
Role of miRNA-1 and miRNA-21 in Acute Myocardial Ischemia-Reperfusion Injury and Their Potential as Therapeutic Strategy


Abstract
:1. Introduction
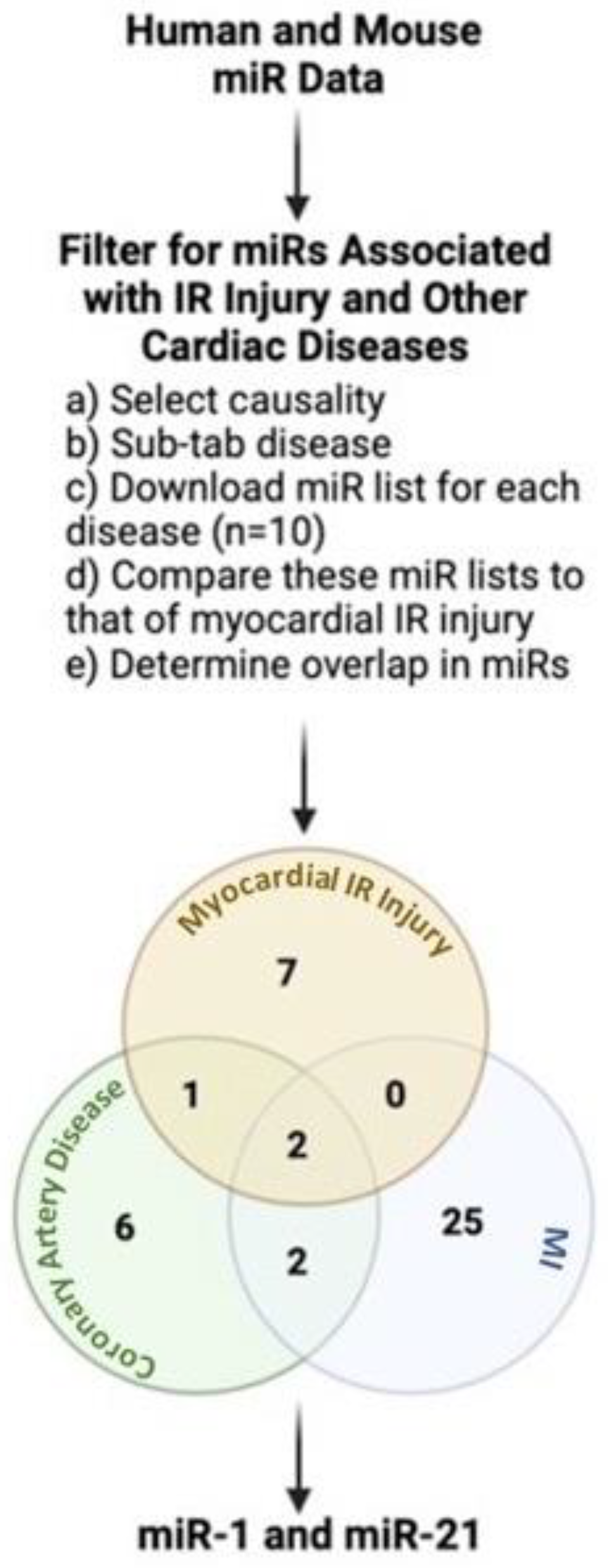
2. miRNAs: A Link between IR Injury and Other Cardiac Diseases in Humans
2.1. Role of Cardiac miR-1 in IR Injury
2.2. Role of Cardiac miR-21 in IR Injury

{kind=link}
{kind=link}
| miR | Findings | Function | Therapeutic Potential | Reference |
|---|---|---|---|---|
| miR-1 | ↓ miR-1 in rodent hearts in response to IR injury. | ↑ Bcl-2 after IR (HR) injury | miR-1 inhibition ↑ Bcl-2 and ↓ IA/AAR and cell apoptosis after IR(HR) injury | [31] |
| ↑ miR-1 in rodent hearts in response to IR injury | ↓ Bcl-2 and Cnx43 after IR (HR) injury | miR-1 mimic ↓ Bcl-2 and Cnx43 in H9C2 cells subjected to HR injury; Telmisartan ↑ Bcl-2 and Cnx43 and ↓ miR-1 after IR (HR) injury | [32] | |
| ↑ miR-1 in rodent hearts in response to IR injury or MI | ↓ KCNJ2/Kir2.1 and GJA1/Cx43 in ischemic myocardium | miR-1 overexpression ↓ KCNJ2/Kir2.1 and GJA1/Cx43 after MI; sEHIs reversed the effects | [33] | |
| ↓ miR-1 in infarcted human hearts in response to MI | N/A | N/A | [41,42] | |
| ↓ miR-1 in H9c2 cells in response to HR | ↑ Bcl-2 after IR (HR) injury | miR-1 inhibition ↑ Bcl-2 and ↓ IA/AAR and cell apoptosis after IR(HR) injury | [31] | |
| ↑ miR-1 in neonatal cardiac myocytes in response to HR | ↑ apoptosis and ↓ Bcl-2 after HR injury | miR-1 mimic ↑ apoptosis and ↓ Bcl-2 in neonatal rat cardiomyocytes subjected to HR injury; H2S reverses the effects | [34] | |
| ↑ miR-1 in remote myocardium compared to infarcted zone or healthy hearts in infarcted human hearts | N/A | N/A | [43] | |
| ↑levels of serum miR-1 after acute MI in pigs and humans | N/A | N/A | [44] | |
| miR-1 overexpression worsened cardiac I/R injury in transgenic mice | ↑ LDH, CK levels, caspase-3 expression, apoptosis and cardiac infarct area after IR (HR) injury | miR-1 overexpression exacerbate IR (HR) injury by ↑ LDH, CK levels, caspase-3 expression, apoptosis and cardiac infarct area | [35] | |
| miR-1 inhibition protects against IR (HR) injury in rodents and cardiomyocytes | ↑ LDH, CK levels, caspase-3 expression, apoptosis and cardiac infarct area after IR (HR) injury | LNA-antimiR-1 attenuated IR (HR) by ↑ PKCε and HSP60 | [35] | |
| miR-1 inhibition protects against IR (HR) injury in rodents and H9c2 cells | ↑ Bcl-2 after IR (HR) injury | miR-1 inhibition ↑ Bcl-2 and ↓ IA/AAR and cell apoptosis after IR(HR) injury | [31] | |
