
Nanocarrier-Based Delivery of SN22 as a Tocopheryl Oxamate Prodrug Achieves Rapid Tumor Regression and Extends Survival in High-Risk Neuroblastoma Models

 ,
,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
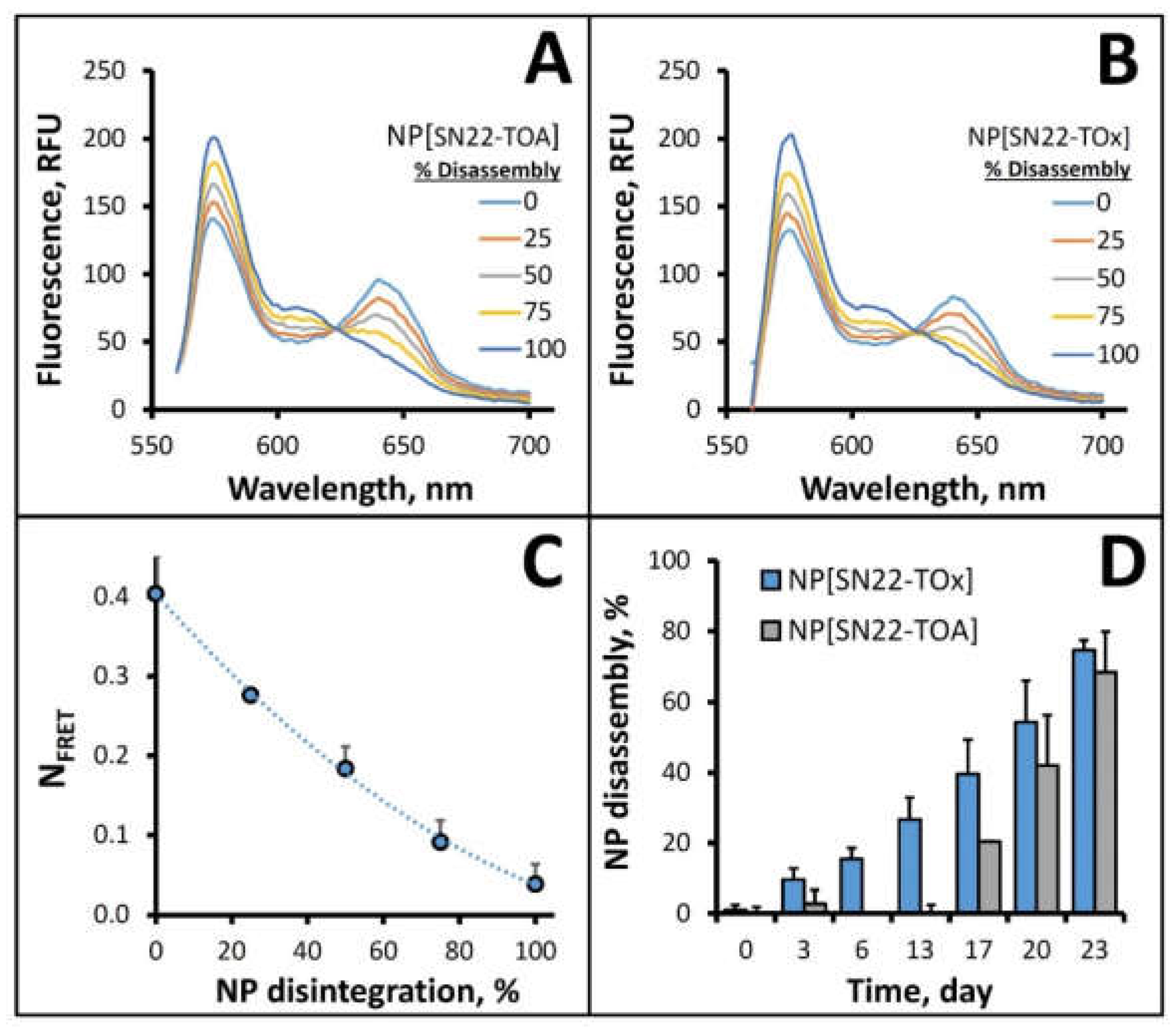
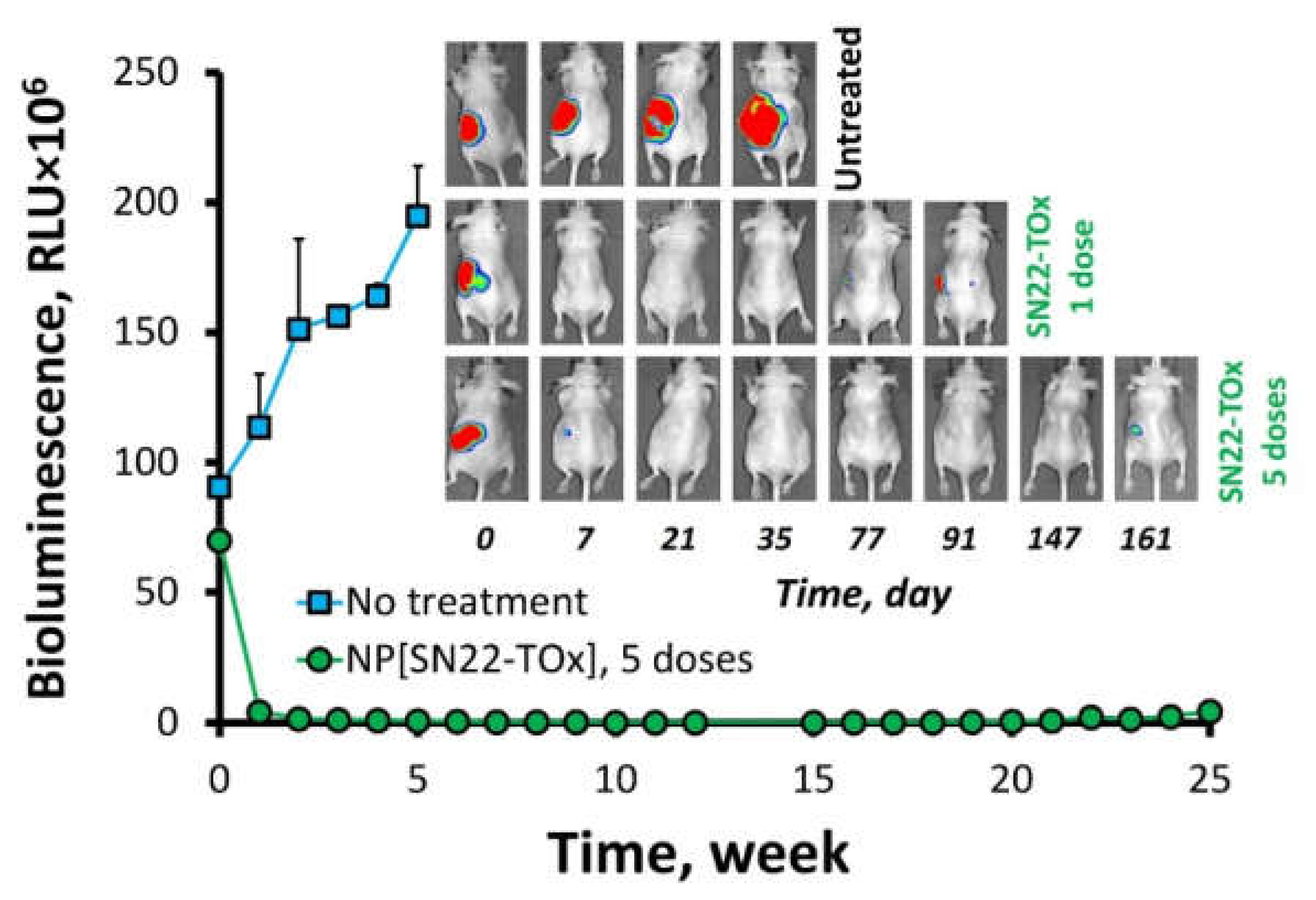
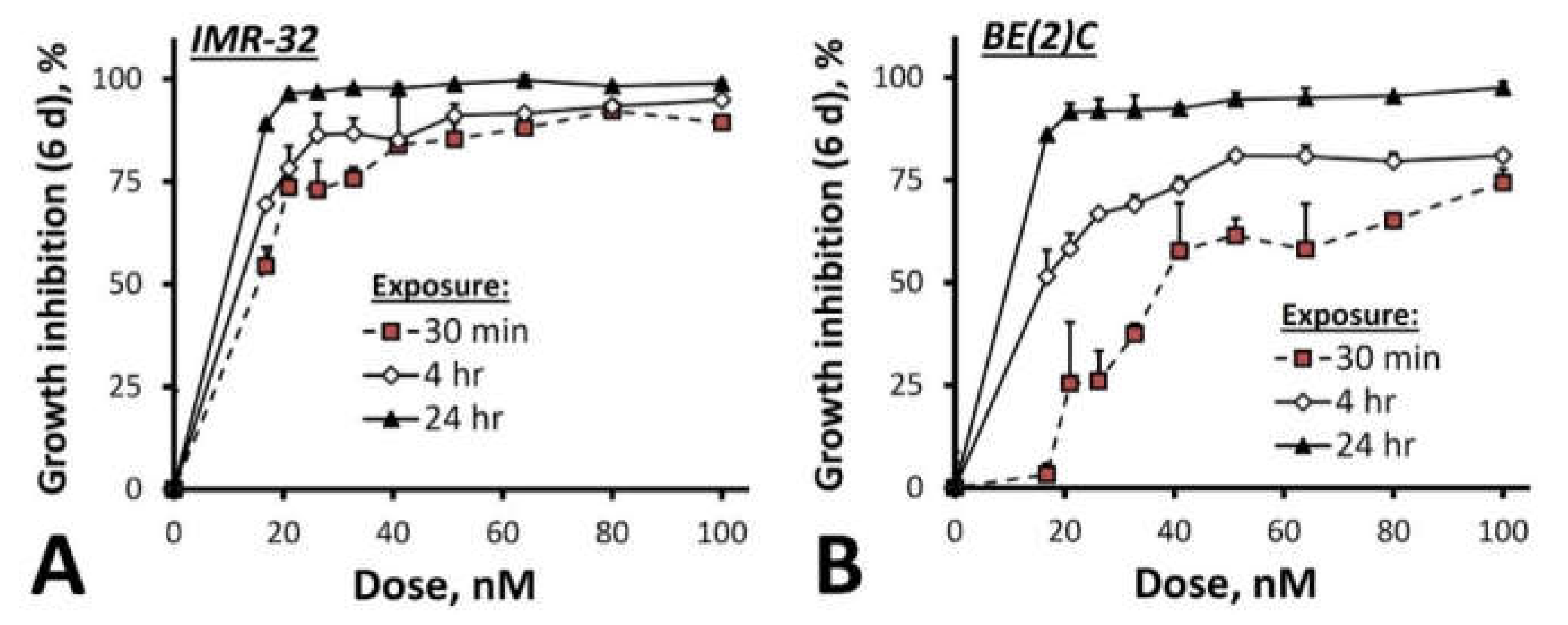
2. Results
3. Discussion
4. Materials and Methods
4.1. Prodrug Synthesis
4.2. NP[Prodrug] Formulation and Characterization
4.3. Cell Culture Studies
4.4. Therapeutic Efficacy Studies in Preclinical Models of High-Risk NB
4.5. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Johnsen, J.I.; Dyberg, C.; Wickstrom, M. Neuroblastoma—A neural crest derived embryonal malignancy. Front. Mol. Neurosci. 2019, 12, 9. [Google Scholar] [CrossRef] [PubMed]
- London, W.B.; Bagatell, R.; Weigel, B.J.; Fox, E.; Guo, D.; Van Ryn, C.; Naranjo, A.; Park, J.R. Historical time to disease progression and progression-free survival in patients with recurrent/refractory neuroblastoma treated in the modern era on Children’s Oncology Group early-phase trials. Cancer 2017, 123, 4914–4923. [Google Scholar] [CrossRef] [PubMed]
- Moreno, L.; Rubie, H.; Varo, A.; Le Deley, M.C.; Amoroso, L.; Chevance, A.; Garaventa, A.; Gambart, M.; Bautista, F.; Valteau-Couanet, D.; et al. Outcome of children with relapsed or refractory neuroblastoma: A meta-analysis of ITCC/SIOPEN European phase II clinical trials. Pediatr. Blood Cancer 2017, 64, 25–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Chen, S.S.; Clarke, S.; Veschi, V.; Thiele, C.J. Targeting MYCN in Pediatric and Adult Cancers. Front. Oncol. 2020, 10, 623679. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Weiss, W.A. Neuroblastoma and MYCN. Cold Spring Harb. Perspect. Med. 2013, 3, a014415. [Google Scholar] [CrossRef]
- Molenaar, J.J.; Ebus, M.E.; Geerts, D.; Koster, J.; Lamers, F.; Valentijn, L.J.; Westerhout, E.M.; Versteeg, R.; Caron, H.N. Inactivation of CDK2 is synthetically lethal to MYCN over-expressing cancer cells. Proc. Natl. Acad. Sci. USA 2009, 106, 12968–12973. [Google Scholar] [CrossRef] [Green Version]
