role of the NLRP3 Inflammasome in Preeclampsia. Front Endocrinol. 2020, 11, 80. [Google Scholar] [CrossRef] [PubMed]
- Jabbour, M.N.; Elder, J.B.; Samuelson, C.G.; Khashabi, S.; Hofman, F.M.; Giannotta, S.L.; Liu, C.Y. Aberrant angiogenic characteristics of human brain arteriovenous malformation endothelial cells. Neurosurgery 2009, 64, 139–146; discussion 146–148. [Google Scholar] [CrossRef]
- Ding, X.; Gu, R.; Zhang, M.; Ren, H.; Shu, Q.; Xu, G.; Wu, H. Microglia enhanced the angiogenesis, migration and proliferation of co-cultured RMECs. BMC Ophthal. Mol. 2018, 18, 249. [Google Scholar] [CrossRef]
- Nikolaev, S.I.; Vetiska, S.; Bonilla, X.; Boudreau, E.; Jauhiainen, S.; Rezai Jahromi, B.; Khyzha, N.; DiStefano, P.V.; Suutarinen, S.; Kiehl, T.R.; et al. Somatic Activating KRAS Mutations in Arteriovenous Malformations of the Brain. N. Engl. J. Med. 2018, 378, 250–261. [Google Scholar] [CrossRef]
- Cai, J.; Pardali, E.; Sanchez-Duffhues, G.; ten Dijke, P. BMP signaling in vascular diseases. FEBS Lett. 2012, 586, 1993–2002. [Google Scholar] [CrossRef] [PubMed]
- Pardali, E.; Goumans, M.J.; ten Dijke, P. Signaling by members of the TGF-beta family in vascular morphogenesis and disease. Trends Cell Biol. 2010, 20, 556–567. [Google Scholar] [CrossRef]
- Pardali, E.; Ten Dijke, P. TGFbeta signaling and cardiovascular diseases. Int. J. Biol. Sci. 2012, 8, 195–213. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Yao, J.; Radparvar, M.; Blazquez-Medela, A.M.; Guihard, P.J.; Jumabay, M.; Bostrom, K.I. Reducing Jagged 1 and 2 levels prevents cerebral arteriovenous malformations in matrix Gla protein deficiency. Proc. Natl. Acad. Sci. USA 2013, 110, 19071–19076. [Google Scholar] [CrossRef] [Green Version]
- Barbosa Do Prado, L.; Han, C.; Oh, S.P.; Su, H. Recent Advances in Basic Research for Brain Arteriovenous Malformation. Int. J. Mol. Sci. 2019, 20, 5324. [Google Scholar] [CrossRef] [Green Version]
- ZhuGe, Q.; Zhong, M.; Zheng, W.; Yang, G.Y.; Mao, X.; Xie, L.; Chen, G.; Chen, Y.; Lawton, M.T.; Young, W.L.; et al. Notch-1 signalling is activated in brain arteriovenous malformations in humans. Brain 2009, 132, 3231–3241. [Google Scholar] [CrossRef] [Green Version]
- Darby, S.; Sahadevan, K.; Khan, M.M.; Robson, C.N.; Leung, H.Y.; Gnanapragasam, V.J. Loss of Sef (similar expression to FGF) expression is associated with high grade and metastatic prostate cancer. Oncogene 2006, 25, 4122–4127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Girondel, C.; Levesque, K.; Langlois, M.J.; Pasquin, S.; Saba-El-Leil, M.K.; Rivard, N.; Friesel, R.; Servant, M.J.; Gauchat, J.F.; Lesage, S.; et al. Loss of interleukin-17 receptor D promotes chronic inflammation-associated tumorigenesis. Oncogene 2021, 40, 452–464. [Google Scholar] [CrossRef] [PubMed]
- Ku, Y.H.; Cho, B.J.; Kim, M.J.; Lim, S.; Park, Y.J.; Jang, H.C.; Choi, S.H. Rosiglitazone increases endothelial cell migration and vascular permeability through Akt phosphorylation. BMC Pharmacol. Toxicol. 2017, 18, 62. [Google Scholar] [CrossRef] [PubMed]
- Lamalice, L.; Le Boeuf, F.; Huot, J. Endothelial cell migration during angiogenesis. Circ. Res. 2007, 100, 782–794. [Google Scholar] [CrossRef]
- Lusis, A.J. Atherosclerosis. Nature 2000, 407, 233–241. [Google Scholar] [CrossRef]
- Goumans, M.J.; Lebrin, F.; Valdimarsdottir, G. Controlling the angiogenic switch: A balance between two distinct TGF-b receptor signaling pathways. Trends CardioVasc. Med. 2003, 13, 301–307. [Google Scholar] [CrossRef]