| miR-21 | ↓ miR-21 in infarct areas of mouse IR model | ↑ apoptosis and PDCD4, Bax/Bcl-2 and cleaved caspase-3/caspase-3 ratio after IR injury | miR-21 mimics ↓ apoptosis by inhibiting PDCD4 in cardiomyocytes subjected to OGD/R | [70] |
| diverse time-dependent changes in circulating miR-21 in post-MI patients | N/A | N/A | [13] | |
| miR-21 protected cultured cardiac myocytes against HR-induced apoptosis via its target PDCD4 | ↑ apoptosis and PDCD4, Bax/Bcl-2 and cleaved caspase-3/caspase-3 ratio after IR injury | miR-21 mimics ↓ apoptosis by inhibiting PDCD4 in cardiomyocytes subjected to OGD/R | [70] | |
| miR-21 protected cultured cardiac myocytes against HR-induced apoptosis via its target PDCD4 | ↑ apoptosis in cardiomyocytes treated with H2O2 | pre-miR-21 ↓ H2O2-induced apoptosis of cardiomyocytes; overexpression of PDCD4 inhibited pre-miR-21 mediated protective effect | [71] |
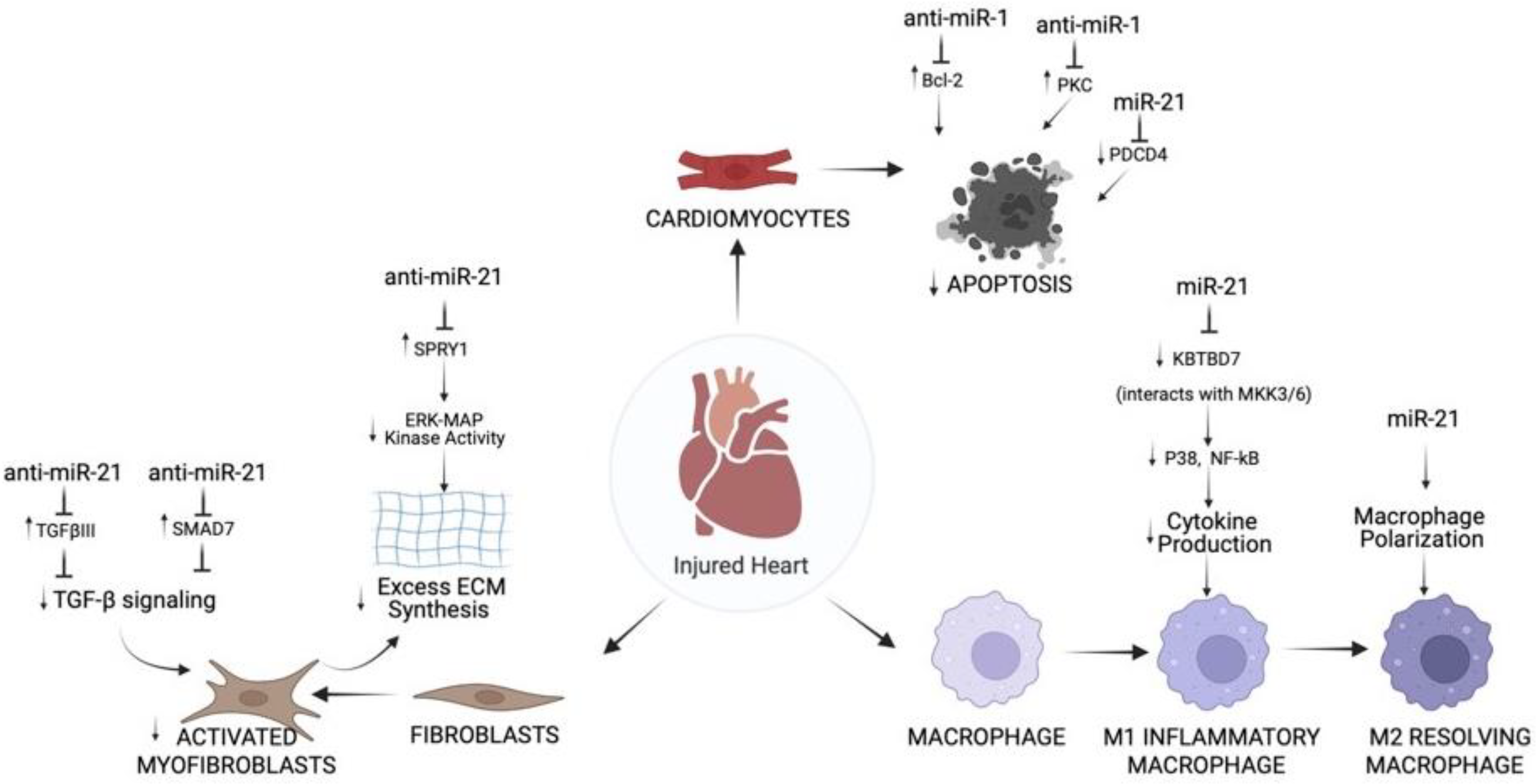
3. Role of miR-1 and miR-21 in Different Cell Types of IR Injured Hearts
3.1. Single-Cell Sequencing Data on IR Injury
3.2. Role of miR-1 and miR-21 in Cardiac Cell Types Post-MI
3.2.1. Cardiomyocytes
3.2.2. Fibroblasts
3.2.3. Immune Cells
3.3. Studies Integrating mRNA Expression Data from Single-Cell with miRNAs Evident in IR Injury
4. Therapeutic Potential of miRNAs in IR Injury
4.1. Modes of miRNA Therapy Delivery
4.2. Potential Benefits of Delivering miRNA Therapies Post-MI
5. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| MI | myocardial infarction |
| PCI | percutaneous coronary intervention |
| IR | ischemia-reperfusion |
| miRNAs | microRNAs |
| miR-1 | miRNA-1 |
| miR-21 | miRNA-21 |
| CVD | cardiovascular disease |
| Cx43 | connexin 43 |
| Irx5 | iroquois homeobox domain 5 |
| KCND2 | potassium voltage-gated channel subfamily D member 2 |
| EPCs | endothelial progenitor cells |
| ROS | reactive oxygen species |
| PDCD4 | programmed cell death 4 |
| scRNAseq | single-cell RNA sequencing |
| M1 | pro-inflammatory macrophage |
| M2 | anti-inflammatory macrophage |
| PKCε | protein kinase C epsilon |
| TGF-β | transforming growth factor-β |
| SMADs | decapentaplegic homologs |
| TGFβIII | TGF-β receptor III |
| DAMP | damage-associated molecular patterns |
| LNA | locked nucleic acids |
| OME | 2′-O-methyl group |
| AAV | adeno-associated viruses |
| MSC-EV | mesenchymal stem cell-derived extracellular vesicles |
| miR-133 | miRNA-133 |
References
- Fan, Z.X.; Yang, J. The Role of microRNAs in Regulating Myocardial Ischemia Reperfusion Injury. Saudi. Med. J. 2015, 36, 787–793. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention. Heart Disease Facts. Available online: https://www.cdc.gov/heartdisease/facts.htm (accessed on 26 January 2022).