- Porro, A.; Haber, M.; Diolaiti, D.; Iraci, N.; Henderson, M.; Gherardi, S.; Valli, E.; Munoz, M.A.; Xue, C.; Flemming, C.; et al. Direct and coordinate regulation of ATP-binding cassette transporter genes by Myc factors generates specific transcription signatures that significantly affect the chemoresistance phenotype of cancer cells. J. Biol. Chem. 2010, 285, 19532–19543. [Google Scholar] [CrossRef] [Green Version]
- Hirschmann-Jax, C.; Foster, A.E.; Wulf, G.G.; Nuchtern, J.G.; Jax, T.W.; Gobel, U.; Goodell, M.A.; Brenner, M.K. A distinct “side population” of cells with high drug efflux capacity in human tumor cells. Proc. Natl. Acad. Sci. USA 2004, 101, 14228–14233. [Google Scholar] [CrossRef] [Green Version]
- Acosta, S.; Lavarino, C.; Paris, R.; Garcia, I.; de Torres, C.; Rodriguez, E.; Beleta, H.; Mora, J. Comprehensive characterization of neuroblastoma cell line subtypes reveals bilineage potential similar to neural crest stem cells. BMC Dev. Biol. 2009, 9, 12. [Google Scholar] [CrossRef] [Green Version]
- Cialfi, S.; McDowell, H.P.; Altavista, P.; Boldrini, R.; Bosco, S.; Clerico, A.; Jenkner, A.; Kokai, G.; Pizer, B.; Dominici, C. ABC drug transporter gene expression in neuroblastoma. J. Clin. Oncol. 2010, 28, 9524. [Google Scholar] [CrossRef]
- Bates, S.E.; Medina-Perez, W.Y.; Kohlhagen, G.; Antony, S.; Nadjem, T.; Robey, R.W.; Pommier, Y. ABCG2 mediates differential resistance to SN-38 (7-ethyl-10-hydroxycamptothecin) and homocamptothecins. J. Pharmacol. Exp. Ther. 2004, 310, 836–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schellens, J.H.; Maliepaard, M.; Scheper, R.J.; Scheffer, G.L.; Jonker, J.W.; Smit, J.W.; Beijnen, J.H.; Schinkel, A.H. Transport of topoisomerase I inhibitors by the breast cancer resistance protein. Potential clinical implications. Ann. N. Y. Acad. Sci. 2000, 922, 188–194. [Google Scholar] [CrossRef]
- Thomas, A.; Pommier, Y. Targeting topoisomerase I in the era of precision medicine. Clin. Cancer Res. 2019, 25, 6581–6589. [Google Scholar] [CrossRef] [PubMed]
