
Development of a Gene-Activated Scaffold Incorporating Multifunctional Cell-Penetrating Peptides for pSDF-1α Delivery for Enhanced Angiogenesis in Tissue Engineering Applications

 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
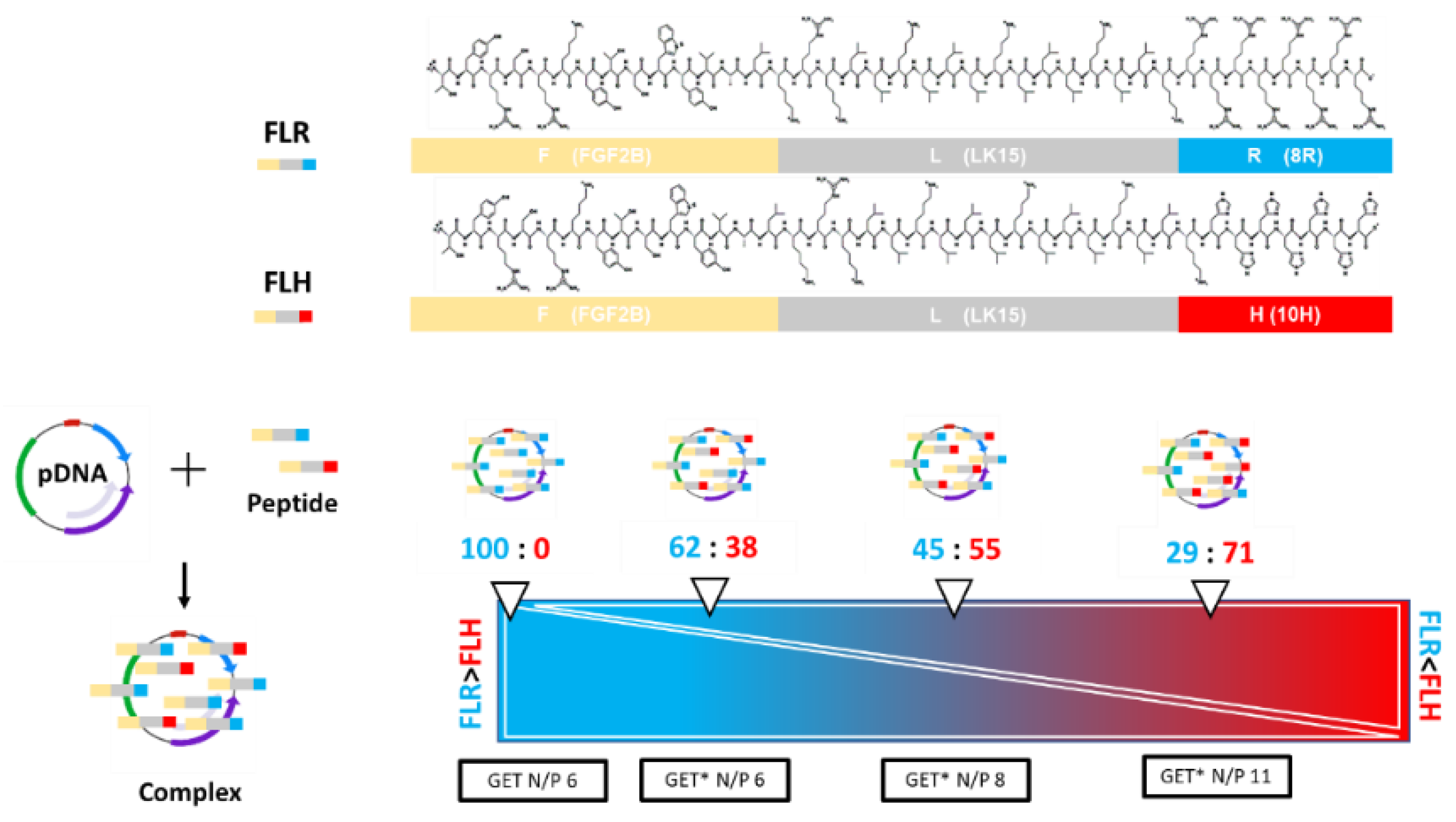
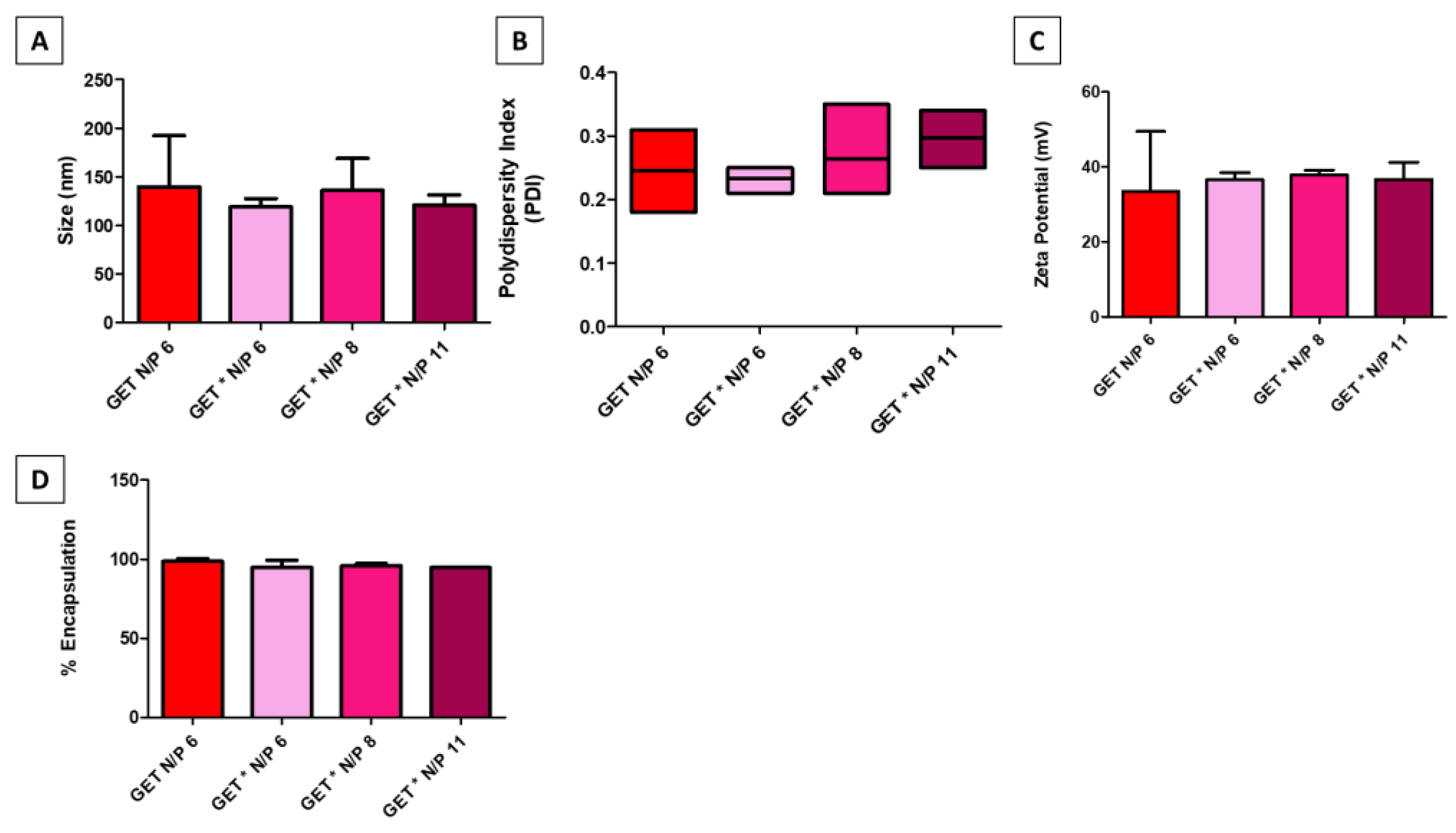
2.1. GET*-pDNA Nanoparticles Physico-Chemical Properties
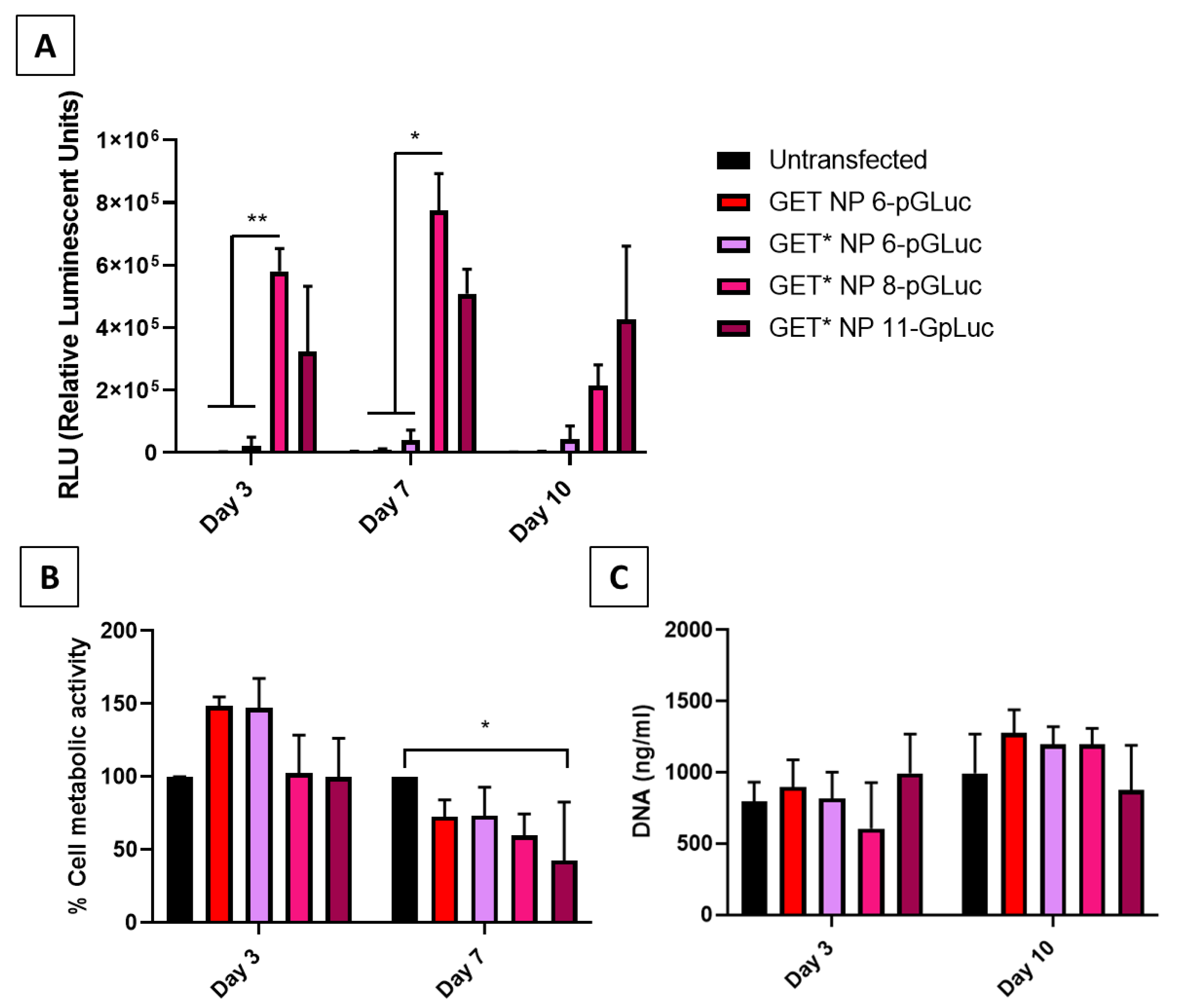
2.2. 2D Comparison of GET and GET* Nanoparticles in MSCs
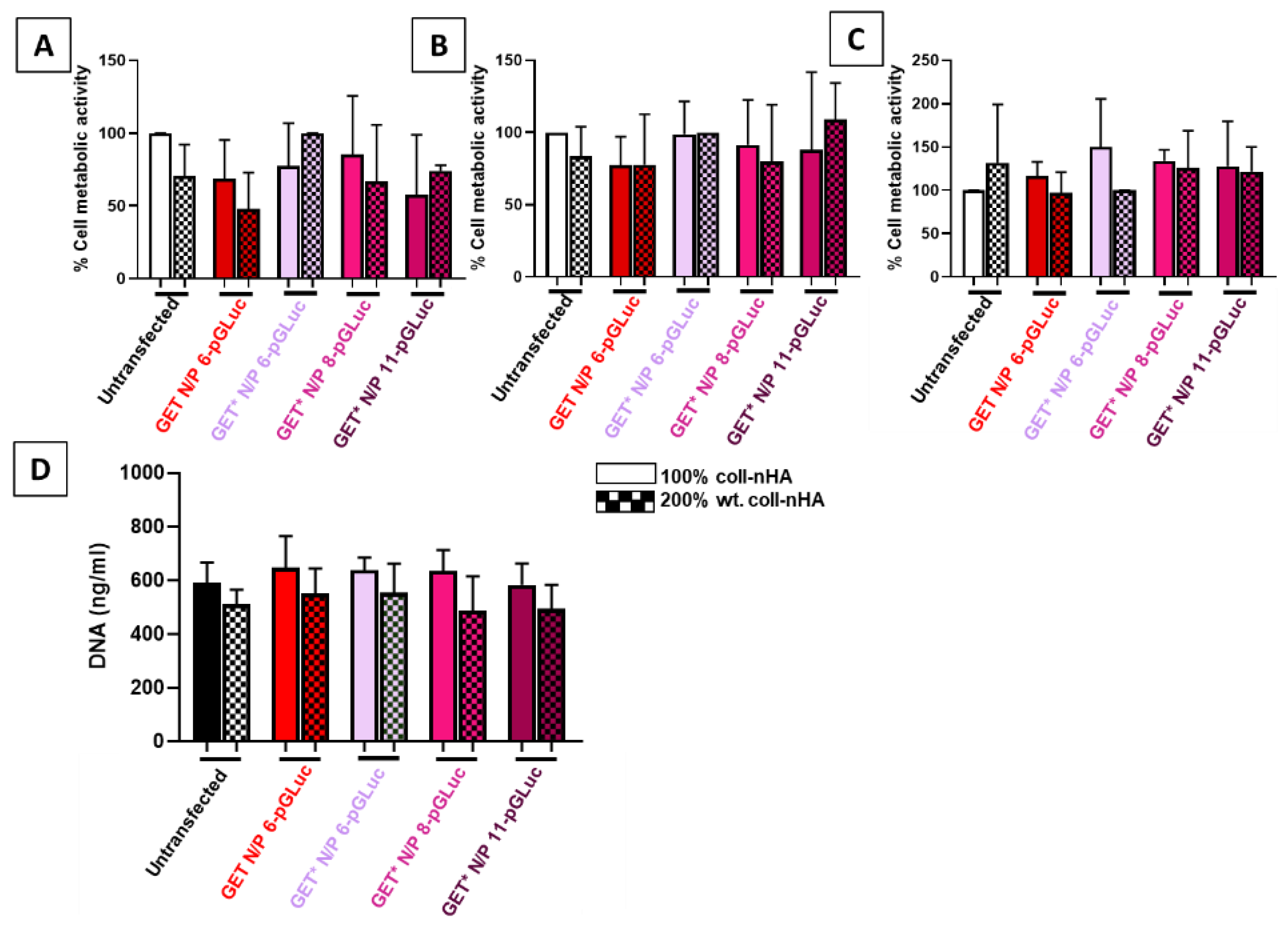
2.3. Incorporation of GET* Nanoparticles into Coll–nHA Scaffolds and Assessment of Biocompatibility
2.4. Measurement of GET* Coll–nHA Scaffold Transfection Efficiency
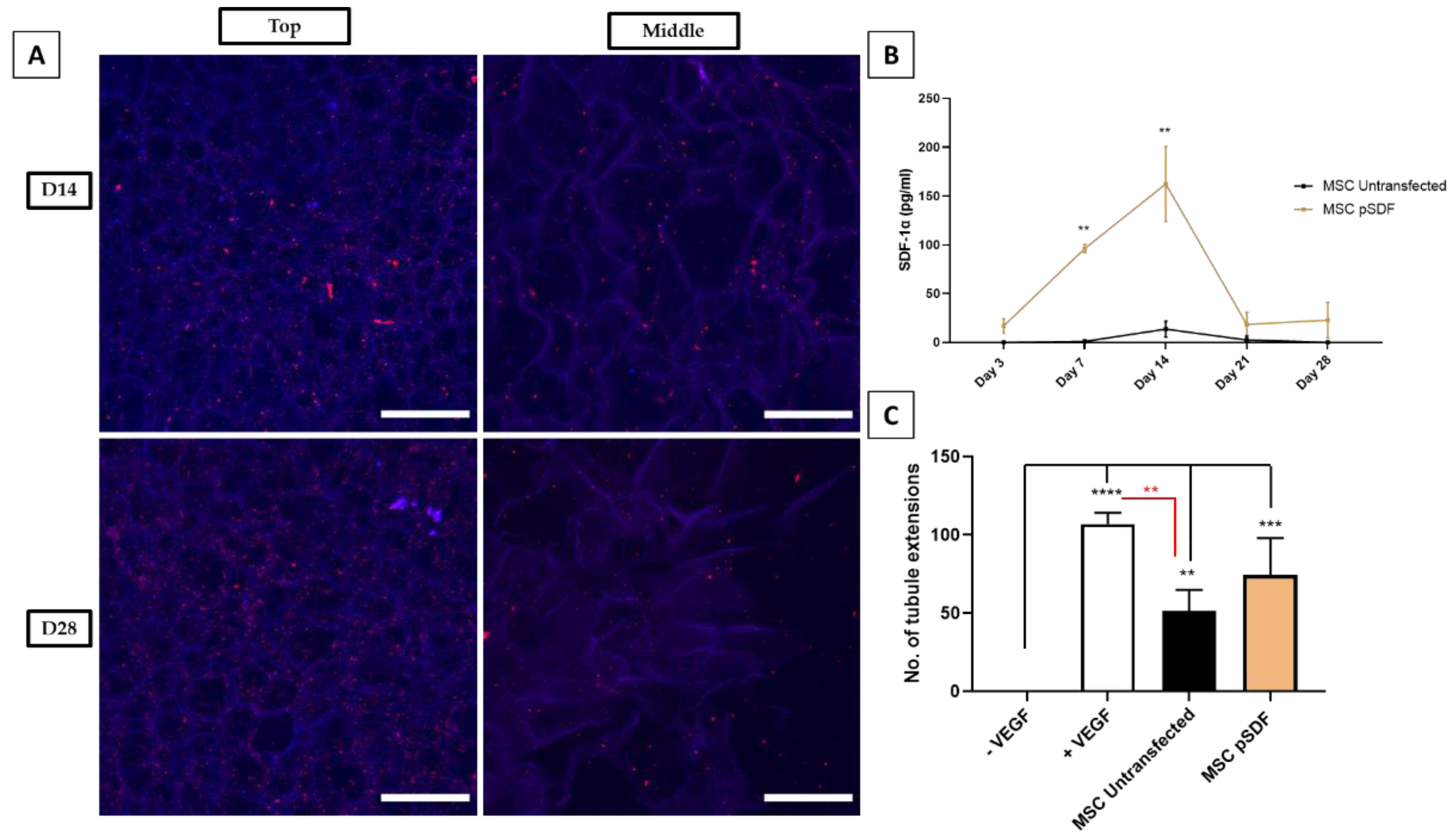
2.5. Characterisation of the Stability and Angiogenic Potential of GET* N/P 8 Scaffolds
3. Discussion
4. Materials and Methods
4.1. Nanoparticle Formulation
4.1.1. Plasmid Propagation
4.1.2. GET and GET* Nanoparticle Formulation
4.2. Physico-Chemical Characterisation
4.2.1. Size, Polydispersity Index and Zeta Potential
4.2.2. pDNA Encapsulation Efficiency
4.3. Mesenchymal Stem Cell Isolation and Culture
4.4. Optimisation of GET*-pDNA Delivery in 2D Monolayer
4.4.1. rMSC Transfection with pGLuc
4.4.2. Cytocompatibility of GET*-pDNA Nanoparticles
4.5. Scaffold-Based Optimsation of GET* Nanoparticle Delivery
4.5.1. Collagen–Nanohydroxyapatite Scaffold Formulation
4.5.2. Scaffold-Based Transfection of rMSCs
4.5.3. Cytocompatibility of GET* Gene-Activated Scaffolds
4.6. Assessment of Gene-Activated Scaffold Stability and Therapeutic Efficacy
4.6.1. Confocal Imaging of Cy3-Tagged GET* N/P 8 Nanoparticles on Scaffolds
4.6.2. Measurement of SDF-1α Expression and Angiogenesis
4.7. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Marsell, R.; Einhorn, T.A. The biology of fracture healing. Injury 2011, 42, 551–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sivaraj, K.K.; Adams, R.H. Blood vessel formation and function in bone. Development 2016, 143, 2706–2715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sen, M.; Miclau, T. Autologous iliac crest bone graft: Should it still be the gold standard for treating nonunions? Injury 2007, 38, S75–S80. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, F.J. Biomaterials & scaffolds for tissue engineering. Mater. Today 2011, 14, 88–95. [Google Scholar] [CrossRef]
- Carletti, E.; Motta, A.; Migliaresi, C. Scaffolds for tissue engineering and 3D cell culture. Methods Mol. Biol. 2011, 695, 17–39. [Google Scholar]