- Virani, S.S.; Alonso, A.; Benjamin, E.J.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Delling, F.N.; et al. American Heart Association Council on Epidemiology and Prevention Statistics Committee and Stroke Statistics Subcommittee. Heart Disease and Stroke Statistics-2020 Update: A Report From the American Heart Association. Circulation 2020, 141, e139–e596. [Google Scholar] [CrossRef] [PubMed]
- Hausenloy, D.J.; Yellon, D.M. Myocardial ischemia-reperfusion injury: A neglected therapeutic target. J. Clin. Investig. 2013, 123, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Yang, C. Clinical Manifestations and Basic Mechanisms of Myocardial Ischemia/Reperfusion Injury. Tzu Chi Med. J. 2018, 30, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Braunwald, E.; Kloner, R.A. Myocardial reperfusion: A double-edged sword? J. Clin. Investig. 1985, 76, 1713–1719. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.I.; Miyazono, K. Emerging Complexity of microRNA Generation Cascade. J. Biochem. 2011, 149, 15–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, L.; Xiong, X.; Liu, Y.; Wang, J. miRNA-1: Functional roles and dysregulation in heart disease. Mol. Biosyst. 2014, 10, 2775–2782. [Google Scholar] [CrossRef]
- Kabekkodu, S.P.; Shukla, V.; Varghese, V.K.; D′ Souza, J.; Chakrabarty, S.; Satyamoorthy, K. Clustered miRNAs and their role in biological functions and diseases. Biol. Rev. Camb. Philos. Soc. 2018, 93, 1955–1986. [Google Scholar] [CrossRef]
- Colpaert, R.M.; Calore, M. MicroRNAs in cardiac diseases. Cells 2019, 8, 737. [Google Scholar] [CrossRef] [Green Version]
- Kukreja, R.C.; Yin, C.; Salloum, F.N. MicroRNAs: New Players in Cardiac Injury and Protection. Mol. Pharmacol. 2011, 80, 558–564. [Google Scholar] [CrossRef] [Green Version]
- Ye, Y.; Perez-Polo, J.R.; Qian, J.; Birnbaum, Y. The role of microRNA in modulating myocardial ischemia-reperfusion injury. Physiol. Genom. 2011, 43, 534–542. [Google Scholar] [CrossRef]
- Major, J.L.; Bagchi, R.A.; Pires da Silva, J. Application of microRNA Database Mining in Biomarker Discovery and Identification of Therapeutic Targets for Complex Disease. Methods Protoc. 2020, 4, 5. [Google Scholar] [CrossRef]
- Laterza, O.F.; Lim, L.; Garrett-Engele, P.W.; Vlasakova, K.; Muniappa, N.; Tanaka, W.K.; Johnson, J.M.; Sina, J.F.; Fare, T.L.; Sistare, F.D.; et al. Plasma MicroRNAs as sensitive and specific biomarkers of tissue injury. Clin. Chem. 2009, 55, 1977–1983. [Google Scholar] [CrossRef] [Green Version]
- Weiss, J.B.; Eisenhardt, S.U.; Stark, G.B.; Bode, C.; Moser, M.; Grundmann, S. MicroRNAs in ischemia-reperfusion injury. Am. J. Cardiovasc. Dis. 2012, 2, 237–247. [Google Scholar]
- D′alessandra, Y.; Devanna, P.; Limana, F.; Straino, S.; Di Carlo, A.; Brambilla, P.G.; Rubino, M.; Carena, M.C.; Spazzafumo, L.; De Simone, M.; et al. Circulating microRNAs are new and sensitive biomarkers of myocardial infarction. Eur. Heart J. 2010, 31, 2765–2773. [Google Scholar] [CrossRef] [Green Version]
- Zhou, S.S.; Jin, J.P.; Wang, J.Q.; Zhang, Z.G.; Freedman, J.H.; Zheng, Y.; Cai, L. miRNAS in cardiovascular diseases: Potential biomarkers, therapeutic targets and challenges. Acta Pharmacol. Sin. 2018, 39, 1073–1084. [Google Scholar] [CrossRef] [Green Version]
- Bian, B.; Yu, X.F.; Wang, G.Q.; Teng, T.M. Role of miRNA-1 in regulating connexin 43 in ischemia-reperfusion heart injury: A rat model. Cardiovasc. Pathol. 2017, 27, 37–42. [Google Scholar] [CrossRef]
- Lu, M.; Zhang, Q.; Deng, M.; Miao, J.; Guo, Y.; Gao, W.; Cui, Q. An analysis of human microRNA and disease associations. PLoS ONE 2008, 3, e3420. [Google Scholar] [CrossRef] [Green Version]
- Huang, Z.; Shi, J.; Gao, Y.; Cui, C.; Zhang, S.; Li, J.; Zhou, Y.; Cui, Q. HMDD v3.0: A database for experimentally supported human microRNA-disease associations. Nucleic Acids Res. 2019, 47, D1013–D1017. [Google Scholar] [CrossRef] [Green Version]
- Hausenloy, D.J.; Bøtker, H.E.; Condorelli, G.; Ferdinandy, P.; Garcia-Dorado, D.; Heusch, G.; Lecour, S.; Van Laake, L.W.; Madonna, R.; Ruiz-Meana, M.; et al. Translating cardioprotection for patient benefit: Position paper from the Working Group of Cellular Biology of the Heart of the European Society of Cardiology. Cardiovasc. Res. 2013, 98, 7–27. [Google Scholar] [CrossRef] [Green Version]
- Neri, M.; Riezzo, I.; Pascale, N.; Pomara, C.; Turillazzi, E. Ischemia/Reperfusion Injury following Acute Myocardial Infarction: A Critical Issue for Clinicians and Forensic Pathologists. Mediat. Inflamm. 2017, 2017, 7018393. [Google Scholar] [CrossRef]