- Zage, P.E. Novel Therapies for Relapsed and Refractory Neuroblastoma. Children 2018, 5, 148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kunimoto, T.; Nitta, K.; Tanaka, T.; Uehara, N.; Baba, H.; Takeuchi, M.; Yokokura, T.; Sawada, S.; Miyasaka, T.; Mutai, M. Antitumor activity of a new camptothecin derivative, SN-22, against various murine tumors. J. Pharmacobiodyn. 1987, 10, 148–151. [Google Scholar] [CrossRef] [PubMed]
- Nagata, H.; Kaneda, N.; Furuta, T.; Sawada, S.; Yokokura, T.; Miyasaka, T.; Fukada, M.; Notake, K. Action of 7-ethylcamptothecin on tumor cells and its disposition in mice. Cancer Treat. Rep. 1987, 71, 341–348. [Google Scholar]
- Sawada, S.; Yokokura, T.; Miyasaka, T. Synthesis and antitumor activity of A-ring or E-lactone modified water-soluble prodrugs of 20(S)-camptothecin, including development of irinotecan hydrochloride trihydrate (CPT-11). Curr. Pharm. Design. 1995, 1, 113–132. [Google Scholar]
- Kunimoto, T.; Nitta, K.; Tanaka, T.; Uehara, N.; Baba, H.; Takeuchi, M.; Yokokura, T.; Sawada, S.; Miyasaka, T.; Mutai, M. Antitumor activity of 7-ethyl-10-[4-(1-piperidino)-1-piperidino]carbonyloxy-camptothecin, a novel water-soluble derivative of camptothecin, against murine tumors. Cancer Res. 1987, 47, 5944–5947. [Google Scholar]
- Venditto, V.J.; Simanek, E.E. Cancer therapies utilizing the camptothecins: A review of the in vivo literature. Mol. Pharm. 2010, 7, 307–349. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Carbonero, R.; Supko, J.G. Current perspectives on the clinical experience, pharmacology, and continued development of the camptothecins. Clin. Cancer. Res. 2002, 8, 641–661. [Google Scholar]
- Slatter, J.G.; Schaaf, L.J.; Sams, J.P.; Feenstra, K.L.; Johnson, M.G.; Bombardt, P.A.; Cathcart, K.S.; Verburg, M.T.; Pearson, L.K.; Compton, L.D.; et al. Pharmacokinetics, metabolism, and excretion of irinotecan (CPT-11) following I.V. infusion of [(14)C]CPT-11 in cancer patients. Drug Metab. Dispos. 2000, 28, 423–433. [Google Scholar] [PubMed]
- Gandhi, Y.A.; Morris, M.E. Structure-activity relationships and quantitative structure-activity relationships for breast cancer resistance protein (ABCG2). AAPS J. 2009, 11, 541–552. [Google Scholar] [CrossRef]
