
Genome-Wide Prediction of Transcription Start Sites in Conifers

 ,
,  and
and 
Abstract
:1. Introduction
2. Results
2.1. Prediction of TSS in Four Conifer Species
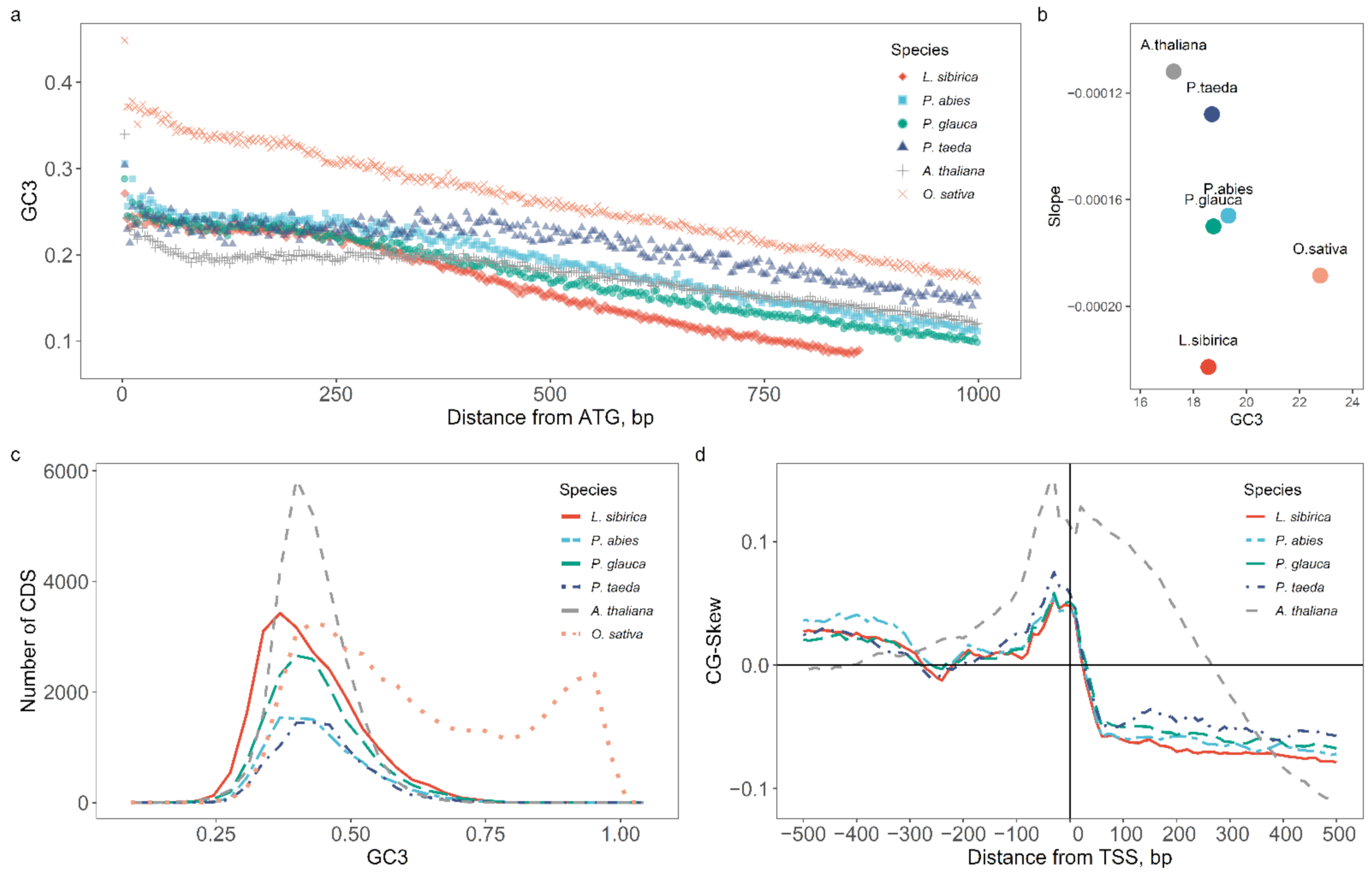
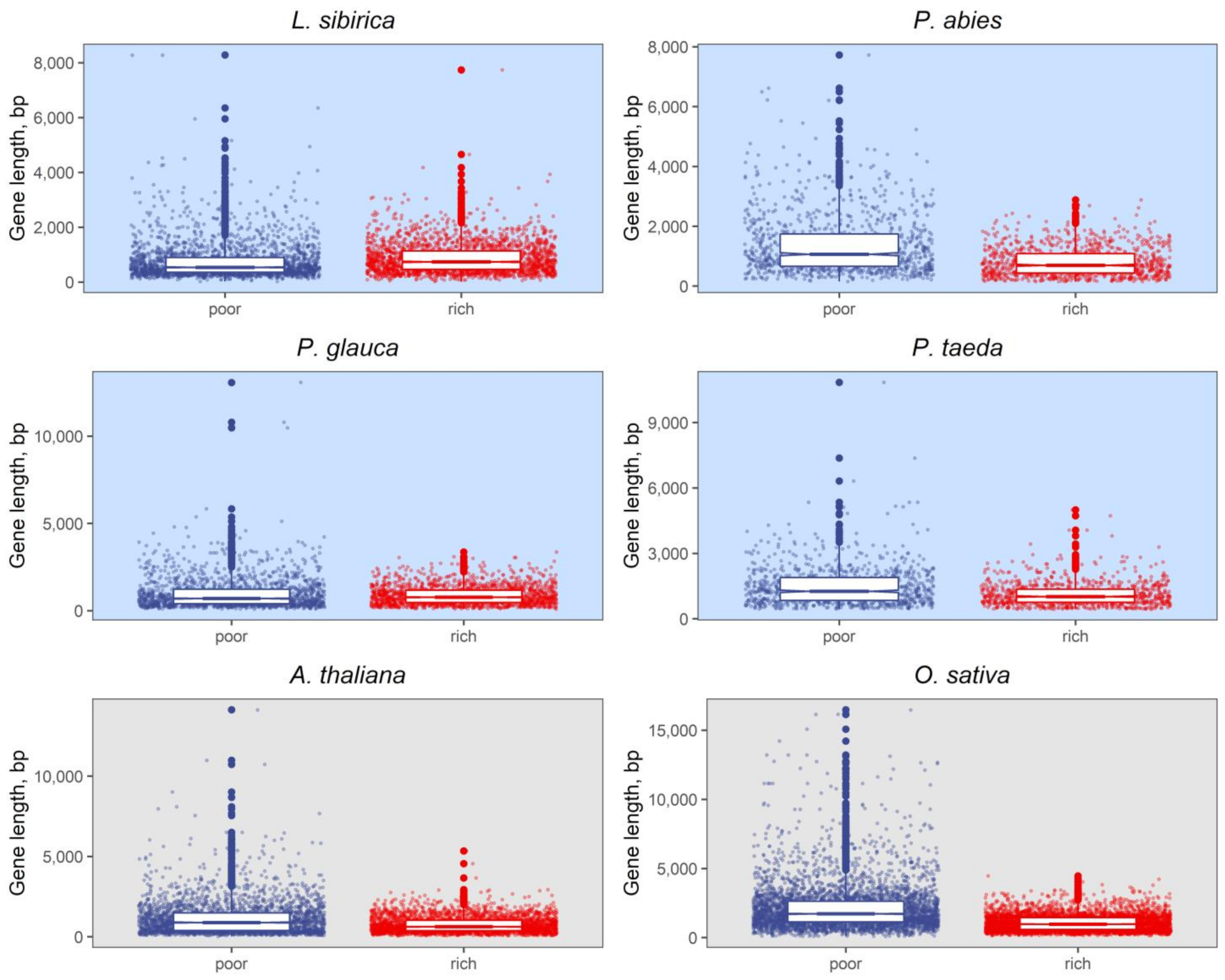
2.2. Nucleotide Composition Analysis of Promoter and Coding Sequences
3. Discussion
3.1. Prediction of TSS
3.2. Nucleotide Composition of Promoters and Coding Regions
4. Materials and Methods
4.1. Genome Assemblies and Annotations
4.2. Gene Filtering
4.3. Prediction of TSS
4.4. Nucleotide Composition Analysis
4.5. Genome Visualization
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tatarinova, T.; Kryshchenko, A.; Triska, M.; Hassan, M.; Murphy, D.; Neely, M.; Schumitzky, A. NPEST: A nonparametric method and a database for transcription start site prediction. Quant. Biol. 2013, 1, 261–271. [Google Scholar] [CrossRef] [PubMed]
- Reyes, A.; Huber, W. Alternative start and termination sites of transcription drive most transcript isoform differences across human tissues. Nucleic Acids Res. 2018, 46, 582–592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juven-Gershon, T.; Kadonaga, J.T. Regulation of gene expression via the core promoter and the basal transcriptional machinery. Dev. Biol. 2010, 339, 225–229. [Google Scholar] [CrossRef] [Green Version]
- Alexandrov, N.N.; Troukhan, M.E.; Brover, V.V.; Tatarinova, T.; Flavell, R.B.; Feldmann, K.A. Features of Arabidopsis genes and genome discovered using full-length cDNAs. Plant Mol. Biol. 2006, 60, 69–85. [Google Scholar] [CrossRef]
- Alexandrov, N.N.; Brover, V.V.; Freidin, S.; Troukhan, M.E.; Tatarinova, T.V.; Zhang, H.; Swaller, T.J.; Lu, Y.-P.; Bouck, J.; Flavell, R.B.; et al. Insights into corn genes derived from large-scale cDNA sequencing. Plant Mol. Biol. 2009, 69, 179–194. [Google Scholar] [CrossRef] [Green Version]
- Troukhan, M.; Tatarinova, T.; Bouck, J.; Flavell, R.B.; Alexandrov, N.N. Genome-wide discovery of cis-elements in promoter sequences using gene expression. OMICS 2009, 13, 139–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tatarinova, T.V.; Chekalin, E.; Nikolsky, Y.; Bruskin, S.; Chebotarov, D.; McNally, K.L.; Alexandrov, N. Nucleotide diversity analysis highlights functionally important genomic regions. Sci. Rep. 2016, 6, 35730. [Google Scholar] [CrossRef]
- Triska, M.; Ivliev, A.; Nikolsky, Y.; Tatarinova, T.V. Analysis of cis-regulatory elements in gene co-expression networks in cancer. Methods Mol. Biol. 2017, 1613, 291–310. [Google Scholar] [CrossRef]
- Roy, A.L.; Singer, D.S. Core promoters in transcription: Old problem, new insights. Trends Biochem. Sci. 2015, 40, 165–171. [Google Scholar] [CrossRef] [Green Version]