- Rosamond, W.D.; Chambless, L.E.; Heiss, G.; Mosley, T.H.; Coresh, J.; Whitsel, E.; Wagenknecht, L.; Ni, H.; Folsom, A.R. Twenty-two-year trends in incidence of myocardial infarction, coronary heart disease mortality, and case fatality in 4 US communities, 1987–2008. Circulation 2012, 125, 1848–1857. [Google Scholar] [CrossRef] [Green Version]
- Thygesen, K.; Alpert, J.S.; Jaffe, A.S.; Chaitman, B.R.; Bax, J.J.; Morrow, D.A.; White, H.D. Executive Group on behalf of the Joint European Society of Cardiology (ESC)/American College of Cardiology (ACC)/American Heart Association (AHA)/World Heart Federation (WHF) Task Force for the Universal Definition of Myocardial Infarction. Fourth Universal Definition of Myocardial Infarction (2018). J. Am. Coll. Cardiol. 2018, 72, 2231–2264. [Google Scholar]
- Baines, C.P. How and when do myocytes die during ischemia and reperfusion: The late phase. J. Cardiovasc. Pharmacol. Ther. 2011, 16, 239–243. [Google Scholar] [CrossRef]
- Prasad, A.; Stone, G.W.; Holmes, D.R.; Gersh, B. Reperfusion injury, microvascular dysfunction, and cardioprotection: The “dark side” of reperfusion. Circulation 2009, 120, 2105–2112. [Google Scholar] [CrossRef]
- Turer, A.T.; Hill, J.A. Pathogenesis of myocardial ischemia-reperfusion injury and rationale for therapy. Am. J. Cardiol. 2010, 106, 360–368. [Google Scholar] [CrossRef] [Green Version]
- Dirksen, M.T.; Laarman, G.J.; Simoons, M.L.; Duncker, D.J. Reperfusion injury in humans: A review of clinical trials on reperfusion injury inhibitory strategies. Cardiovasc. Res. 2007, 74, 343–355. [Google Scholar] [CrossRef]
- McCafferty, K.; Forbes, S.; Thiemermann, C.; Yaqoob, M.M. The challenge of translating ischemic conditioning from animal models to humans: The role of comorbidities. Dis. Model Mech. 2014, 7, 1321–1333. [Google Scholar] [CrossRef] [Green Version]
- Pan, Z.; Sun, X.; Ren, J.; Li, X.; Gao, X.; Lu, C.; Zhang, Y.; Sun, H.; Wang, Y.; Wang, H.; et al. miR-1 exacerbates cardiac ischemia-reperfusion injury in mouse models. PLoS ONE 2012, 7, e50515. [Google Scholar] [CrossRef]
- Zhai, C.; Tang, G.; Peng, L.; Hu, H.; Qian, G.; Wang, S.; Yao, J.; Zhang, X.; Fang, Y.; Yang, S. Inhibition of microRNA-1 attenuates hypoxia/re-oxygenation-induced apoptosis of cardiomyocytes by directly targeting Bcl-2 but not GADD45Beta. Am. J. Transl. Res. 2015, 7, 1952–1962. [Google Scholar]
- Trotta, M.C.; Ferraro, B.; Messina, A.; Panarese, I.; Gulotta, E.; Nicoletti, G.F.; D’Amico, M.; Pieretti, G. Telmisartan cardioprotects from the ischaemic/hypoxic damage through a miR-1-dependent pathway. J. Cell. Mol. Med. 2019, 23, 6635–6645. [Google Scholar] [CrossRef] [Green Version]
- Gui, Y.J.; Yang, T.; Liu, Q.; Liao, C.X.; Chen, J.Y.; Wang, Y.T.; Hu, J.H.; Xu, D.Y. Soluble epoxide hydrolase inhibitors, t-AUCB, regulated microRNA-1 and its target genes in myocardial infarction mice. Oncotarget 2017, 8, 94635–94649. [Google Scholar] [CrossRef] [Green Version]
- Kang, B.; Hong, J.; Xiao, J.; Zhu, X.; Ni, X.; Zhang, Y.; He, B.; Wang, Z. Involvement of miR-1 in the protective effect of hydrogen sulfide against cardiomyocyte apoptosis induced by ischemia/reperfusion. Mol. Biol. Rep. 2014, 41, 6845–6853. [Google Scholar] [CrossRef]
- Rao, P.K.; Toyama, Y.; Chiang, H.R.; Gupta, S.; Bauer, M.; Medvid, R.; Reinhardt, F.; Liao, R.; Krieger, M.; Jaenisch, R.; et al. Loss of cardiac microRNA-mediated regulation leads to dilated cardiomyopathy and heart failure. Circ. Res. 2009, 105, 585–594. [Google Scholar] [CrossRef] [Green Version]
- Shan, Z.X.; Lin, Q.X.; Deng, C.Y.; Zhu, J.N.; Mai, L.P.; Liu, J.L.; Fu, Y.H.; Liu, X.Y.; Li, Y.X.; Zhang, Y.Y.; et al. miR-1/miR-206 regulate Hsp60 expression contributing to glucose-mediated apoptosis in cardiomyocytes. FEBS Lett. 2010, 584, 3592–3600. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.Y.; Song, Y.H.; Geng, Y.J.; Lin, Q.X.; Shan, Z.X.; Lin, S.G.; Li, Y. Glucose induces apoptosis of cardiomyocytes via microRNA-1 and IGF-1. Biochem. Biophys. Res. Commun. 2008, 376, 548–552. [Google Scholar] [CrossRef]
- Tang, Y.; Zheng, J.; Sun, Y.; Wu, Z.; Liu, Z.; Huang, G. MicroRNA-1 regulates cardiomyocyte apoptosis by targeting Bcl-2. Int. Heart J. 2009, 50, 377–387. [Google Scholar] [CrossRef] [Green Version]
- Zhu, W.S.; Guo, W.; Zhu, J.N.; Tang, C.M.; Fu, Y.H.; Lin, Q.X.; Tan, N.; Shan, Z.X. Hsp90aa1: A novel target gene of miR-1 in cardiac ischemia/reperfusion injury. Sci. Rep. 2016, 6, 24498. [Google Scholar] [CrossRef] [Green Version]
- He, B.; Xiao, J.; Ren, A.J.; Zhang, Y.F.; Zhang, H.; Chen, M.; Xie, B.; Gao, X.G.; Wang, Y.W. Role of mmiR-1 and miR-133a in myocardial ischemic post conditioning. J. Biomed. Sci. 2011, 18, 22. [Google Scholar] [CrossRef] [Green Version]
