
Medicated Scaffolds Prepared with Hydroxyapatite/Streptomycin Nanoparticles Encapsulated into Polylactide Microfibers

 , , , ,
, , , ,  and
and
Abstract
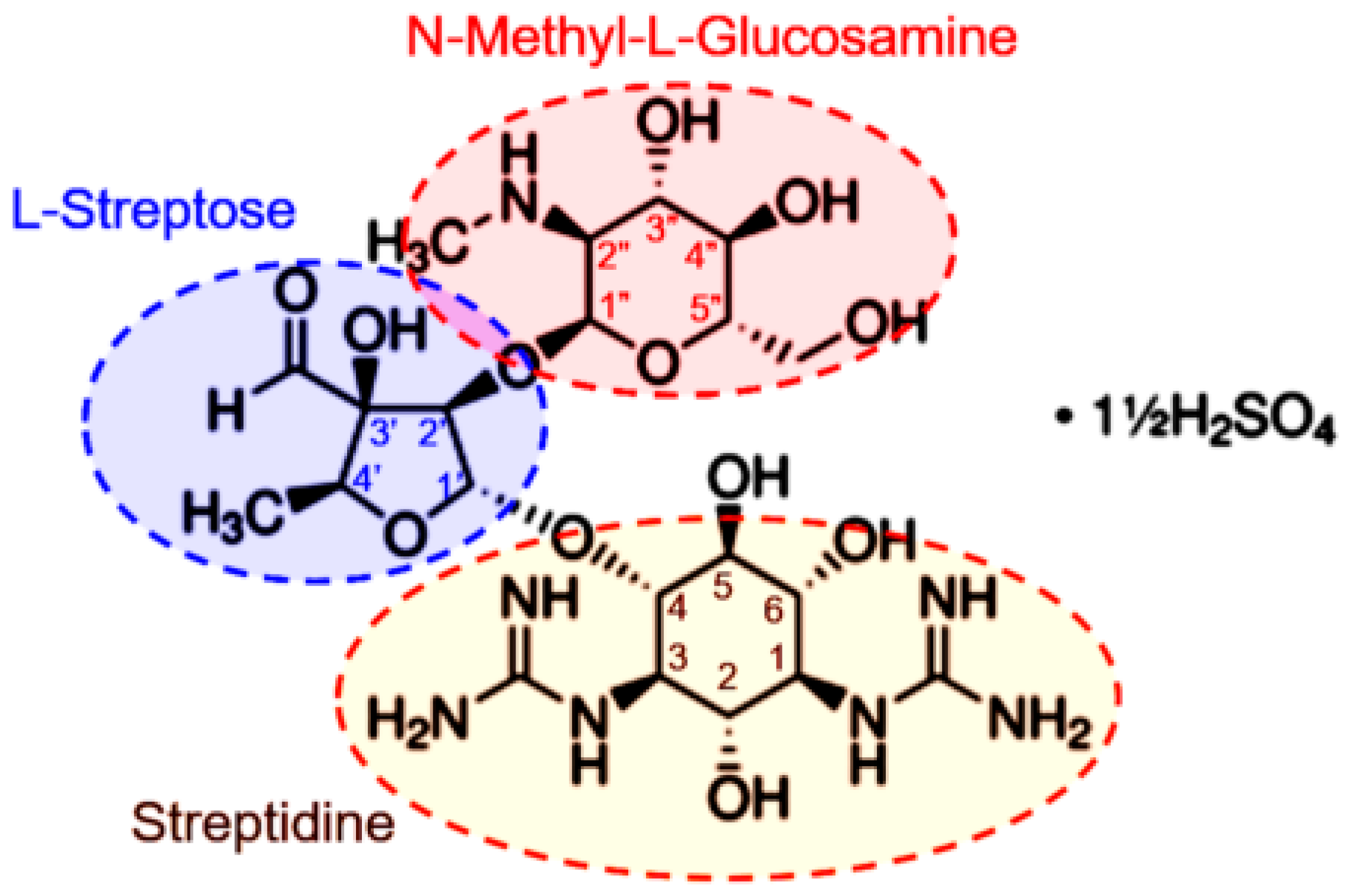
:1. Introduction
2. Results and Discussion
2.1. Characterization of Hydroxyapatite Particles
2.2. Preparation of Electrospun PLA Matrices Loaded with Nanoparticles Incorporating STR
3. Materials and Methods
3.1. Materials
3.2. Synthesis of Amorphous (ACP) and Crystalline (cHAp) Hydroxyapatite
3.3. Hydroxyapatite Nanoparticles with Encapsulated STR
3.4. Adsorption of STR onto ACP and cHAp Nanoparticles
3.5. Electrospun PLA Microfibers Loaded with HAp Nanoparticles
3.6. Characterization
3.6.1. Scanning Electronic Microscopy (SEM)
3.6.2. UV-Vis Spectroscopy Analysis
3.6.3. Fourier Transform Infrared (FTIR) Spectroscopy
3.6.4. Dynamic Light Scattering (DLS) and Z-Potential
3.6.5. Wettability
3.6.6. Thermal Degradation
3.6.7. X-ray Diffraction (XRD)
3.7. Encapsulation or Adsorption Efficiency
3.8. Release of Streptomycin from Loaded Nanoparticles
3.9. Antibacterial Activity
3.10. Cytotoxicity Evaluation
3.11. FTIR Microspectroscopy
3.12. FTIR Microspectra Data Analysis
3.13. Statistical Analysis
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jarcho, M.; Kay, J.F.; Gumar, K.I.; Doremus, R.H.; Drobeck, H.P. Tissue, cellular and subcellular events at a bone ceramic hydroxyapatite interface. J. Biosci. Bioeng. 1977, 1, 79–92. [Google Scholar]
- Bagambisa, F.B.; Joos, U. Preliminary studies on the phenomenological behaviors of osteoblasts cultured on hydroxyapatite ceramics. Biomaterials 1990, 11, 50–56. [Google Scholar] [CrossRef]
- Wang, M. Developing bioactive composite materials for tissue replacement. Biomaterials 2003, 23, 2133–2151. [Google Scholar] [CrossRef]
- Chen, Q.Z.; Wong, C.T.; Lu, W.W.; Cheung, K.M.C.; Leong, J.C.Y.; Luk, K.D.K. Strengthening mechanisms of bone-bonding to crystalline hydroxyapatite in vivo. Biomaterials 2004, 25, 4243–4254. [Google Scholar] [CrossRef]
- Jaffe, W.L.; Scott, D.F. Total hip arthroplasty with hydroxyapatite-coated prosthesis. J. Bone Jt. Surg Am. 1996, 78, 1918–1934. [Google Scholar] [CrossRef]
- Oonishi, H. Orthopedic applications of hydroxyapatite. Biomaterials 1991, 12, 171–178. [Google Scholar] [CrossRef]
- Dorozhkin, S.V. Multiphasic calcium orthophosphate (CaPO4) bioceramics and their biomedical applications. Ceram. Int 2016, 42, 6529–6554. [Google Scholar] [CrossRef]
- Santos, M.H.; de Oliveira, M.; de Freitas Souza, P.; Mansur, H.S.; Vasconcelos, W.L. Synthesis control and characterization of hydroxyapatite prepared by wet precipitation process. Mater. Res. 2004, 7, 625–630. [Google Scholar] [CrossRef]