- Park, J.Y.; Park, S.H.; Kim, M.G.; Park, S.H.; Yoo, T.H.; Kim, M.S. Biomimetic scaffolds for bone tissue engineering. Adv. Exp. Med. Biol. 2018, 1064, 109–121. [Google Scholar]
- Wubneh, A.; Tsekoura, E.K.; Ayranci, C.; Uludağ, H. Current state of fabrication technologies and materials for bone tissue engineering. Acta Biomater. 2018, 80, 1–30. [Google Scholar] [CrossRef]
- De Misquita, M.R.D.O.F.; Bentini, R.; Goncalves, F. The performance of bone tissue engineering scaffolds in in vivo animal models: A systematic review. J. Biomater. Appl. 2016, 31, 625–636. [Google Scholar] [CrossRef]
- Lyons, F.G.; Gleeson, J.P.; Partap, S.; Coghlan, K.; O’Brien, F.J. Novel Microhydroxyapatite Particles in a Collagen Scaffold: A Bioactive Bone Void Filler? Clin. Orthop. Relat. Res. 2014, 472, 1318–1328. [Google Scholar] [CrossRef] [Green Version]
- David, F.; Levingstone, T.J.; Schneeweiss, W.; de Swarte, M.; Jahns, H.; Gleeson, J.P.; O’Brien, F.J. Enhanced bone healing using collagen-hydroxyapatite scaffold implantation in the treatment of a large multiloculated mandibular aneurysmal bone cyst in a thoroughbred filly. J. Tissue Eng. Regen. Med. 2015, 9, 1193–1199. [Google Scholar] [CrossRef]
- Ryan, A.; Gleeson, J.P.; Matsiko, A.; Thompson, E.M.; O’Brien, F.J. Effect of different hydroxyapatite incorporation methods on the structural and biological properties of porous collagen scaffolds for bone repair. J. Anat. 2014, 227, 732–745. [Google Scholar] [CrossRef] [PubMed]
- Cunniffe, G.M.; Curtin, C.M.; Thompson, E.M.; Dickson, G.R.; O’Brien, F.J. Content-Dependent Osteogenic Response of Nanohydroxyapatite: An in Vitro and in Vivo Assessment within Collagen-Based Scaffolds. ACS Appl. Mater. Interfaces 2016, 8, 23477–23488. [Google Scholar] [CrossRef] [PubMed]
- Cunniffe, G.M.; Dickson, G.R.; Partap, S.; Stanton, K.; O’Brien, F.J. Development and characterisation of a collagen nano-hydroxyapatite composite scaffold for bone tissue engineering. J. Mater. Sci. Mater. Electron. 2010, 21, 2293–2298. [Google Scholar] [CrossRef] [PubMed]
- Sanz-Herrera, J.A.; Reina-Romo, E. Cell-biomaterial mechanical interaction in the framework of tissue engineering: Insights, computational modeling and perspectives. Int. J. Mol. Sci. 2011, 12, 8217–8244. [Google Scholar] [CrossRef]
- Murphy, C.M.; O’Brien, F.J.; Little, D.G.; Schindeler, A. Cell-scaffold interactions in the bone tissue engineering triad. Eur. Cells Mater. 2013, 26, 120–132. [Google Scholar] [CrossRef]