- Boštjančič, E.; Zidar, N.; Štajer, D.; Glavač, D. MicroRNAs miR-1, miR-133a, miR-133b and miR-208 are dysregulated in human myocardial infarction. Cardiology 2010, 115, 163–169. [Google Scholar] [CrossRef]
- Pinchi, E.; Frati, P.; Aromatario, M.; Cipolloni, L.; Fabbri, M.; La Russa, R.; Maiese, A.; Neri, M.; Santurro, A.; Scopetti, M.; et al. miR-1, miR-499 and miR-208 are sensitive markers to diagnose sudden death due to early acute myocardial infarction. J. Cell. Mol. Med. 2019, 23, 6005–6016. [Google Scholar] [CrossRef] [Green Version]
- Boštjančič, E.; Zidar, N.; Štajer, D.; Glavač, D. MicroRNA miR-1 is up-regulated in remote myocardium in patients with myocardial infarction. Folia Biol. 2010, 56, 27–31. [Google Scholar]
- Kuwabara, Y.; Ono, K.; Horie, T.; Nishi, H.; Nagao, K.; Kinoshita, M.; Watanabe, S.; Baba, O.; Kojima, Y.; Shizuta, S.; et al. Increased microRNA-1 and microRNA-133a levels in serum of patients with cardiovascular disease indicate myocardial damage. Circ. Cardiovasc. Genet. 2011, 4, 446–454. [Google Scholar] [CrossRef]
- Qipshidze Kelm, N.; Piell, K.M.; Wang, E.; Cole, M.P. MicroRNAs as predictive biomarkers for myocardial injury in aged mice following myocardial infarction. J. Cell. Physiol. 2018, 233, 5214–5221. [Google Scholar] [CrossRef]
- van der Weg, K.; Prinzen, F.W.; Gorgels, A.P. Editor′s Choice- Reperfusion cardiac arrhythmias and their relation to reperfusion-induced cell death. Eur. Heart J. Acute Cardiovasc. Care 2019, 8, 142–152. [Google Scholar] [CrossRef] [Green Version]
- Majidi, M.; Kosinski, A.S.; Al-Khatib, S.M.; Lemmert, M.E.; Smolders, L.; van Weert, A.; Reiber, J.H.; Tzivoni, D.; Bär, F.W.; Wellens, H.J.; et al. Reperfusion ventricular arrhythmia ‘bursts’ in TIMI 3 flow restoration with primary angioplasty for anterior ST-elevation myocardial infarction: A more precise definition of reperfusion arrhythmias. Europace 2008, 10, 988–997. [Google Scholar] [CrossRef]
- Majidi, M.; Kosinski, A.S.; Al-Khatib, S.M.; Lemmert, M.E.; Smolders, L.; van Weert, A.; Reiber, J.H.; Tzivoni, D.; Bär, F.W.; Wellens, H.J.; et al. Reperfusion ventricular arrhythmia ‘bursts’ predict larger infarct size despite TIMI 3 flow restoration with primary angioplasty for anterior ST-elevation myocardial infarction. Eur. Heart J. 2009, 30, 757–764. [Google Scholar] [CrossRef] [Green Version]
- Majidi, M.; Kosinski, A.S.; Al-Khatib, S.M.; Smolders, L.; Cristea, E.; Lansky, A.J.; Stone, G.W.; Mehran, R.; Gibbons, R.J.; Crijns, H.J.; et al. Implications of ventricular arrhythmia “bursts” with normal epicardial flow, myocardial blush, and ST-segment recovery in anterior ST-elevation myocardial infarction reperfusion: A biosignature of direct myocellular injury “downstream of downstream”. Eur. Heart J. Acute Cardiovasc. Care. 2015, 4, 51–59. [Google Scholar] [CrossRef]
- van der Weg, K.; Kuijt, W.J.; Bekkers, S.C.; Tijssen, J.G.; Green, C.L.; Lemmert, M.E.; Krucoff, M.W.; Gorgels, A.P. Reperfusion ventricular arrhythmia bursts identify larger infarct size in spite of optimal epicardial and microvascular reperfusion using cardiac magnetic resonance imaging. Eur. Heart J. Acute Cardiovasc. Care 2018, 7, 246–256. [Google Scholar] [CrossRef] [Green Version]
- van der Weg, K.; Kuijt, W.J.; Tijssen, J.G.; Bekkers, S.C.; Haeck, J.D.; Green, C.L.; Lemmert, M.E.; de Winter, R.J.; Gorgels, A.P.; Krucoff, M.W. Prospective evaluation of where reperfusion ventricular arrhythmia “bursts” fit into optimal reperfusion in STEMI. Int. J. Cardiol. 2015, 195, 136–142. [Google Scholar] [CrossRef] [Green Version]
- van der Weg, K.; Majidi, M.; Haeck, J.D.; Tijssen, J.G.; Green, C.L.; Koch, K.T.; Kuijt, W.J.; Krucoff, M.W.; Gorgels, A.P.; de Winter, R.J. Ventricular arrhythmia burst is an independent indicator of larger infarct size even in optimal reperfusion in STEMI. J. Electrocardiol. 2016, 49, 345–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engelen, D.J.; Gressin, V.; Krucoff, M.W.; Theuns, D.A.; Green, C.; Cheriex, E.C.; Maison-Blanche, P.; Dassen, W.R.; Wellens, H.J.; Gorgels, A.P. Usefulness of frequent arrhythmias after epicardial recanalization in anterior wall acute myocardial infarction as a marker of cellular injury leading to poor recovery of left ventricular function. Am. J. Cardiol. 2003, 92, 1143–1149. [Google Scholar] [CrossRef] [PubMed]