- Manuel, C.M.; Ferraz, M.P.; Monteiro, F.J. Nanoapatite and microporous structures of hydroxyapatite. In Proceedings of the 17th European Society of Biomaterials, Barcelona, Spain, 11–15 September 2002; Volume 153, p. 17. [Google Scholar]
- Chai, C.S.; Ben-Nissan, B. Bioactive nanocrystalline sol-gel hydroxyapatite coatings. J. Mater. Sci. Mater. Med. 1999, 10, 465–469. [Google Scholar] [CrossRef]
- Manafi, S.A.; Joughehdoust, S. Synthesis of hydroxyapatite nanostructure by hydrothermal condition for biomedical application. Iran. J. Pharm. Sci. 2009, 5, 89–94. [Google Scholar]
- Kimura, I. Synthesis of hydroxyapatite by interfacial reaction in a multiple emulsion. Adv. Mater Sci Eng. 2007, 2007, 071284. [Google Scholar] [CrossRef] [Green Version]
- Tas, A.C. Synthesis of biomimetic Ca-hydroxyapatite powders at 37 degrees C in synthetic body fluids. Biomaterials 2000, 21, 1429–1438. [Google Scholar] [PubMed]
- Thamaraiselvi, T.V.; Prabakaran, K.; Rajeswari, S. Synthsis of hydroxyapatite that mimic bone mineralogy. Trends Biomater Artif Org. 2006, 19, 81–83. [Google Scholar]
- Zhang, C.; Yang, J.; Quan, Z.; Yang, P.; Li, C.; Hou, Z.; Lin, J. Hydroxyapatite nano- and microcrystals with multiform morphologies: Controllable synthesis and luminescence properties. Cryst Growth Des. 2009, 9, 2725–2733. [Google Scholar] [CrossRef]
- Allen, T.M.; Cullis, P.R. Drug delivery systems: Entering the mainstream. Science 2004, 303, 1818–1822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartus, R.T.; Tracy, M.A.; Emerich, D.F.; Zale, S. Sustained delivery of proteins for novel therapeutic products. Science 1998, 281, 1161–1162. [Google Scholar] [CrossRef] [PubMed]
- Couvreur, P.; Puisieux, F. Nano- and microparticles for the delivery of polypeptides and proteins. Adv. Drug Deliv. Rev. 1993, 10, 141–162. [Google Scholar] [CrossRef]
- Langer, R. New methods of drug delivery. Science 1990, 249, 1527–1533. [Google Scholar] [CrossRef]
- Thomas, S.C.; Sharma, H.; Mishra, P.K.; Talegaonkar, S. Ceramic nanoparticles: Fabrication methods and applications in drug delivery. Curr. Pharm. Des. 2015, 21, 6165–6188. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.C.; Sharma, H.; Rawat, P.; Verma, A.; Leekha, A.; Kumar, V.; Tyagi, A.; Gurjar, B.S.; Iqbal, Z.; Talegaonkar, S. Synergistic anticancer efficacy of bendamustine hydrochloride loaded bioactive hydroxyapatite nanoparticles: In-vitro, ex-vivo and in-vivo evaluation. Colloids Surf. B Biointerface 2016, 146, 852–860. [Google Scholar] [CrossRef] [PubMed]
- Vallet-Regí, M.; Balas, F.; Arcos, D. Mesoporous materials for drug delivery. Angew. Chem. Int. Ed. 2007, 46, 7548–7558. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Tanaka, Y.; Czernuszka, J.T. Encapsulation and release of a hydrophobic drug from hydroxyapatite coated liposomes. Biomaterials 2007, 28, 2687–2694. [Google Scholar] [CrossRef]