- ten Dijke, P.; Arthur, H.M. Extracellular control of TGFbeta signalling in vascular development and disease. Nat. Rev. Mol. Cell Biol. 2007, 8, 857–869. [Google Scholar] [CrossRef] [PubMed]
- McAllister, K.A.; Grogg, K.M.; Johnson, D.W.; Gallione, C.J.; Baldwin, M.A.; Jackson, C.E.; Helmbold, E.A.; Markel, D.S.; McKinnon, W.C.; Murrell, J.; et al. Endoglin, a TGF-beta binding protein of endothelial cells, is the gene for hereditary haemorrhagic telangiectasia type 1. Nat. Genet. 1994, 8, 345–351. [Google Scholar] [CrossRef] [Green Version]
- Satomi, J.; Mount, R.J.; Toporsian, M.; Paterson, A.D.; Wallace, M.C.; Harrison, R.V.; Letarte, M. Cerebral vascular abnormalities in a murine model of hereditary hemorrhagic telangiectasia. Stroke 2003, 34, 783–789. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Pan, C.C.; Bloodworth, J.C.; Nixon, A.B.; Theuer, C.; Hoyt, D.G.; Lee, N.Y. Antibody-directed coupling of endoglin and MMP-14 is a key mechanism for endoglin shedding and deregulation of TGF-beta signaling. Oncogene 2014, 33, 3970–3979. [Google Scholar] [CrossRef]
- Hashimoto, T.; Wen, G.; Lawton, M.T.; Boudreau, N.J.; Bollen, A.W.; Yang, G.Y.; Barbaro, N.M.; Higashida, R.T.; Dowd, C.F.; Halbach, V.V.; et al. Abnormal expression of matrix metalloproteinases and tissue inhibitors of metalloproteinases in brain arteriovenous malformations. Stroke 2003, 34, 925–931. [Google Scholar] [CrossRef]
- Hawinkels, L.J.; Kuiper, P.; Wiercinska, E.; Verspaget, H.W.; Liu, Z.; Pardali, E.; Sier, C.F.; ten Dijke, P. Matrix metalloproteinase-14 (MT1-MMP)-mediated endoglin shedding inhibits tumor angiogenesis. Cancer Res. 2010, 70, 4141–4150. [Google Scholar] [CrossRef] [Green Version]
- Rathouska, J.; Jezkova, K.; Nemeckova, I.; Nachtigal, P. Soluble endoglin, hypercholesterolemia and endothelial dysfunction. Atherosclerosis 2015, 243, 383–388. [Google Scholar] [CrossRef] [PubMed]
- Venkatesha, S.; Toporsian, M.; Lam, C.; Hanai, J.; Mammoto, T.; Kim, Y.M.; Bdolah, Y.; Lim, K.H.; Yuan, H.T.; Libermann, T.A.; et al. Soluble endoglin contributes to the pathogenesis of preeclampsia. Nat. Med. 2006, 12, 642–649. [Google Scholar] [CrossRef]
- Lawera, A.; Tong, Z.; Thorikay, M.; Redgrave, R.E.; Cai, J.; van Dinther, M.; Morrell, N.W.; Afink, G.B.; Charnock-Jones, D.S.; Arthur, H.M.; et al. Role of soluble endoglin in BMP9 signaling. Proc. Natl. Acad. Sci. USA 2019, 116, 17800–17808. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Novoa, J.M.; Bernabeu, C. The physiological role of endoglin in the cardiovascular system. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, H959–H974. [Google Scholar] [CrossRef] [Green Version]
- Cheng, P.; Ma, L.; Shaligram, S.; Walker, E.J.; Yang, S.T.; Tang, C.; Zhu, W.; Zhan, L.; Li, Q.; Zhu, X.; et al. Effect of elevation of vascular endothelial growth factor level on exacerbation of hemorrhage in mouse brain arteriovenous malformation. J. Neurosurg. 2019, 132, 1566–1573. [Google Scholar] [CrossRef] [Green Version]
- Juraskova, B.; Andrys, C.; Holmerova, I.; Solichova, D.; Hrnciarikova, D.; Vankova, H.; Vasatko, T.; Krejsek, J. Transforming growth factor beta and soluble endoglin in the healthy senior and in Alzheimer’s disease patients. J. Nutr. Health Aging 2010, 14, 758–761. [Google Scholar] [CrossRef] [PubMed]