- Belevych, A.E.; Sansom, S.E.; Terentyeva, R.; Ho, H.T.; Nishijima, Y.; Martin, M.M.; Jindal, H.K.; Rochira, J.A.; Kunitomo, Y.; Abdellatif, M.; et al. MicroRNA-1 and -133 increase arrhythmogenesis in heart failure by dissociating phosphatase activity from RyR2 complex. PLoS ONE 2011, 6, e28324. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.F.; Ding, Y.J.; Shen, Y.W.; Xue, A.M.; Xu, H.M.; Luo, C.L.; Li, B.X.; Liu, Y.L.; Zhao, Z.Q. MicroRNA-1 represses Cx43 expression in viral myocarditis. Mol. Cell. Biochem. 2012, 362, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Curcio, A.; Torella, D.; Iaconetti, C.; Pasceri, E.; Sabatino, J.; Sorrentino, S.; Giampà, S.; Micieli, M.; Polimeni, A.; Henning, B.J.; et al. MicroRNA-1 downregulation increases connexin 43 displacement and induces ventricular tachyarrhythmias in rodent hypertrophic hearts. PLoS ONE 2013, 8, e70158. [Google Scholar] [CrossRef] [Green Version]
- Liao, C.; Gui, Y.; Guo, Y.; Xu, D. The regulatory function of microRNA-1 in arrhythmias. Mol. Biosyst. 2016, 12, 328–333. [Google Scholar] [CrossRef]
- Schulz, R.; Boengler, K.; Totzeck, A.; Luo, Y.; Garcia-Dorado, D.; Heusch, G. Connexin 43 in ischemic pre- and postconditioning. Heart Fail. Rev. 2007, 12, 261–266. [Google Scholar] [CrossRef]
- Bian, B.; Yu, X.; Wang, Q.; Teng, T.; Nie, J. Atorvastatin protects myocardium against ischemia-reperfusion arrhythmia by increasing Connexin 43 expression: A rat model. Eur. J. Pharmacol. 2015, 768, 13–20. [Google Scholar] [CrossRef]
- Myers, R.; Timofeyev, V.; Li, N.; Kim, C.; Ledford, H.A.; Sirish, P.; Lau, V.; Zhang, Y.; Fayyaz, K.; Singapuri, A.; et al. Feedback mechanisms for cardiac-specific microRNAs and cAMP signaling in electrical remodeling. Circ. Arrhythm. Electrophysiol. 2015, 8, 942–950. [Google Scholar] [CrossRef] [Green Version]
- Yin, C.; Salloum, F.N.; Kukreja, R.C. A novel role of microRNA in late preconditioning: Upregulation of endothelial nitric oxide synthase and heat shock protein 70. Circ. Res. 2009, 104, 572–575. [Google Scholar] [CrossRef]
- Yin, C.; Wang, X.; Kukreja, R.C. Endogenous microRNAs induced by heat-shock reduce myocardial infarction following ischemia-reperfusion in mice. FEBS Lett. 2008, 582, 4137–4142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, R.; Cheng, Y.; Yue, J.; Yang, J.; Liu, X.; Chen, H.; Dean, D.B.; Zhang, C. MicroRNA expression signature and antisense-mediated depletion reveal an essential role of MicroRNA in vascular neointimal lesion formation. Circ. Res. 2007, 100, 1579–1588. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Ji, R.; Yue, J.; Yang, J.; Liu, X.; Chen, H.; Dean, D.B.; Zhang, C. MicroRNAs are aberrantly expressed in hypertrophic heart: Do they play a role in cardiac hypertrophy? Am. J. Pathol. 2007, 170, 1831–1840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thum, T.; Gross, C.; Fiedler, J.; Fischer, T.; Kissler, S.; Bussen, M.; Galuppo, P.; Just, S.; Rottbauer, W.; Frantz, S.; et al. MicroRNA-21 contributes to myocardial disease by stimulating MAP kinase signalling in fibroblasts. Nature 2008, 456, 980–984. [Google Scholar] [CrossRef]
- Butt, M.; Dwivedi, G.; Blann, A.; Khair, O.; YH Lip, G. Endothelial dysfunction: Methods of assessment & implications for cardiovascular diseases. Curr. Pharm. Des. 2010, 16, 3442–3454. [Google Scholar]
- Zuo, K.; Li, M.; Zhang, X.; Lu, C.; Wang, S.; Zhi, K.; He, B. MiR-21 suppresses endothelial progenitor cell proliferation by activating the TGFβ signaling pathway via downregulation of WWP1. Int. J. Clin. Exp. Pathol. 2015, 8, 414–422. [Google Scholar]
- Yong, K.; Dogra, G.; Boudville, N.; Chan, D.; Adams, L.; Ching, H.; Lim, E.M.; Lim, W.H. Interleukin-12 is associated with arterial stiffness in healthy individuals. Am. J. Hypertens. 2013, 26, 159–162. [Google Scholar] [CrossRef] [Green Version]
- Cao, J.; Zhang, K.; Zheng, J.; Dong, R. MicroRNA-146a and -21 cooperate to regulate vascular smooth muscle cell proliferation via modulation of the Notch signaling pathway. Mol. Med. Rep. 2015, 11, 2889–2895. [Google Scholar] [CrossRef] [Green Version]
- Gu, H.; Liu, Z.; Li, Y.; Xie, Y.; Yao, J.; Zhu, Y.; Xu, J.; Dai, Q.; Zhong, C.; Zhu, H.; et al. Serum-Derived Extracellular Vesicles Protect Against Acute Myocardial Infarction by Regulating miR-21/PDCD4 Signaling Pathway. Front. Physiol. 2018, 9, 348. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.; Liu, X.; Zhang, S.; Lin, Y.; Yang, J.; Zhang, C. MicroRNA-21 protects against the H(2)O(2)-induced injury on cardiac myocytes via its target gene PDCD4. J. Mol. Cell. Cardiol. 2009, 47, 5–14. [Google Scholar] [CrossRef] [Green Version]