- Wang, X.; Wang, Y.; Gou, W.; Lu, Q.; Peng, J.; Lu, S. Role of mesenchymal stem cells in bone regeneration and fracture repair: A review. Int. Orthop. 2013, 37, 2491–2498. [Google Scholar] [CrossRef]
- Bruder, S.P.; Fink, D.J.; Caplan, A.I. Mesenchymal stem cells in bone development, bone repair, and skeletal regeneration therapy. J. Cell. Biochem. 1994, 56, 283–294. [Google Scholar] [CrossRef]
- Caplan, A.I. Mesenchymal stem cells. J. Orthop. Res. 1991, 9, 641–650. [Google Scholar] [CrossRef]
- Bruder, S.P.; Jaiswal, N.; Ricalton, N.S.; Mosca, J.D.; Kraus, K.H.; Kadiyala, S. Mesenchymal Stem Cells in Osteobiology and Applied Bone Regeneration. Clin. Orthop. Relat. Res. 1998, 355, S247–S256. [Google Scholar] [CrossRef]
- Griffin, M.; Iqbal, S.A.; Bayat, A. Exploring the application of mesenchymal stem cells in bone repair and regeneration. J. Bone Jt. Surgery. Br. Vol. 2011, 93, 427–434. [Google Scholar] [CrossRef] [Green Version]
- Granero-Moltó, F.; Weis, J.A.; Miga, M.I.; Landis, B.; Myers, T.J.; O’Rear, L.; Longobardi, L.; Jansen, E.D.; Mortlock, D.P.; Spagnoli, A. Regenerative Effects of Transplanted Mesenchymal Stem Cells in Fracture Healing. Stem Cells 2009, 27, 1887–1898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seong, J.M.; Kim, B.-C.; Park, J.-H.; Kwon, I.K.; Mantalaris, A.; Hwang, Y.-S. Stem cells in bone tissue engineering. Biomed. Mater. 2010, 5, 062001. [Google Scholar] [CrossRef] [PubMed]
- Watson, L.; Elliman, S.J.; Coleman, C.M. From isolation to implantation: A concise review of mesenchymal stem cell therapy in bone fracture repair. Stem Cell Res. Ther. 2014, 5, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watt, S.M.; Gullo, F.; Van Der Garde, M.; Markeson, D.; Camicia, R.; Khoo, C.P.; Zwaginga, J.J. The angiogenic properties of mesenchymal stem/stromal cells and their therapeutic potential. Br. Med Bull. 2013, 108, 25–53. [Google Scholar] [CrossRef]
- Perez, J.R.; Kouroupis, D.; Li, D.J.; Best, T.M.; Kaplan, L.; Correa, D. Tissue Engineering and Cell-Based Therapies for Fractures and Bone Defects. Front. Bioeng. Biotechnol. 2018, 6, 105. [Google Scholar] [CrossRef] [Green Version]
- Hacobian, A.; Posa-Markaryan, K.; Sperger, S.; Stainer, M.; Hercher, D.; Feichtinger, G.; Schuh, C.M.; Redl, H. Improved osteogenic vector for non-viral gene therapy. Eur. Cells Mater. 2016, 31, 191–204. [Google Scholar] [CrossRef] [Green Version]
- Spiller, K.L.; Vunjak-Novakovic, G. Clinical translation of controlled protein delivery systems for tissue engineering. Drug Deliv. Transl. Res. 2013, 5, 101. [Google Scholar] [CrossRef] [Green Version]