- Saita, E.; Miura, K.; Suzuki-Sugihara, N.; Miyata, K.; Ikemura, N.; Ohmori, R.; Ikegami, Y.; Kishimoto, Y.; Kondo, K.; Momiyama, Y. Plasma Soluble Endoglin Levels Are Inversely Associated With the Severity of Coronary Atherosclerosis-Brief Report. Arterioscler Thromb. Vasc. Biol. 2017, 37, 49–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blazquez-Medela, A.M.; Garcia-Ortiz, L.; Gomez-Marcos, M.A.; Recio-Rodriguez, J.I.; Sanchez-Rodriguez, A.; Lopez-Novoa, J.M.; Martinez-Salgado, C. Increased plasma soluble endoglin levels as an indicator of cardiovascular alterations in hypertensive and diabetic patients. BMC Med. 2010, 8, 86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallardo-Vara, E.; Tual-Chalot, S.; Botella, L.M.; Arthur, H.M.; Bernabeu, C. Soluble endoglin regulates expression of angiogenesis-related proteins and induction of arteriovenous malformations in a mouse model of hereditary hemorrhagic telangiectasia. Dis. Model Mech. 2018, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dudiki, T.; Meller, J.; Mahajan, G.; Liu, H.; Zhevlakova, I.; Stefl, S.; Witherow, C.; Podrez, E.; Kothapalli, C.R.; Byzova, T.V. Microglia control vascular architecture via a TGFbeta1 dependent paracrine mechanism linked to tissue mechanics. Nat. Commun. 2020, 11, 986. [Google Scholar] [CrossRef] [Green Version]
- Cheadle, L.; Rivera, S.A.; Phelps, J.S.; Ennis, K.A.; Stevens, B.; Burkly, L.C.; Lee, W.A.; Greenberg, M.E. Sensory Experience Engages Microglia to Shape Neural Connectivity through a Non-Phagocytic Mechanism. Neuron 2020, 108, 451–468.e459. [Google Scholar] [CrossRef]
- Hagemeyer, N.; Hanft, K.M.; Akriditou, M.A.; Unger, N.; Park, E.S.; Stanley, E.R.; Staszewski, O.; Dimou, L.; Prinz, M. Microglia contribute to normal myelinogenesis and to oligodendrocyte progenitor maintenance during adulthood. Acta Neuropathol. 2017, 134, 441–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chitu, V.; Biundo, F.; Shlager, G.G.L.; Park, E.S.; Wang, P.; Gulinello, M.E.; Gokhan, S.; Ketchum, H.C.; Saha, K.; DeTure, M.A.; et al. Microglial Homeostasis Requires Balanced CSF-1/CSF-2 Receptor Signaling. Cell Rep. 2020, 30, 3004–3019.e3005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diaz-Aparicio, I.; Paris, I.; Sierra-Torre, V.; Plaza-Zabala, A.; Rodriguez-Iglesias, N.; Marquez-Ropero, M.; Beccari, S.; Huguet, P.; Abiega, O.; Alberdi, E.; et al. Microglia Actively Remodel Adult Hippocampal Neurogenesis through the Phagocytosis Secretome. J. Neurosci. 2020, 40, 1453–1482. [Google Scholar] [CrossRef]
- Sierra-Torre, V.; Plaza-Zabala, A.; Bonifazi, P.; Abiega, O.; Diaz-Aparicio, I.; Tegelberg, S.; Lehesjoki, A.E.; Valero, J.; Sierra, A. Microglial phagocytosis dysfunction in the dentate gyrus is related to local neuronal activity in a genetic model of epilepsy. Epilepsia 2020, 61, 2593–2608. [Google Scholar] [CrossRef] [PubMed]
- Willis, E.F.; MacDonald, K.P.A.; Nguyen, Q.H.; Garrido, A.L.; Gillespie, E.R.; Harley, S.B.R.; Bartlett, P.F.; Schroder, W.A.; Yates, A.G.; Anthony, D.C.; et al. Repopulating Microglia Promote Brain Repair in an IL-6-Dependent Manner. Cell 2020, 180, 833–846.e816. [Google Scholar] [CrossRef] [PubMed]
- Bisht, K.; Okojie, K.A.; Sharma, K.; Lentferink, D.H.; Sun, Y.Y.; Chen, H.R.; Uweru, J.O.; Amancherla, S.; Calcuttawala, Z.; Campos-Salazar, A.B.; et al. Capillary-associated microglia regulate vascular structure and function through PANX1-P2RY12 coupling in mice. Nat. Commun. 2021, 12, 5289. [Google Scholar] [CrossRef]