- Gryshkova, V.; Fleming, A.; McGhan, P.; De Ron, P.; Fleurance, R.; Valentin, J.P.; da Costa, A.N. miR-21-5p as a potential biomarker of inflammatory infiltration in the heart upon acute drug-induced cardiac injury in rats. Toxicol. Lett. 2018, 286, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Zi, M.; Jin, J.; Prehar, S.; Oceandy, D.; Kimura, T.E.; Lei, M.; Neyses, L.; Weston, A.H.; Cartwright, E.J.; et al. Cardiac-specific deletion of mkk4 reveals its role in pathological hypertrophic remodeling but not in physiological cardiac growth. Circ. Res. 2009, 104, 905–914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minamino, T.; Yujiri, T.; Terada, N.; Taffet, G.E.; Michael, L.H.; Johnson, G.L.; Schneider, M.D. MEKK1 is essential for cardiac hypertrophy and dysfunction induced by Gq. Proc. Natl. Acad. Sci. USA 2002, 99, 3866–3871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orogo, A.M.; Gustafsson, Å.B. Cell death in the myocardium: My heart won’t go on. IUBMB Life 2013, 65, 651–656. [Google Scholar] [CrossRef]
- Liu, B.; Wang, B.; Zhang, X.; Lock, R.; Nash, T.; Vunjak-Novakovic, G. Cell type-specific microRNA therapies for myocardial infarction. Sci. Transl. Med. 2021, 13, eabd0914. [Google Scholar] [CrossRef]
- Prabhu, S.D.; Frangogiannis, N.G. The Biological Basis for Cardiac Repair After Myocardial Infarction: From Inflammation to Fibrosis. Circ. Res. 2016, 119, 91–112. [Google Scholar] [CrossRef]
- Epelman, S.; Liu, P.P.; Mann, D.L. Role of innate and adaptive immune mechanisms in cardiac injury and repair. Nat. Rev. Immunol. 2015, 15, 117–129. [Google Scholar] [CrossRef]
- Forte, E.; Furtado, M.B.; Rosenthal, N. The interstitium in cardiac repair: Role of the immune-stromal cell interplay. Nat. Rev. Cardiol. 2018, 15, 601–616. [Google Scholar] [CrossRef]
- Frangogiannis, N.G. The inflammatory response in myocardial injury, repair, and remodelling. Nat. Rev. Cardiol. 2014, 11, 255–265. [Google Scholar] [CrossRef] [Green Version]
- Psarras, S.; Beis, D.; Nikouli, S.; Tsikitis, M.; Capetanaki, Y. Three in a Box: Understanding Cardiomyocyte, Fibroblast, and Innate Immune Cell Interactions to Orchestrate Cardiac Repair Processes. Front. Cardiovasc. Med. 2019, 6, 32. [Google Scholar] [CrossRef]
- Molenaar, B.; van Rooij, E. Single-Cell Sequencing of the Mammalian Heart. Circ. Res. 2018, 129, 1033–1035. [Google Scholar] [CrossRef] [PubMed]
- Molenaar, B.; Timmer, L.T.; Droog, M.; Perini, I.; Versteeg, D.; Kooijman, L.; Monshouwer-Kloots, J.; de Ruiter, H.; Gladka, M.M.; van Rooij, E. Single-cell transcriptomics following ischemic injury identifies a role for B2M in cardiac repair. Commun. Biol. 2021, 4, 146. [Google Scholar] [CrossRef] [PubMed]
- Mouton, A.J.; DeLeon-Pennell, K.Y.; Gonzalez, O.J.R.; Flynn, E.R.; Freeman, T.C.; Saucerman, J.J.; Garrett, M.R.; Ma, Y.; Harmancey, R.; Lindsey, M.L. Mapping macrophage polarization over the myocardial infarction time continuum. Basic Res. Cardiol. 2018, 113, 26. [Google Scholar] [CrossRef] [Green Version]
- Mouton, A.J.; Ma, Y.; Gonzalez, O.J.R.; Daseke, M.J.; Flynn, E.R.; Freeman, T.C.; Garrett, M.R.; DeLeon-Pennell, K.Y.; Lindsey, M.L. Fibroblast polarization over the myocardial infarction time continuum shifts roles from inflammation to angiogenesis. Basic Res. Cardiol. 2019, 114, 6. [Google Scholar] [CrossRef] [Green Version]
- Farbehi, N.; Patrick, R.; Dorison, A.; Xaymardan, M.; Janbandhu, V.; Wystub-Lis, K.; Ho, J.W.; Nordon, R.E.; Harvey, R.P. Single-cell expression profiling reveals dynamic flux of cardiac stromal, vascular and immune cells in health and injury. Elife. 2019, 8, e43882. [Google Scholar] [CrossRef]
- Nomura, S.; Satoh, M.; Fujita, T.; Higo, T.; Sumida, T.; Ko, T.; Yamaguchi, T.; Tobita, T.; Naito, A.T.; Ito, M.; et al. Cardiomyocyte gene programs encoding morphological and functional signatures in cardiac hypertrophy and failure. Nat. Commun. 2018, 9, 4435. [Google Scholar] [CrossRef]
- Kannan, S.; Miyamoto, M.; Lin, B.L.; Zhu, R.; Murphy, S.; Kass, D.A.; Andersen, P.; Kwon, C. Large Particle Fluorescence-Activated Cell Sorting Enables High-Quality Single-Cell RNA Sequencing and Functional Analysis of Adult Cardiomyocytes. Circ. Res. 2019, 125, 567–569. [Google Scholar] [CrossRef]
- Chevalier, M.; Vermij, S.H.; Wyler, K.; Gillet, L.; Keller, I.; Abriel, H. Transcriptomic analyses of murine ventricular cardiomyocytes. Sci. Data 2018, 5, 180170. [Google Scholar] [CrossRef] [Green Version]
- Yellon, D.M.; Hausenloy, D.J. Myocardial reperfusion injury. N. Engl. J. Med. 2007, 357, 1121–1135. [Google Scholar] [CrossRef]
- Lijnen, P.J.; Petrov, V.V.; Fagard, R.H. Induction of cardiac fibrosis by transforming growth factor-beta(1). Mol. Genet. Metab. 2000, 71, 418–435. [Google Scholar] [CrossRef]
- Ye, H.; Cai, P.C.; Zhou, Q.; Ma, W.L. Transforming growth factor-β1 suppresses the up-regulation of matrix metalloproteinase-2 by lung fibroblasts in response to tumor necrosis factor-α. Wound Repair Regen. 2011, 19, 392–399. [Google Scholar] [CrossRef] [PubMed]