- Sandelin, A.; Carninci, P.; Lenhard, B.; Ponjavic, J.; Hayashizaki, Y.; Hume, D.A. Mammalian RNA polymerase II core promoters: Insights from genome-wide studies. Nat. Rev. Genet. 2007, 8, 424–436. [Google Scholar] [CrossRef]
- Lagrange, T.; Kapanidis, A.N.; Tang, H.; Reinberg, D.; Ebright, R.H. New core promoter element in RNA polymerase II-dependent transcription: Sequence-specific DNA binding by transcription factor IIB. Genes Dev. 1998, 12, 34–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, W.; Roberts, S.G.E. A core promoter element downstream of the TATA box that is recognized by TFIIB. Genes Dev. 2005, 19, 2418–2423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kadonaga, J.T. The DPE, a core promoter element for transcription by RNA polymerase II. Exp. Mol. Med. 2002, 34, 259–264. [Google Scholar] [CrossRef]
- Burke, T.W.; Kadonaga, J.T. Drosophila TFIID binds to a conserved downstream basal promoter element that is present in many TATA-box-deficient promoters. Genes Dev. 1996, 10, 711–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, D.-H.; Gershenzon, N.; Gupta, M.; Ioshikhes, I.P.; Reinberg, D.; Lewis, B.A. Functional characterization of core promoter elements: The downstream core element is recognized by TAF1. Mol. Cell Biol. 2005, 25, 9674–9686. [Google Scholar] [CrossRef] [Green Version]
- Franco-Zorrilla, J.M.; Solano, R. Identification of plant transcription factor target sequences. Biochim. Biophys. Acta Gene Regul. Mech. 2017, 1860, 21–30. [Google Scholar] [CrossRef]
- Morton, T.; Petricka, J.; Corcoran, D.L.; Li, S.; Winter, C.M.; Carda, A.; Benfey, P.N.; Ohler, U.; Megraw, M. Paired-end analysis of transcription start sites in Arabidopsis reveals plant-specific promoter signatures. Plant Cell 2014, 26, 2746–2760. [Google Scholar] [CrossRef] [Green Version]
- Bianchi, V.J.; Rubio, M.; Trainotti, L.; Verde, I.; Bonghi, C.; Martínez-Gómez, P. Prunus transcription factors: Breeding perspectives. Front. Plant Sci. 2015, 6, 443. [Google Scholar] [CrossRef] [Green Version]
- Narang, V.; Sung, W.-K.; Mittal, A. Computational modeling of oligonucleotide positional densities for human promoter prediction. Artif. Intell. Med. 2005, 35, 107–119. [Google Scholar] [CrossRef]
- Shahmuradov, I.A.; Umarov, R.K.; Solovyev, V.V. TSSPlant: A new tool for prediction of plant Pol II promoters. Nucleic Acids Res. 2017, 45, e65. [Google Scholar] [CrossRef]
- Pachganov, S.; Murtazalieva, K.; Zarubin, A.; Sokolov, D.; Chartier, D.R.; Tatarinova, T.V. TransPrise: A novel machine learning approach for eukaryotic promoter prediction. PeerJ 2019, 7, e7990. [Google Scholar] [CrossRef] [PubMed]