- Schatz, A.; Waksman, S.A. Effect of streptomycin and other antibiotic substances upon Mycobacterium tuberculosis and related organisms. Proc. Soc. Exp. Biol. Med. 1944, 57, 244–248. [Google Scholar] [CrossRef]
- Murray, J.F. A century of tuberculosis. Am. J. Respir Crit Care Med. 2004, 169, 1181–1186. [Google Scholar] [CrossRef]
- Schatz, A.; Bugle, E.; Waksman, S.A. Streptomycin, a substance exhibiting antibiotic activity against Gram-positive and Gram-negative bacteria. Proc. Soc. Exp. Biol. Med. 1944, 55, 66–69. [Google Scholar] [CrossRef]
- Ramirez, M.S.; Nikolaidis, N.; Tolmasky, M.E. Rise and dissemination of aminoglycoside resistance: The aac(6’)-Ib paradigm. Front. Microbiol. 2013, 4, 121. [Google Scholar] [CrossRef] [Green Version]
- Becker, B.; Cooper, M.A. Aminoglycoside antibiotics in the 21st century. ACS Chem. Biol. 2012, 8, 105–115. [Google Scholar] [CrossRef]
- Edson, R.S.; Terrell, C.L. The aminoglycosides. Mayo Clin. Proc. 1991, 66, 1158–1164. [Google Scholar] [CrossRef]
- Lamb, R.; Ozsvari, B.; Lisanti, C.L.; Tanowitz, H.B.; Howell, A.; Martinez-Outschoorn, U.E.; Sotgia, F.; Lisanti, M.P. Antibiotics that target mitochondria effectively eradicate cancer stem cells, across multiple tumor types: Treating cancer like an infectious disease. Oncotarget 2015, 6, 4569–4584. [Google Scholar] [CrossRef] [Green Version]
- Lamb, R.; Harrison, H.; Hulit, J.; Smith, D.L.; Lisanti, M.P.; Sotgia, F. Mitochondria as new therapeutic targets for erradicating cancer stem cells: Qualitative proteomics and functional validation via MCT1/2 inhibition. Oncotarget 2014, 5, 11029–11037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibaldi, M.; Feldman, S. Establishment of sink conditions in dissolution rate determinations. Theoretical considerations and application to nondisintegrating dosage forms. J. Pharm. Sci. 1967, 56, 1238–1242. [Google Scholar] [CrossRef]
- Wagner, J.G. Interpretation of percent dissolved-time plots derived from in vitro testing of conventional tablets and capsules. J. Pharm. Sci. 1969, 58, 1253–1257. [Google Scholar] [CrossRef]
- Higuchi, T. Rate of release of medicaments from ointment bases containing drugs in suspension. J. Pharm. Sci. 1961, 50, 874–875. [Google Scholar] [CrossRef] [PubMed]
- Higuchi, T. Mechanism of sustained-action medication. Theoretical analysis of rate of release of solid drugs dispersed in solid matrices. J. Pharm. Sci. 1963, 52, 1145–1149. [Google Scholar] [CrossRef]
- Llorens, E.; Calderón, S.; del Valle, L.J.; Puiggalí, J. Polybiguanide (PHMB) loaded in PLA scaffolds displaying high hydrophobic, biocompatibility and antibacterial properties. Mat. Sci. Eng. C-Mater. 2015, 50, 74–84. [Google Scholar] [CrossRef] [PubMed]