- Bender, E. Regulating the gene-therapy revolution. Nature 2018, 564, S20–S22. [Google Scholar] [CrossRef]
- Arjmand, B.; Larijani, B.; Hosseini, M.S.; Payab, M.; Gilany, K.; Goodarzi, P.; Roudsari, P.P.; Baharvand, M.A.; Mohammadi, N.S.H. The Horizon of Gene Therapy in Modern Medicine: Advances and Challenges. Adv. Exp. Med. Biol. 2020, 1247, 33–64. [Google Scholar] [CrossRef]
- Park, K.S.; Sun, X.; Aikins, M.E.; Moon, J.J. Non-viral COVID-19 vaccine delivery systems. Adv. Drug Deliv. Rev. 2021, 169, 137–151. [Google Scholar] [CrossRef]
- Vogel, A.B.; Kanevsky, I.; Che, Y.; Swanson, K.A.; Muik, A.; Vormehr, M.; Kranz, L.M.; Walzer, K.C.; Hein, S.; Güler, A.; et al. BNT162b vaccines protect rhesus macaques from SARS-CoV-2. Nature 2021, 592, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Milane, L.; Amiji, M. Clinical approval of nanotechnology-based SARS-CoV-2 mRNA vaccines: Impact on translational nanomedicine. Drug Deliv. Transl. Res. 2021, 11, 1309–1315. [Google Scholar] [CrossRef]
- Walther, W.; Stein, U. Viral vectors for gene transfer: A review of their use in the treatment of human diseases. Drugs 2000, 60, 249–271. [Google Scholar] [CrossRef] [PubMed]
- Elsabahy, M.; Nazarali, A.; Foldvari, M. Non-viral nucleic acid delivery: Key challenges and future directions. Curr. Drug Deliv. 2011, 8, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Thorne, B.; Takeya, R.; Vitelli, F.; Swanson, X. Gene therapy. Adv. Biochem. Eng. Biotechnol. 2018, 165, 351–399. [Google Scholar] [PubMed]
- Patil, S.; Gao, Y.-G.; Lin, X.; Li, Y.; Dang, K.; Tian, Y.; Zhang, W.-J.; Jiang, S.-F.; Qadir, A.; Qian, A.-R. The Development of Functional Non-Viral Vectors for Gene Delivery. Int. J. Mol. Sci. 2019, 20, 5491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gresch, O.; Altrogge, L. Transfection of Difficult-to-Transfect Primary Mammalian Cells. In Protein Expression in Mammalian Cells; Humana Press: Totowa, NJ, USA, 2012; pp. 65–74. [Google Scholar] [CrossRef]
- Raftery, R.M.; Tierney, E.G.; Curtin, C.M.; Cryan, S.A.; O’Brien, F.J. Development of a gene-activated scaffold platform for tissue engineering applications using chitosan-pDNA nanoparticles on collagen-based scaffolds. J. Control. Release 2015, 210, 84–94. [Google Scholar] [CrossRef]
- Tierney, E.G.; Duffy, G.P.; Cryan, S.A.; Curtin, C.M.; O’Brien, F.J. Non-viral gene-activated matrices: Next generation constructs for bone repair. Organogenesis 2013, 9, 22–28. [Google Scholar] [CrossRef] [Green Version]