- De Medeiros Oliveira, M.; Bonadio, I.; Lie de Melo, A.; Mendes Souza, G.; Durham, A.M. TSSFinder—Fast and accurate ab initio prediction of the core promoter in eukaryotic genomes. Brief. Bioinform. 2021, 22, bbab198. [Google Scholar] [CrossRef]
- Ibraheem, O.; Botha, C.E.J.; Bradley, G. In silico analysis of cis-acting regulatory elements in 5′ regulatory regions of sucrose transporter gene families in rice (Oryza sativa Japonica) and Arabidopsis thaliana. Comput. Biol. Chem. 2010, 34, 268–283. [Google Scholar] [CrossRef] [PubMed]
- Duraisamy, G.S.; Mishra, A.K.; Kocabek, T.; Matoušek, J. Identification and characterization of promoters and cis-regulatory elements of genes involved in secondary metabolites production in hop (Humulus lupulus L.). Comput. Biol. Chem. 2016, 64, 346–352. [Google Scholar] [CrossRef] [PubMed]
- Wong, D.C.J.; Lopez Gutierrez, R.; Gambetta, G.A.; Castellarin, S.D. Genome-wide analysis of cis-regulatory element structure and discovery of motif-driven gene co-expression networks in grapevine. DNA Res. 2017, 24, 311–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumari, S.; Ware, D. Genome-wide computational prediction and analysis of core promoter elements across plant monocots and dicots. PLoS ONE 2013, 8, e79011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gan, Y.; Guan, J.; Zhou, S. A Comparison study on feature selection of DNA structural properties for promoter prediction. BMC Bioinform. 2012, 13, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanhere, A.; Bansal, M. Structural properties of promoters: Similarities and differences between prokaryotes and eukaryotes. Nucleic Acids Res. 2005, 33, 3165–3175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Triska, M.; Solovyev, V.; Baranova, A.; Kel, A.; Tatarinova, T.V. Nucleotide patterns aiding in prediction of eukaryotic promoters. PLoS ONE 2017, 12, e0187243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yella, V.R.; Kumar, A.; Bansal, M. Identification of putative promoters in 48 eukaryotic genomes on the basis of DNA free energy. Sci. Rep. 2018, 8, 4520. [Google Scholar] [CrossRef] [Green Version]
- Kozobay-Avraham, L.; Hosid, S.; Bolshoy, A. Curvature distribution in prokaryotic genomes. Silico Biol. 2004, 4, 361–375. [Google Scholar]
- Kumar, A.; Bansal, M. Unveiling DNA structural features of promoters associated with various types of TSSs in prokaryotic transcriptomes and their role in gene expression. DNA Res. 2017, 24, 25–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandey, S.P.; Krishnamachari, A. Computational analysis of plant RNA Pol-II Promoters. Biosystems 2006, 83, 38–50. [Google Scholar] [CrossRef] [PubMed]