- Romero-Montero, A.; Aguirre-Díaz, I.S.; Puiggalí, J.; del Valle, L.J.; Gimeno, M. Self-assembly of supramolecular chemoenzymatic poly-L-phenylalanine. Polym. Chem. 2021, 12, 1199–1209. [Google Scholar] [CrossRef]
- Fawzy, A.; Abdallah, M.; Alqarni, N. Oxidative degradation of neomycin and streptomycin by cerium (IV) in sulphuric and perchloric acid solutions. J. Mol. Liq. 2020, 312, 113439. [Google Scholar] [CrossRef]
- Kuhlmann, I. The prophylactic use of antibiotics in cell culture. Cytotechnology 1995, 19, 95–105. [Google Scholar] [CrossRef]
- Heilman, D.H. Cytotoxicity of streptomycin and streptothricin. P Soc. Exp. Biol. Med. 1945, 60, 365–367. [Google Scholar] [CrossRef]
- Meneghel, J.; Passot, S.; Jamme, F.; Lefrançois, S.; Lieben, P.; Dumas, P.; Fonseca, F. FTIR micro-spectroscopy using synchrotron-based and thermal source-based radiation for probing live bacteria. Anal. Bioanal. Chem. 2020, 412, 7049–7061. [Google Scholar] [CrossRef] [PubMed]
- Zawlik, I.; Kaznowska, E.; Cebulski, J.; Kolodziej, M.; Depciuch, J.; Vongsvivut, J.; Cholewa, M. FPA-FTIR microspectroscopy for monitoring chemotherapy efficacy in triple-negative breast cancer. Sci. Rep. 2016, 6, 37333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zelig, U.; Kapelushnik, J.; Moreh, R.; Mordechai, S.; Nathan, I. Diagnosis of cell death by means of infrared spectroscopy. Biophys. J. 2009, 97, 2107–2114. [Google Scholar] [CrossRef] [Green Version]
- Gasparri, F.; Muzio, M. Monitoring of apoptosis of HL60 cells by Fourier-transform infrared spectroscopy. Biochem. J. 2003, 369, 239–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gioacchini, G.; Notarstefano, V.; Sereni, E.; Zacà, C.; Coticchio, G.; Giorgini, E.; Vaccari, L.; Carnevali, O.; Borini, A. Does the molecular and metabolic profile of human granulosa cells correlate with oocyte fate? New insights by Fourier transform infrared microspectroscopy analysis. Mol. Hum. Reprod. 2018, 24, 521–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Dai, W.; Liu, Z.; Liu, J.; Cheng, J.; Li, Y.; Li, X.; Hu, J.; Lü, J. Single-Cell Infrared Microspectroscopy Quantifies Dynamic Heterogeneity of Mesenchymal Stem Cells during Adipogenic Differentiation. Anal. Chem. 2021, 93, 671–676. [Google Scholar] [CrossRef]
- Donelli, G.; Francolini, I.; Ruggeri, V.; Guaglianone, E.; D’Ilario, L.; Piozzi, A. Pore formers promoted reléase of an antifungal drug from functionalized polyurethanes to inhibit Candida colonization. J. Appl. Microbiol. 2006, 100, 615–622. [Google Scholar] [CrossRef] [Green Version]