- Curtin, C.M.; Tierney, E.G.; McSorley, K.; Cryan, S.-A.; Duffy, G.P.; O’Brien, F.J. Combinatorial Gene Therapy Accelerates Bone Regeneration: Non-Viral Dual Delivery of VEGF and BMP2 in a Collagen-Nanohydroxyapatite Scaffold. Adv. Healthc. Mater. 2015, 4, 223–227. [Google Scholar] [CrossRef]
- Castaño, I.M.; Curtin, C.; Duffy, G.; O’Brien, F.J. Next generation bone tissue engineering: Non-viral miR-133a inhibition using collagen-nanohydroxyapatite scaffolds rapidly enhances osteogenesis. Sci. Rep. 2016, 6, 27941. [Google Scholar] [CrossRef]
- Dixon, J.E.; Osman, G.; Morris, G.E.; Markides, H.; Rotherham, M.; Bayoussef, Z.; El Haj, A.J.; Denning, C.; Shakesheff, K.M. Highly efficient delivery of functional cargoes by the synergistic effect of GAG binding motifs and cell-penetrating peptides. Proc. Natl. Acad. Sci. USA 2016, 113, E291–E299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raftery, R.; Walsh, D.P.; Ferreras, L.B.; Castaño, I.M.; Chen, G.; Lemoine, M.; Osman, G.; Shakesheff, K.; Dixon, J.E.; O’Brien, F.J. Highly versatile cell-penetrating peptide loaded scaffold for efficient and localised gene delivery to multiple cell types: From development to application in tissue engineering. Biomaterials 2019, 216, 119277. [Google Scholar] [CrossRef] [PubMed]
- Castaño, I.M.; Curtin, C.M.; Shaw, G.; Murphy, J.M.; Duffy, G.P.; O’Brien, F.J. A novel collagen-nanohydroxyapatite microRNA-activated scaffold for tissue engineering applications capable of efficient delivery of both miR-mimics and antagomiRs to human mesenchymal stem cells. J. Control. Release 2015, 200, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Castaño, I.M.; Curtin, C.M.; Duffy, G.P.; O’Brien, F.J. Harnessing an Inhibitory Role of miR-16 in Osteogenesis by Human Mesenchymal Stem Cells for Advanced Scaffold-Based Bone Tissue Engineering. Tissue Eng. Part A 2019, 25, 24–33. [Google Scholar] [CrossRef]
- Tierney, E.G.; Duffy, G.P.; Hibbitts, A.J.; Cryan, S.A.; O’Brien, F.J. The development of non-viral gene-activated matrices for bone regeneration using polyethyleneimine (PEI) and collagen-based scaffolds. J. Control. Release 2012, 158, 304–311. [Google Scholar] [CrossRef]
- Ritchie, A.G.A.; Clarke, P.; Dixon, J. ATW August 2020. Anim. Technol. Welf. 2020, 19, 166–168. [Google Scholar]
- Ferreras, L.A.B.; Scott, D.; Reina, S.V.; Roach, P.; Torres, T.E.; Goya, G.F.; Shakesheff, K.M.; Dixon, J.E. Enhanced Cellular Transduction of Nanoparticles Resistant to Rapidly Forming Plasma Protein Coronas. Adv. Biosyst. 2020, 4, 2000162. [Google Scholar] [CrossRef]