- Li, A.H.; Liu, P.P.; Villarreal, F.J.; Garcia, R.A. Dynamic changes in myocardial matrix and relevance to disease: Translational perspectives. Circ. Res. 2014, 114, 916–927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, W.; Li, X.; Li, C.; Wan, L.; Shi, H.; Song, X.; Liu, X.; Chen, X.; Zhang, C.; Shan, H.; et al. TGFBR3, a potential negative regulator of TGF-β signaling, protects cardiac fibroblasts from hypoxia-induced apoptosis. J. Cell. Physiol. 2011, 226, 2586–2594. [Google Scholar] [CrossRef]
- Liang, H.; Zhang, C.; Ban, T.; Liu, Y.; Mei, L.; Piao, X.; Zhao, D.; Lu, Y.; Chu, W.; Yang, B. A novel reciprocal loop between microRNA-21 and TGFβRIII is involved in cardiac fibrosis. Int. J. Biochem. Cell. Biol. 2012, 44, 2152–2160. [Google Scholar] [CrossRef]
- Yang, L.; Wang, B.; Zhou, Q.; Wang, Y.; Liu, X.; Liu, Z.; Zhan, Z. MicroRNA-21 prevents excessive inflammation and cardiac dysfunction after myocardial infarction through targeting KBTBD7. Cell Death Dis. 2018, 9, 769. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, M.M.; Pedersen, J.S. miRNA activity inferred from single cell mRNA expression. Sci. Rep. 2021, 11, 9170. [Google Scholar] [CrossRef]
- Oliveira-Carvalho, V.; Da Silva, M.M.F.; Guimaraes, G.V.; Bacal, F.; Bocchi, E.A. MicroRNAs: New players in heart failure. Mol. Biol. Rep. 2013, 40, 2663–2670. [Google Scholar] [CrossRef]
- Siebert, V.; Allencherril, J.; Ye, Y.; Wehrens, X.H.; Birnbaum, Y. The Role of Non-coding RNAs in Ischemic Myocardial Reperfusion Injury. Cardiovasc. Drugs Ther. 2019, 33, 489–498. [Google Scholar] [CrossRef]
- van Rooij, E. The art of microRNA research. Circ. Res. 2011, 108, 219–234. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, Z.; Gemeinhart, R.A. Progress in microRNA delivery. J. Control. Release 2013, 172, 962–974. [Google Scholar] [CrossRef] [Green Version]
- Simonson, B.; Das, S. MicroRNA Therapeutics: The Next Magic Bullet? Mini. Rev. Med. Chem. 2015, 15, 467–474. [Google Scholar] [CrossRef]
- Kamps, J.A.; Krenning, G. Micromanaging cardiac regeneration: Targeted delivery of microRNAs for cardiac repair and regeneration. World J. Cardiol. 2016, 8, 163–179. [Google Scholar] [CrossRef]
- van Rooij, E.; Purcell, A.L.; Levin, A.A. Developing microRNA therapeutics. Circ. Res. 2012, 110, 496–507. [Google Scholar] [CrossRef]
- Manoharan, M. 2′-carbohydrate modifications in antisense oligonucleotide therapy: Importance of conformation, configuration and conjugation. Biochim. Biophys. Acta 1999, 1489, 117–130. [Google Scholar] [CrossRef]
- Prakash, T.P.; Bhat, B. 2’-Modified oligonucleotides for antisense therapeutics. Curr. Top. Med. Chem. 2007, 7, 641–649. [Google Scholar] [CrossRef]
- Stenvang, J.; Kauppinen, S. MicroRNAs as targets for antisense-based therapeutics. Expert Opin. Biol. Ther. 2008, 8, 59–81. [Google Scholar] [CrossRef]
- Mathiyalagan, P.; Sahoo, S. Exosomes-Based Gene Therapy for MicroRNA Delivery. Methods Mol. Biol. 2017, 1521, 139–152. [Google Scholar]
- Campani, V.; De Rosa, G.; Misso, G.; R Zarone, M.; Grimaldi, A. Lipid Nanoparticles to Deliver miRNA in Cancer. Curr. Pharm. Biotechnol. 2016, 17, 741–749. [Google Scholar] [CrossRef]
- Qiu, G.; Zheng, G.; Ge, M.; Wang, J.; Huang, R.; Shu, Q.; Xu, J. Mesenchymal stem cell-derived extracellular vesicles affect disease outcomes via transfer of microRNAs. Stem Cell Res. Ther. 2018, 9, 320. [Google Scholar] [CrossRef]
- Bejerano, T.; Etzion, S.; Elyagon, S.; Etzion, Y.; Cohen, S. Nanoparticle Delivery of miRNA-21 Mimic to Cardiac Macrophages Improves Myocardial Remodeling after Myocardial Infarction. Nano Lett. 2018, 18, 5885–5891. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jayawardena, E.; Medzikovic, L.; Ruffenach, G.; Eghbali, M. Role of miRNA-1 and miRNA-21 in Acute Myocardial Ischemia-Reperfusion Injury and Their Potential as Therapeutic Strategy. Int. J. Mol. Sci. 2022, 23, 1512. https://doi.org/10.3390/ijms23031512
Jayawardena E, Medzikovic L, Ruffenach G, Eghbali M. Role of miRNA-1 and miRNA-21 in Acute Myocardial Ischemia-Reperfusion Injury and Their Potential as Therapeutic Strategy. International Journal of Molecular Sciences. 2022; 23(3):1512. https://doi.org/10.3390/ijms23031512
Chicago/Turabian StyleJayawardena, Eranthi, Lejla Medzikovic, Gregoire Ruffenach, and Mansoureh Eghbali. 2022. "Role of miRNA-1 and miRNA-21 in Acute Myocardial Ischemia-Reperfusion Injury and Their Potential as Therapeutic Strategy" International Journal of Molecular Sciences 23, no. 3: 1512. https://doi.org/10.3390/ijms23031512
APA StyleJayawardena, E., Medzikovic, L., Ruffenach, G., & Eghbali, M. (2022). Role of miRNA-1 and miRNA-21 in Acute Myocardial Ischemia-Reperfusion Injury and Their Potential as Therapeutic Strategy. International Journal of Molecular Sciences, 23(3), 1512. https://doi.org/10.3390/ijms23031512