- Santos, A.; Calvet, L.; Terrier-Lacombe, M.J.; Larsen, A.; Benard, J.; Pondarre, C.; Aubert, G.; Morizet, J.; Lavelle, F.; Vassal, G. In vivo treatment with CPT-11 leads to differentiation of neuroblastoma xenografts and topoisomerase I alterations. Cancer Res. 2004, 64, 3223–3229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vassal, G.; Terrier-Lacombe, M.J.; Bissery, M.C.; Venuat, A.M.; Gyergyay, F.; Benard, J.; Morizet, J.; Boland, I.; Ardouin, P.; Bressac-de-Paillerets, B.; et al. Therapeutic activity of CPT-11, a DNA-topoisomerase I inhibitor, against peripheral primitive neuroectodermal tumour and neuroblastoma xenografts. Br. J. Cancer 1996, 74, 537–545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vassal, G.; Giammarile, F.; Brooks, M.; Geoerger, B.; Couanet, D.; Michon, J.; Stockdale, E.; Schell, M.; Geoffray, A.; Gentet, J.C.; et al. A phase II study of irinotecan in children with relapsed or refractory neuroblastoma: A European cooperation of the Societe Francaise d’Oncologie Pediatrique (SFOP) and the United Kingdom Children Cancer Study Group (UKCCSG). Eur. J. Cancer 2008, 44, 2453–2460. [Google Scholar] [CrossRef] [PubMed]
- Yoshikawa, M.; Ikegami, Y.; Hayasaka, S.; Ishii, K.; Ito, A.; Sano, K.; Suzuki, T.; Togawa, T.; Yoshida, H.; Soda, H.; et al. Novel camptothecin analogues that circumvent ABCG2-associated drug resistance in human tumor cells. Int. J. Cancer 2004, 110, 921–927. [Google Scholar] [CrossRef]
- Nakagawa, H.; Saito, H.; Ikegami, Y.; Aida-Hyugaji, S.; Sawada, S.; Ishikawa, T. Molecular modeling of new camptothecin analogues to circumvent ABCG2-mediated drug resistance in cancer. Cancer Lett. 2006, 234, 81–89. [Google Scholar] [CrossRef]
- Zamboni, W.C.; Torchilin, V.; Patri, A.K.; Hrkach, J.; Stern, S.; Lee, R.; Nel, A.; Panaro, N.J.; Grodzinski, P. Best practices in cancer nanotechnology: Perspective from NCI nanotechnology alliance. Clin. Cancer Res. 2012, 18, 3229–3241. [Google Scholar] [CrossRef] [Green Version]
- Perrault, S.D.; Walkey, C.; Jennings, T.; Fischer, H.C.; Chan, W.C. Mediating tumor targeting efficiency of nanoparticles through design. Nano. Lett. 2009, 9, 1909–1915. [Google Scholar] [CrossRef]