- Zuo, Y.-C.; Li, Q.-Z. Identification of TATA and TATA-less promoters in plant genomes by integrating diversity measure, GC-skew and DNA geometric flexibility. Genomics 2011, 97, 112–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tatarinova, T.; Brover, V.; Troukhan, M.; Alexandrov, N. Skew in CG content near the transcription start site in Arabidopsis thaliana. Bioinformatics 2003, 19, i313–i314. [Google Scholar] [CrossRef] [Green Version]
- Fujimori, S.; Washio, T.; Tomita, M. GC-compositional strand bias around transcription start sites in plants and fungi. BMC Genom. 2005, 6, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aerts, S.; Thijs, G.; Dabrowski, M.; Moreau, Y.; De Moor, B. Comprehensive analysis of the base composition around the transcription start site in metazoa. BMC Genom. 2004, 5, 34. [Google Scholar] [CrossRef] [Green Version]
- Carels, N.; Bernardi, G. Two classes of genes in plants. Genetics 2000, 154, 1819–1825. [Google Scholar] [CrossRef] [PubMed]
- Tatarinova, T.V.; Alexandrov, N.N.; Bouck, J.B.; Feldmann, K.A. GC3 biology in corn, rice, sorghum and other grasses. BMC Genom. 2010, 11, 308. [Google Scholar] [CrossRef] [Green Version]
- Clément, Y.; Fustier, M.-A.; Nabholz, B.; Glémin, S. The bimodal distribution of genic GC content is ancestral to monocot species. Genome Biol. Evol. 2014, 7, 336–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serres-Giardi, L.; Belkhir, K.; David, J.; Glémin, S. Patterns and evolution of nucleotide landscapes in seed plants. Plant Cell 2012, 24, 1379–1397. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.-L.; Tatarinova, T.V.; Rosli, R.; Amiruddin, N.; Azizi, N.; Halim, M.A.A.; Sanusi, N.S.N.M.; Jayanthi, N.; Ponomarenko, P.; Triska, M.; et al. Evidence-based gene models for structural and functional annotations of the oil palm genome. Biol. Direct 2017, 12, 21. [Google Scholar] [CrossRef] [Green Version]
- Nystedt, B.; Street, N.R.; Wetterbom, A.; Zuccolo, A.; Lin, Y.-C.; Scofield, D.G.; Vezzi, F.; Delhomme, N.; Giacomello, S.; Alexeyenko, A.; et al. The Norway spruce genome sequence and conifer genome evolution. Nature 2013, 497, 579–584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warren, R.L.; Keeling, C.I.; Yuen, M.M.S.; Raymond, A.; Taylor, G.A.; Vandervalk, B.P.; Mohamadi, H.; Paulino, D.; Chiu, R.; Jackman, S.D.; et al. Improved white spruce (Picea glauca) genome assemblies and annotation of large gene families of conifer terpenoid and phenolic defense metabolism. Plant J. 2015, 83, 189–212. [Google Scholar] [CrossRef] [PubMed]
- Zimin, A.V.; Stevens, K.A.; Crepeau, M.W.; Puiu, D.; Wegrzyn, J.L.; Yorke, J.A.; Langley, C.H.; Neale, D.B.; Salzberg, S.L. An improved assembly of the loblolly pine mega-genome using long-read single-molecule sequencing. GigaScience 2017, 6, giw016. [Google Scholar] [CrossRef]