- Rivas, M.; Pelechà, M.; Franco, L.; Turon, P.; Alemán, C.; del Valle, L.J.; Puiggalí, J. Incorporation of chloramphenicol loaded hydroxyapatite nanoparticles into polylactide. Int. J. Mol. Sci. 2019, 20, 5056. [Google Scholar] [CrossRef] [Green Version]
- Klug, H.; Alexander, L. X-Ray Diffraction Procedures for Polycrystallite and Amorphous Materials, 2nd ed.; John Wiley and Sons: Hoboken, NJ, USA, 1974. [Google Scholar]
- Landi, E.; Tampieri, A.; Celotti, G.; Sprio, S. Densification behaviour and mechanisms of synthetic hydroxyapatites. J. Eur. Ceram Soc. 2000, 20, 2377–2387. [Google Scholar] [CrossRef]
- Olmo, C.; Franco, L.; del Valle, L.J.; Puiggalí, J. Biodegradable polylactide scaffolds with pharmacological activity by means of ultrasound micromolding technology. Appl. Sci. 2020, 10, 3106. [Google Scholar] [CrossRef]
- Keridou, I.; Franco, L.; Martínez, J.C.; Turon, P.; del Valle, L.J.; Puiggalí, J. Electrospun scaffolds for wound healing applications from poly(4-hydroxybutyrate): A biobased and biodegradable linear polymer with high elastomeric properties. J. Appl. Polym. Sci. 2022, 139, e51447. [Google Scholar] [CrossRef]
- Rivas, M.; del Valle, L.J.; Rodríguez-Rivero, A.M.; Turon, P.; Puiggalí, J.; Alemán, C. Loading of Antibiotic into biocoated hydroxyapatite nanoparticles: Smart antitumor platforms with regulated release. ACS Biomater. Sci. Eng. 2018, 4, 3234–3245. [Google Scholar] [CrossRef] [Green Version]
- Yousef, I.; Bréard, J.; SidAhmed-Adrar, N.; Maâmer-Azzabi, A.; Marchal, C.; Dumas, P.; Le Naour, F. Infrared spectral signatures of CDCP1-induced effects in colon carcinoma cells. Analyst 2011, 136, 5162–5168. [Google Scholar] [CrossRef] [PubMed]
- Dowling, L.M.; Roach, P.; Rutter, A.V.; Yousef, I.; Pillai, S.; Latham, D.; van Pittius, D.G.; Sulé-Suso, J. Optimization of sample preparation using glass slides for spectral pathology. Appl. Spectrosc. 2021, 75, 343–350. [Google Scholar] [CrossRef]
- Baker, M.J.; Trevisan, J.; Bassan, P.; Bhargava, R.; Butler, H.J.; Dorling, K.M.; Fielden, P.R.; Fogarty, S.W.; Fullwood, N.J.; Heys, K.A.; et al. Using fourier transform IR spectroscopy to analyze biological materials. Nat. Protoc 2014, 9, 1771–1791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martínez-Rovira, I.; Seksek, O.; Dokic, I.; Brons, S.; Abdollahi, A.; Yousef, I. Study of the intracellular nanoparticle-based radio sensitization mechanisms in F98 glioma cells treated with charged particle therapy through synchrotron-based infrared microspectroscopy. Analyst 2020, 145, 2345–2356. [Google Scholar] [CrossRef] [PubMed] [Green Version]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| STR Loaded Path [*] | ACP | cHAp | ||
|---|---|---|---|---|
| EE (%) | DL (%) | EE (%) | DL (%) | |