- Grossman, J.H.; Crist, R.M.; Clogston, J.D. Early Development Challenges for Drug Products Containing Nanomaterials. AAPS J. 2016. [Google Scholar] [CrossRef]
- Alferiev, I.S.; Iyer, R.; Croucher, J.L.; Adamo, R.F.; Zhang, K.; Mangino, J.L.; Kolla, V.; Fishbein, I.; Brodeur, G.M.; Levy, R.J.; et al. Nanoparticle-mediated delivery of a rapidly activatable prodrug of SN-38 for neuroblastoma therapy. Biomaterials 2015, 51, 22–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forrest, M.L.; Yanez, J.A.; Remsberg, C.M.; Ohgami, Y.; Kwon, G.S.; Davies, N.M. Paclitaxel prodrugs with sustained release and high solubility in poly(ethylene glycol)-b-poly(epsilon-caprolactone) micelle nanocarriers: Pharmacokinetic disposition, tolerability, and cytotoxicity. Pharm. Res. 2008, 25, 194–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lundberg, B.B.; Risovic, V.; Ramaswamy, M.; Wasan, K.M. A lipophilic paclitaxel derivative incorporated in a lipid emulsion for parenteral administration. J. Control. Release 2003, 86, 93–100. [Google Scholar] [CrossRef]
- Nguyen, F.; Alferiev, I.; Guan, P.; Guerrero, D.T.; Kolla, V.; Moorthy, G.S.; Chorny, M.; Brodeur, G.M. Enhanced intratumoral delivery of SN38 as a tocopherol oxyacetate prodrug using nanoparticles in a neuroblastoma xenograft model. Clin. Cancer Res. 2018, 24, 2585–2593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taft, R.W. Polar and steric substituent constants for aliphatic and o-benzoate groups from rates of esterification and hydrolysis of esters. J. Am. Chem. Soc. 1952, 74, 3120–3128. [Google Scholar] [CrossRef]
- Lawson, K.A.; Anderson, K.; Menchaca, M.; Atkinson, J.; Sun, L.; Knight, V.; Gilbert, B.E.; Conti, C.; Sanders, B.G.; Kline, K. Novel vitamin E analogue decreases syngeneic mouse mammary tumor burden and reduces lung metastasis. Mol. Cancer Ther. 2003, 2, 437–444. [Google Scholar]
- Neuzil, J.; Zhao, M.; Ostermann, G.; Sticha, M.; Gellert, N.; Weber, C.; Eaton, J.W.; Brunk, U.T. Alpha-tocopheryl succinate, an agent with in vivo anti-tumour activity, induces apoptosis by causing lysosomal instability. Biochem. J. 2002, 362 Pt 3, 709–715. [Google Scholar] [CrossRef] [PubMed]