- Kuzmin, D.A.; Feranchuk, S.I.; Sharov, V.V.; Cybin, A.N.; Makolov, S.V.; Putintseva, Y.A.; Oreshkova, N.V.; Krutovsky, K.V. Stepwise large genome assembly approach: A case of Siberian larch (Larix sibirica Ledeb). BMC Bioinform. 2019, 20, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holt, C.; Yandell, M. MAKER2: An annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinform. 2011, 12, 491. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, N. LEAFY, a pioneer transcription factor in plants: A mini-review. Front. Plant Sci. 2021, 12, 701406. [Google Scholar] [CrossRef]
- Papuga, J.; Hoffmann, C.; Dieterle, M.; Moes, D.; Moreau, F.; Tholl, S.; Steinmetz, A.; Thomas, C. Arabidopsis LIM proteins: A family of actin bundlers with distinct expression patterns and modes of regulation. Plant Cell 2010, 22, 3034–3052. [Google Scholar] [CrossRef] [Green Version]
- Wong, G.K.-S.; Wang, J.; Tao, L.; Tan, J.; Zhang, J.; Passey, D.A.; Yu, J. Compositional gradients in Gramineae genes. Genome Res. 2002, 12, 851–856. [Google Scholar] [CrossRef] [Green Version]
- Biłas, R.; Szafran, K.; Hnatuszko-Konka, K.; Kononowicz, A.K. Cis-regulatory elements used to control gene expression in plants. Plant Cell Tiss. Organ. Cult. 2016, 127, 269–287. [Google Scholar] [CrossRef] [Green Version]
- Messing, J.; Geraghty, D.; Heidecker, G.; Hu, N.-T.; Kridl, J.; Rubenstein, I. Plant gene structure. In Genetic Engineering of Plants: An Agricultural Perspective; Kosuge, T., Meredith, C.P., Hollaender, A., Wilson, C.M., Eds.; Springer: Boston, MA, USA, 1983; pp. 211–227. ISBN 978-1-4684-4544-2. [Google Scholar]
- Porto, M.S.; Pinheiro, M.P.N.; Batista, V.G.L.; dos Santos, R.C.; de Albuquerque Melo Filho, P.; de Lima, L.M. Plant promoters: An approach of structure and function. Mol. Biotechnol. 2014, 56, 38–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhadi, S.R.; Krom, N.; Ramakrishna, W. Genome-wide comparative analysis of putative bidirectional promoters from rice, Arabidopsis and Populus. Gene 2009, 429, 65–73. [Google Scholar] [CrossRef]
- Krom, N.; Ramakrishna, W. Comparative analysis of divergent and convergent gene pairs and their expression patterns in rice, Arabidopsis, and Populus. Plant Physiol. 2008, 147, 1763–1773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, Y.Y.; Yoshioka, Y.; Hyakumachi, M.; Obokata, J. Characteristics of core promoter types with respect to gene structure and expression in Arabidopsis thaliana. DNA Res. 2011, 18, 333–342. [Google Scholar] [CrossRef] [Green Version]