- Ferreras, L.A.B.; Chan, S.Y.; Reina, S.V.; Dixon, J.E. Rapidly Transducing and Spatially Localized Magnetofection Using Peptide-Mediated Non-Viral Gene Delivery Based on Iron Oxide Nanoparticles. ACS Appl. Nano Mater. 2021, 4, 167–181. [Google Scholar]
- Kusumbe, A.P.; Ramasamy, S.K.; Adams, R.H. Coupling of angiogenesis and osteogenesis by a specific vessel subtype in bone. Nature 2014, 507, 323–328. [Google Scholar] [CrossRef]
- Knight, M.N.; Hankenson, K.D. Mesenchymal Stem Cells in Bone Regeneration. Adv. Wound Care 2013, 2, 306. [Google Scholar] [CrossRef] [Green Version]
- Šponer, P.; Kučera, T.; Diaz-Garcia, D.; Filip, S. The role of mesenchymal stem cells in bone repair and regeneration. Eur. J. Orthop. Surg. Traumatol. 2014, 24, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Laiva, A.L.; O’Brien, F.J.; Keogh, M.B. SDF-1α gene-activated collagen scaffold drives functional differentiation of human Schwann cells for wound healing applications. Biotechnol. Bioeng. 2020, 118, 725–736. [Google Scholar] [CrossRef] [PubMed]
- Hosogane, N.; Huang, Z.; Rawlins, B.A.; Liu, X.; Boachie-Adjei, O.; Boskey, A.; Zhu, W. Stromal derived factor-1 regulates bone morphogenetic protein 2-induced osteogenic differentiation of primary mesenchymal stem cells. Int. J. Biochem. Cell Biol. 2010, 42, 1132–1141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janko, M.; Sahm, J.; Schaible, A.; Brune, J.C.; Bellen, M.; Schröder, K.; Seebach, C.; Marzi, I.; Henrich, D. Comparison of three different types of scaffolds preseeded with human bone marrow mononuclear cells on the bone healing in a femoral critical size defect model of the athymic rat. J. Tissue Eng. Regen. Med. 2018, 12, 653–666. [Google Scholar] [CrossRef] [PubMed]
- Cipitria, A.; Boettcher, K.; Schoenhals, S.; Garske, D.S.; Schmidt-Bleek, K.; Ellinghaus, A.; Dienelt, A.; Peters, A.; Mehta, M.; Madl, C.M.; et al. In-situ tissue regeneration through SDF-1α driven cell recruitment and stiffness-mediated bone regeneration in a critical-sized segmental femoral defect. Acta Biomater. 2017, 60, 50–63. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Li, X.; Li, J.; Zhong, L.; Chen, X.; Chen, S. SDF-1 mediates mesenchymal stem cell recruitment and migration via the SDF-1/CXCR4 axis in bone defect. J. Bone Miner. Metab. 2021, 39, 126–138. [Google Scholar] [CrossRef]
- Tong, H.; Wang, C.; Shi, Q.; Fernandes, J.C.; Dai, K.; Tang, G.; Zhang, X.; Huang, Y. Polyethylenimine600-β-cyclodextrin: A promising nanopolymer for nonviral gene delivery of primary mesenchymal stem cells. Int. J. Nanomed. 2013, 8, 1935–1946. [Google Scholar] [CrossRef] [Green Version]