- Férriz, J.M.; Vinšová, J. Prodrug design of phenolic drugs. Curr. Pharm. Des. 2010, 16, 2033–2052. [Google Scholar] [CrossRef]
- Moriguchi, I.; Hirono, S.; Liu, Q.; Nakagome, I.; Matsushita, Y. Simple method of calculating octanol/water partition coefficient. Chem. Pharm. Bull. 1992, 40, 127–130. [Google Scholar] [CrossRef] [Green Version]
- Chorny, M.; Fishbein, I.; Danenberg, H.D.; Golomb, G. Lipophilic drug loaded nanospheres prepared by nanoprecipitation: Effect of formulation variables on size, drug recovery and release kinetics. J. Control. Release 2002, 83, 389–400. [Google Scholar] [CrossRef]
- Chorny, M.; Fishbein, I.; Danenberg, H.D.; Golomb, G. Study of the drug release mechanism from tyrphostin AG-1295-loaded nanospheres by in situ and external sink methods. J. Control. Release 2002, 83, 401–414. [Google Scholar] [CrossRef]
- Lantsman, K.; Tombes, R.M. CaMK-II oligomerization potential determined using CFP/YFP FRET. Biochim. Biophys. Acta 2005, 1746, 45–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tumilowicz, J.J.; Nichols, W.W.; Cholon, J.J.; Greene, A.E. Definition of a continuous human cell line derived from neuroblastoma. Cancer Res. 1970, 30, 2110–2118. [Google Scholar] [PubMed]
- Keshelava, N.; Zuo, J.J.; Chen, P.; Waidyaratne, S.N.; Luna, M.C.; Gomer, C.J.; Triche, T.J.; Reynolds, C.P. Loss of p53 function confers high-level multidrug resistance in neuroblastoma cell lines. Cancer Res. 2001, 61, 6185–6193. [Google Scholar]
- Polunin, Y.; Alferiev, I.S.; Brodeur, G.M.; Voronov, A.; Chorny, M. Environment-Sensitive Polymeric Micelles Encapsulating SN-38 Potently Suppress Growth of Neuroblastoma Cells Exhibiting Intrinsic and Acquired Drug Resistance. ACS. Pharmacol. Transl. Sci. 2021, 4, 240–247. [Google Scholar] [CrossRef] [PubMed]
- Morgenstern, D.A.; Baruchel, S.; Irwin, M.S. Current and future strategies for relapsed neuroblastoma: Challenges on the road to precision therapy. J. Pediatr. Hematol. Oncol. 2013, 35, 337–347. [Google Scholar] [CrossRef]