- Portales-Casamar, E.; Thongjuea, S.; Kwon, A.T.; Arenillas, D.; Zhao, X.; Valen, E.; Yusuf, D.; Lenhard, B.; Wasserman, W.W.; Sandelin, A. JASPAR 2010: The greatly expanded open-access database of transcription factor binding profiles. Nucleic Acids Res. 2010, 38, D105–D110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, F.; Yang, D.-C.; Meng, Y.-Q.; Jin, J.; Gao, G. PlantRegMap: Charting functional regulatory maps in plants. Nucleic Acids Res. 2020, 48, D1104–D1113. [Google Scholar] [CrossRef]
- Wingender, E. The TRANSFAC project as an example of framework technology that supports the analysis of genomic regulation. Brief. Bioinform. 2008, 9, 326–332. [Google Scholar] [CrossRef] [Green Version]
- Wasserman, W.W.; Sandelin, A. Applied bioinformatics for the identification of regulatory elements. Nat. Rev. Genet. 2004, 5, 276–287. [Google Scholar] [CrossRef]
- Liu, C.; Zhang, T. Expansion and stress responses of the AP2/EREBP superfamily in cotton. BMC Genom. 2017, 18, 118. [Google Scholar] [CrossRef] [Green Version]
- Dietz, K.-J.; Vogel, M.O.; Viehhauser, A. AP2/EREBP transcription factors are part of gene regulatory networks and integrate metabolic, hormonal and environmental signals in stress acclimation and retrograde signalling. Protoplasma 2010, 245, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Svingen, T.; Tonissen, K.F. Hox transcription factors and their elusive mammalian gene targets. Heredity 2006, 97, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Noyes, M.B.; Christensen, R.G.; Wakabayashi, A.; Stormo, G.D.; Brodsky, M.H.; Wolfe, S.A. Analysis of homeodomain specificities allows the family-wide prediction of preferred recognition sites. Cell 2008, 133, 1277–1289. [Google Scholar] [CrossRef] [Green Version]
- Guo, M.; Liu, J.-H.; Ma, X.; Luo, D.-X.; Gong, Z.-H.; Lu, M.-H. The plant heat stress transcription factors (HSFs): Structure, regulation, and function in response to abiotic stresses. Front. Plant Sci. 2016, 7, 114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, G.; Mittler, R. Could heat shock transcription factors function as hydrogen peroxide sensors in plants? Ann. Bot. 2006, 98, 279–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prouse, M.B.; Campbell, M.M. Interactions between the R2R3-MYB transcription factor, AtMYB61, and target DNA binding sites. PLoS ONE 2013, 8, e65132. [Google Scholar] [CrossRef]
- Wang, B.; Luo, Q.; Li, Y.; Yin, L.; Zhou, N.; Li, X.; Gan, J.; Dong, A. Structural insights into target DNA recognition by R2R3-MYB transcription factors. Nucleic Acids Res. 2020, 48, 460–471. [Google Scholar] [CrossRef]
- Rangannan, V.; Bansal, M. High-quality annotation of promoter regions for 913 bacterial genomes. Bioinformatics 2010, 26, 3043–3050. [Google Scholar] [CrossRef] [Green Version]