- Ramos-Murillo, A.I.; Rodríguez, E.; Beltrán, K.; Ricaurte, C.; Camacho, B.; Salguero, G.; Godoy-Silva, R.D. Efficient Non-Viral Gene Modification of Mesenchymal Stromal Cells from Umbilical Cord Wharton’s Jelly with Polyethylenimine. Pharmaceutics 2020, 12, 896. [Google Scholar] [CrossRef]
- Pillay, V.; Murugan, K.; Choonara, Y.; Kumar, P.; Bijukumar, D.; du Toit, L. Parameters and characteristics governing cellular internalization and trans-barrier trafficking of nanostructures. Int. J. Nanomed. 2015, 10, 2191–2206. [Google Scholar] [CrossRef] [Green Version]
- Sergeeva, Y.N.; Jung, L.; Weill, C.; Erbacher, P.; Tropel, P.; Félix, O.; Viville, S.; Decher, G. Control of the transfection efficiency of human dermal fibroblasts by adjusting the characteristics of jetPEI®/plasmid complexes/polyplexes through the cation/anion ratio. Colloids Surf. A Physicochem. Eng. Asp. 2018, 550, 193–198. [Google Scholar] [CrossRef]
- Bus, T.; Traeger, A.; Schubert, U.S. The great escape: How cationic polyplexes overcome the endosomal barrier. J. Mater. Chem. B 2018, 6, 6904–6918. [Google Scholar] [CrossRef] [PubMed]
- Cupic, K.I.; Rennick, J.J.; Johnston, A.P.; Such, G.K. Controlling endosomal escape using nanoparticle composition: Current progress and future perspectives. Nanomedicine 2019, 14, 215–223. [Google Scholar] [CrossRef] [PubMed]
- Sawant, R.; Torchilin, V. Intracellular Delivery of Nanoparticles with CPPs. Breast Cancer 2010, 683, 431–451. [Google Scholar] [CrossRef]
- Koren, E.; Torchilin, V.P. Cell-penetrating peptides: Breaking through to the other side. Trends Mol. Med. 2012, 18, 385–393. [Google Scholar] [CrossRef]
- Feng, J.; Tang, L. The Cell-Type Specificity and Endosomal Escape of Cell-Penetrating Peptides. Curr. Pharm. Des. 2014, 21, 1351–1356. [Google Scholar] [CrossRef]
- Salerno, J.C.; Ngwa, V.M.; Nowak, S.J.; Chrestensen, C.A.; Healey, A.N.; McMurry, J.L. Novel cell-penetrating peptide-adaptors effect intracellular delivery and endosomal escape of protein cargos. J. Cell Sci. 2016, 129, 893–897. [Google Scholar] [CrossRef] [Green Version]
- Lecher, J.C.; Nowak, S.; McMurry, J.L. Breaking in and busting out: Cell-penetrating peptides and the endosomal escape problem. Biomol. Concepts 2017, 8, 131–141. [Google Scholar] [CrossRef]