- Liew, S.T.; Yang, L.X. Design, synthesis and development of novel camptothecin drugs. Curr. Pharm. Des. 2008, 14, 1078–1097. [Google Scholar] [CrossRef]
- Keshelava, N.; Groshen, S.; Reynolds, C.P. Cross-resistance of topoisomerase I and II inhibitors in neuroblastoma cell lines. Cancer Chemother. Pharmacol. 2000, 45, 1–8. [Google Scholar] [CrossRef]
- Prasad, K.N.; Kumar, B.; Yan, X.D.; Hanson, A.J.; Cole, W.C. Alpha-tocopheryl succinate, the most effective form of vitamin E for adjuvant cancer treatment: A review. J. Am. Coll. Nutr. 2003, 22, 108–117. [Google Scholar] [CrossRef]
- Zhao, Y.; Neuzil, J.; Wu, K. Vitamin E analogues as mitochondria-targeting compounds: From the bench to the bedside? Mol. Nutr. Food. Res. 2009, 53, 129–139. [Google Scholar] [CrossRef] [Green Version]
- Swettenham, E.; Witting, P.K.; Salvatore, B.A.; Neuzil, J. Alpha-tocopheryl succinate selectively induces apoptosis in neuroblastoma cells: Potential therapy of malignancies of the nervous system? J. Neurochem. 2005, 94, 1448–1456. [Google Scholar] [CrossRef] [PubMed]
- Neuzil, J.; Tomasetti, M.; Zhao, Y.; Dong, L.F.; Birringer, M.; Wang, X.F.; Low, P.; Wu, K.; Salvatore, B.A.; Ralph, S.J. Vitamin E analogs, a novel group of “mitocans,” as anticancer agents: The importance of being redox-silent. Mol. Pharmacol. 2007, 71, 1185–1199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomic-Vatic, A.; Eytina, J.; Chapman, J.; Mahdavian, E.; Neuzil, J.; Salvatore, B.A. Vitamin E amides, a new class of vitamin E analogues with enhanced proapoptotic activity. Int. J. Cancer 2005, 117, 188–193. [Google Scholar] [CrossRef] [PubMed]
- Chang, E.; Ni, J.; Yin, Y.; Lin, C.C.; Chang, P.; James, N.S.; Chemler, S.R.; Yeh, S. Alpha-vitamin E derivative, RRR-alpha-tocopheryloxybutyric acid inhibits the proliferation of prostate cancer cells. Asian. J. Androl. 2007, 9, 31–39. [Google Scholar] [CrossRef]
- Gruber, J.; Staniek, K.; Krewenka, C.; Moldzio, R.; Patel, A.; Bohmdorfer, S.; Rosenau, T.; Gille, L. Tocopheramine succinate and tocopheryl succinate: Mechanism of mitochondrial inhibition and superoxide radical production. Bioorg. Med. Chem. 2014, 22, 684–691. [Google Scholar] [CrossRef] [PubMed]