- Duval, I.; Lachance, D.; Giguère, I.; Bomal, C.; Morency, M.-J.; Pelletier, G.; Boyle, B.; MacKay, J.J.; Séguin, A. Large-scale screening of transcription factor–promoter interactions in spruce reveals a transcriptional network involved in vascular development. J. Exp. Bot. 2014, 65, 2319–2333. [Google Scholar] [CrossRef] [Green Version]
- Sena, J.S.; Lachance, D.; Duval, I.; Nguyen, T.T.A.; Stewart, D.; Mackay, J.; Séguin, A. Functional analysis of the PgCesA3 white spruce cellulose synthase gene promoter in secondary xylem. Front. Plant Sci. 2019, 10, 626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bedon, F.; Levasseur, C.; Grima-Pettenati, J.; Séguin, A.; MacKay, J. Sequence analysis and functional characterization of the promoter of the Picea glauca cinnamyl alcohol dehydrogenase gene in transgenic white spruce plants. Plant Cell Rep. 2009, 28, 787–800. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.; Zhao, X.; Kelly, K.A.; Venn, O.; Higgins, J.D.; Yelina, N.E.; Hardcastle, T.J.; Ziolkowski, P.A.; Copenhaver, G.P.; Franklin, F.C.H.; et al. Arabidopsis meiotic crossover hotspots overlap with H2A.Z nucleosomes at gene promoters. Nat. Genet. 2013, 45, 1327–1336. [Google Scholar] [CrossRef] [Green Version]
- Hellsten, U.; Wright, K.M.; Jenkins, J.; Shu, S.; Yuan, Y.; Wessler, S.R.; Schmutz, J.; Willis, J.H.; Rokhsar, D.S. Fine-scale variation in meiotic recombination in Mimulus inferred from population shotgun sequencing. Proc. Natl. Acad. Sci. USA 2013, 110, 19478–19482. [Google Scholar] [CrossRef] [Green Version]
- Fortes, G.G.; Bouza, C.; Martínez, P.; Sánchez, L. Diversity in isochore structure among cold-blooded vertebrates based on GC content of coding and non-coding sequences. Genetica 2007, 129, 281–289. [Google Scholar] [CrossRef] [PubMed]
- Jørgensen, F.G.; Schierup, M.H.; Clark, A.G. Heterogeneity in regional GC content and differential usage of codons and amino acids in GC-poor and GC-rich regions of the genome of Apis mellifera. Mol. Biol. Evol. 2007, 24, 611–619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lynch, D.B.; Logue, M.E.; Butler, G.; Wolfe, K.H. Chromosomal G + C content evolution in yeasts: Systematic interspecies differences, and GC-poor troughs at centromeres. Genome Biol. Evol. 2010, 2, 572–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neale, D.B.; Wegrzyn, J.L.; Stevens, K.A.; Zimin, A.V.; Puiu, D.; Crepeau, M.W.; Cardeno, C.; Koriabine, M.; Holtz-Morris, A.E.; Liechty, J.D.; et al. Decoding the massive genome of loblolly pine using haploid DNA and novel assembly strategies. Genome Biol. 2014, 15, R59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wegrzyn, J.L.; Liechty, J.D.; Stevens, K.A.; Wu, L.-S.; Loopstra, C.A.; Vasquez-Gross, H.A.; Dougherty, W.M.; Lin, B.Y.; Zieve, J.J.; Martínez-García, P.J.; et al. Unique features of the loblolly pine (Pinus taeda L.) megagenome revealed through sequence annotation. Genetics 2014, 196, 891–909. [Google Scholar] [CrossRef] [Green Version]