| (PO4)3− dissolution, (ps) | 21.5 | 1.8 | 25.9 | 2.2 |
| Ca2+ dissolution, (cs) | 16.9 | 1.4 | 21.8 | 1.9 |
| Reaction mixture, (rm) | 5.4 | 0.5 | 7.7 | 0.7 |
| Adsorption, (as) | 11.7 | 3.2 | 13.2 | 3.6 |
| Sample, [*] | XC (%) | L (nm) | ||
|---|---|---|---|---|
| ACP | cHAp | ACP | cHAp | |
| Without STR | 24.0 | 78.4 | 11.8 | 28.3 |
| STR in Ca2+ dissolution, (cs) | 20.0 | 76.7 | 8.6 | 24.4 |
| STR in (PO4)3− dissolution, (ps) | 20.0 | 72.2 | 7.8 | 23.8 |
| STR in the reaction mixture, (rm) | 6.8 | 63.2 | 6.2 | 19.9 |
| STR by adsorption, (as) | 9.5 | 66.4 | 6.7 | 21.5 |
| STR Loading, [*] | ACP | cHAp | ||||||
|---|---|---|---|---|---|---|---|---|
| kH (h−0.5) | r | k1 (h−1) | r | kH (h−0.5) | r | k1 (h−1) | r | |
| Ca2+ dissolution, (cs) | 0.239 | 0.982 | 0.047 | 0.991 | 0.095 | 0.936 | 0.040 | 0.997 |
| (PO4)3− dissolution, (ps) | 0.261 | 0.988 | 0.038 | 0.979 | 0.099 | 0.988 | 0.028 | 0.960 |
| Reaction mixture, (rm) | 2.000 | 1.000 | 0.073 | 0.951 | 1.275 | 0.974 | 0.062 | 0.994 |
| Adsorption, (as) | 2.123 | 0.981 | 0.074 | 0.963 | 1.328 | 0.959 | 0.039 | 0.988 |
| Fiber | Release Kinetic Parameters | |||
|---|---|---|---|---|
| kH (h−0.5) | r | k1 (h−1) | r | |
| PLA/ACPps | 0.641 | 0.999 | 0.029 | 0.960 |
| PLA/cHApps | 0.544 | 0.997 | 0.020 | 0.961 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kadkhodaie-Elyaderani, A.; de Lama-Odría, M.d.C.; Rivas, M.; Martínez-Rovira, I.; Yousef, I.; Puiggalí, J.; del Valle, L.J. Medicated Scaffolds Prepared with Hydroxyapatite/Streptomycin Nanoparticles Encapsulated into Polylactide Microfibers. Int. J. Mol. Sci. 2022, 23, 1282. https://doi.org/10.3390/ijms23031282
Kadkhodaie-Elyaderani A, de Lama-Odría MdC, Rivas M, Martínez-Rovira I, Yousef I, Puiggalí J, del Valle LJ. Medicated Scaffolds Prepared with Hydroxyapatite/Streptomycin Nanoparticles Encapsulated into Polylactide Microfibers. International Journal of Molecular Sciences. 2022; 23(3):1282. https://doi.org/10.3390/ijms23031282
Chicago/Turabian StyleKadkhodaie-Elyaderani, Amirmajid, Maria del Carmen de Lama-Odría, Manuel Rivas, Immaculada Martínez-Rovira, Ibraheem Yousef, Jordi Puiggalí, and Luis J. del Valle. 2022. "Medicated Scaffolds Prepared with Hydroxyapatite/Streptomycin Nanoparticles Encapsulated into Polylactide Microfibers" International Journal of Molecular Sciences 23, no. 3: 1282. https://doi.org/10.3390/ijms23031282
APA StyleKadkhodaie-Elyaderani, A., de Lama-Odría, M. d. C., Rivas, M., Martínez-Rovira, I., Yousef, I., Puiggalí, J., & del Valle, L. J. (2022). Medicated Scaffolds Prepared with Hydroxyapatite/Streptomycin Nanoparticles Encapsulated into Polylactide Microfibers. International Journal of Molecular Sciences, 23(3), 1282. https://doi.org/10.3390/ijms23031282