- Iadecola, C. The Neurovascular Unit Coming of Age: A Journey through Neurovascular Coupling in Health and Disease. Neuron 2017, 96, 17–42. [Google Scholar] [CrossRef] [Green Version]
- Kisler, K.; Nikolakopoulou, A.M.; Zlokovic, B.V. Microglia have a grip on brain microvasculature. Nat. Commun. 2021, 12, 5290. [Google Scholar] [CrossRef]
- Walker, D.G.; Lue, L.F.; Beach, T.G.; Tooyama, I. Microglial Phenotyping in Neurodegenerative Disease Brains: Identification of Reactive Microglia with an Antibody to Variant of CD105/Endoglin. Cells 2019, 8, 766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gougos, A.; Letarte, M. Primary structure of endoglin, an RGD-containing glycoprotein of human endothelial cells. J. Biol. Chem. 1990, 265, 8361–8364. [Google Scholar] [CrossRef]
- Lastres, P.; Bellon, T.; Cabanas, C.; Sanchez-Madrid, F.; Acevedo, A.; Gougos, A.; Letarte, M.; Bernabeu, C. Regulated expression on human macrophages of endoglin, an Arg-Gly-Asp-containing surface antigen. Eur. J. Immunol. 1992, 22, 393–397. [Google Scholar] [CrossRef]
- O’Connell, P.J.; McKenzie, A.; Fisicaro, N.; Rockman, S.P.; Pearse, M.J.; d’Apice, A.J. Endoglin: A 180-kD endothelial cell and macrophage restricted differentiation molecule. Clin. Exp. Immunol. 1992, 90, 154–159. [Google Scholar] [CrossRef] [PubMed]
- Timis, T.L.; Florian, I.A.; Susman, S.; Florian, I.S. Involvement of Microglia in the Pathophysiology of Intracranial Aneurysms and Vascular Malformations-A Short Overview. Int. J. Mol. Sci. 2021, 22, 6141. [Google Scholar] [CrossRef]
- Al-Shahi, R.; Fang, J.S.; Lewis, S.C.; Warlow, C.P. Prevalence of adults with brain arteriovenous malformations: A community based study in Scotland using capture-recapture analysis. J. Neurol. Neurosurg. Psychiatry 2002, 73, 547–551. [Google Scholar] [CrossRef] [Green Version]
- Kandai, S.; Abdullah, M.S.; Naing, N.N. Angioarchitecture of brain arteriovenous malformations and the risk of bleeding: An analysis of patients in northeastern malaysia. Malays J. Med. Sci. 2010, 17, 44–48. [Google Scholar]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, E.S.; Kim, S.; Yao, D.C.; Savarraj, J.P.J.; Choi, H.A.; Chen, P.R.; Kim, E. Soluble Endoglin Stimulates Inflammatory and Angiogenic Responses in Microglia That Are Associated with Endothelial Dysfunction. Int. J. Mol. Sci. 2022, 23, 1225. https://doi.org/10.3390/ijms23031225
Park ES, Kim S, Yao DC, Savarraj JPJ, Choi HA, Chen PR, Kim E. Soluble Endoglin Stimulates Inflammatory and Angiogenic Responses in Microglia That Are Associated with Endothelial Dysfunction. International Journal of Molecular Sciences. 2022; 23(3):1225. https://doi.org/10.3390/ijms23031225
Chicago/Turabian StylePark, Eun S., Sehee Kim, Derek C. Yao, Jude P. J. Savarraj, Huimahn Alex Choi, Peng Roc Chen, and Eunhee Kim. 2022. "Soluble Endoglin Stimulates Inflammatory and Angiogenic Responses in Microglia That Are Associated with Endothelial Dysfunction" International Journal of Molecular Sciences 23, no. 3: 1225. https://doi.org/10.3390/ijms23031225
APA StylePark, E. S., Kim, S., Yao, D. C., Savarraj, J. P. J., Choi, H. A., Chen, P. R., & Kim, E. (2022). Soluble Endoglin Stimulates Inflammatory and Angiogenic Responses in Microglia That Are Associated with Endothelial Dysfunction. International Journal of Molecular Sciences, 23(3), 1225. https://doi.org/10.3390/ijms23031225