- Birol, I.; Raymond, A.; Jackman, S.D.; Pleasance, S.; Coope, R.; Taylor, G.A.; Yuen, M.M.S.; Keeling, C.I.; Brand, D.; Vandervalk, B.P.; et al. Assembling the 20 Gb white spruce (Picea glauca) genome from whole-genome shotgun sequencing data. Bioinformatics 2013, 29, 1492–1497. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Kel, A.E.; Gössling, E.; Reuter, I.; Cheremushkin, E.; Kel-Margoulis, O.V.; Wingender, E. MATCH: A tool for searching transcription factor binding sites in DNA sequences. Nucleic Acids Res. 2003, 31, 3576–3579. [Google Scholar] [CrossRef] [PubMed] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Assembly and Annotations Parameters | Picea abies [43] | Picea glauca [44] | Pinus taeda [45] | Larix sibirica [46] |
|---|---|---|---|---|
| Estimated genome size, Gbp | 19.57 | 15.79 | 20.15 | 12.03 |
| Assembly length, Gbp | 12.30 | 25.47 | 22.10 | 12.34 |
| Scaffold N50, Kbp | 4.869 | 34.405 | 107.821 | 6.443 |
| GC content, % | 38.81 | 37.08 | 38.06 | 35.41 |
| Repeat content, % | 70.0 | 81.8 | 65–80 * | |
| Total predicted gene models | 58,587 | 103,694 | 36,732 | 50,163 * |
| Average CDS length, bp | 287.21 | 283.56 | 419.81 | 291.01 * |
| Average intron length, bp | 997.94 | 642.73 | 1146.12 | 351.13 * |
| Maximum intron length, bp | 68,268 | 44,113 | 568,968 | 10,152 * |
| RNA/RefSeq supported genes * | 10,434 | 16,839 | 9260 | 23,077 |
| Predicted TSS positions * | 25,889 | 44,651 | 22,633 | 62,420 |
| Unique TSSs filtered by 5′-UTR distribution * | 10,367 | 16,629 | 9149 | 23,016 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bondar, E.I.; Troukhan, M.E.; Krutovsky, K.V.; Tatarinova, T.V. Genome-Wide Prediction of Transcription Start Sites in Conifers. Int. J. Mol. Sci. 2022, 23, 1735. https://doi.org/10.3390/ijms23031735
Bondar EI, Troukhan ME, Krutovsky KV, Tatarinova TV. Genome-Wide Prediction of Transcription Start Sites in Conifers. International Journal of Molecular Sciences. 2022; 23(3):1735. https://doi.org/10.3390/ijms23031735
Chicago/Turabian StyleBondar, Eugeniya I., Maxim E. Troukhan, Konstantin V. Krutovsky, and Tatiana V. Tatarinova. 2022. "Genome-Wide Prediction of Transcription Start Sites in Conifers" International Journal of Molecular Sciences 23, no. 3: 1735. https://doi.org/10.3390/ijms23031735
APA StyleBondar, E. I., Troukhan, M. E., Krutovsky, K. V., & Tatarinova, T. V. (2022). Genome-Wide Prediction of Transcription Start Sites in Conifers. International Journal of Molecular Sciences, 23(3), 1735. https://doi.org/10.3390/ijms23031735