- Meng, Z.; Luan, L.; Kang, Z.; Feng, S.; Meng, Q.; Liu, K. Histidine-enriched multifunctional peptide vectors with enhanced cellular uptake and endosomal escape for gene delivery. J. Mater. Chem. B 2017, 5, 74–84. [Google Scholar] [CrossRef]
- Walrant, A.; Cardon, S.; Burlina, F.; Sagan, S. Membrane Crossing and Membranotropic Activity of Cell-Penetrating Peptides: Dangerous Liaisons? Accounts Chem. Res. 2017, 50, 2968–2975. [Google Scholar] [CrossRef]
- Paris, J.; Román, J.; Manzano, M.; Cabañas, M.; Vallet-Regí, M. Tuning dual-drug release from composite scaffolds for bone regeneration. Int. J. Pharm. 2015, 486, 30–37. [Google Scholar] [CrossRef] [Green Version]
- Hankenson, K.D.; Dishowitz, M.; Gray, C.; Schenker, M. Angiogenesis in bone regeneration. Injury 2011, 42, 556–561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stegen, S.; van Gastel, N.; Carmeliet, G. Bringing new life to damaged bone: The importance of angiogenesis in bone repair and regeneration. Bone 2015, 70, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Malvern Inc. Zeta Potential. 2018. Available online: www.malvern.com (accessed on 15 December 2021).
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Power, R.N.; Cavanagh, B.L.; Dixon, J.E.; Curtin, C.M.; O’Brien, F.J. Development of a Gene-Activated Scaffold Incorporating Multifunctional Cell-Penetrating Peptides for pSDF-1α Delivery for Enhanced Angiogenesis in Tissue Engineering Applications. Int. J. Mol. Sci. 2022, 23, 1460. https://doi.org/10.3390/ijms23031460
Power RN, Cavanagh BL, Dixon JE, Curtin CM, O’Brien FJ. Development of a Gene-Activated Scaffold Incorporating Multifunctional Cell-Penetrating Peptides for pSDF-1α Delivery for Enhanced Angiogenesis in Tissue Engineering Applications. International Journal of Molecular Sciences. 2022; 23(3):1460. https://doi.org/10.3390/ijms23031460
Chicago/Turabian StylePower, Rachael N., Brenton L. Cavanagh, James E. Dixon, Caroline M. Curtin, and Fergal J. O’Brien. 2022. "Development of a Gene-Activated Scaffold Incorporating Multifunctional Cell-Penetrating Peptides for pSDF-1α Delivery for Enhanced Angiogenesis in Tissue Engineering Applications" International Journal of Molecular Sciences 23, no. 3: 1460. https://doi.org/10.3390/ijms23031460
APA StylePower, R. N., Cavanagh, B. L., Dixon, J. E., Curtin, C. M., & O’Brien, F. J. (2022). Development of a Gene-Activated Scaffold Incorporating Multifunctional Cell-Penetrating Peptides for pSDF-1α Delivery for Enhanced Angiogenesis in Tissue Engineering Applications. International Journal of Molecular Sciences, 23(3), 1460. https://doi.org/10.3390/ijms23031460