- Takino, T.; Nakajima, C.; Takakura, Y.; Sezaki, H.; Hashida, M. Controlled biodistribution of highly lipophilic drugs with various parenteral formulations. J. Drug Targets 1993, 1, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Qu, X.; Ando, H. A simple method for reaction rate prediction of ester hydrolysis. J. Mol. Struct. THEOCHEM 2005, 725, 31–37. [Google Scholar] [CrossRef]
- Pastorino, F.; Loi, M.; Sapra, P.; Becherini, P.; Cilli, M.; Emionite, L.; Ribatti, D.; Greenberger, L.M.; Horak, I.D.; Ponzoni, M. Tumor regression and curability of preclinical neuroblastoma models by PEGylated SN38 (EZN-2208), a novel topoisomerase I inhibitor. Clin. Cancer Res. 2010, 16, 4809–4821. [Google Scholar] [CrossRef] [Green Version]
- Cohrs, R.J.; Torelli, S.; Prasad, K.N.; Edwards-Prasad, J.; Sharma, O.K. Effect of vitamin E succinate and a cAMP-stimulating agent on the expression of c-myc and N-myc and H-ras in murine neuroblastoma cells. Int. J. Dev. Neurosci. 1991, 9, 187–194. [Google Scholar] [CrossRef]
- Birringer, M.; EyTina, J.H.; Salvatore, B.A.; Neuzil, J. Vitamin E analogues as inducers of apoptosis: Structure-function relation. Br. J. Cancer 2003, 88, 1948–1955. [Google Scholar] [CrossRef] [Green Version]
- Tengood, J.E.; Alferiev, I.S.; Zhang, K.; Fishbein, I.; Levy, R.J.; Chorny, M. Real-time analysis of composite magnetic nanoparticle disassembly in vascular cells and biomimetic media. Proc. Natl. Acad. Sci. USA 2014, 111, 4245–4250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, Z.; Liu, Y. Reliable and global measurement of fluorescence resonance energy transfer using fluorescence microscopes. Biophys. J. 2001, 81, 2395–2402. [Google Scholar] [CrossRef] [Green Version]
- Chorny, M.; Fishbein, I.; Yellen, B.B.; Alferiev, I.S.; Bakay, M.; Ganta, S.; Adamo, R.; Amiji, M.; Friedman, G.; Levy, R.J. Targeting stents with local delivery of paclitaxel-loaded magnetic nanoparticles using uniform fields. Proc. Natl. Acad. Sci. USA 2010, 107, 8346–8351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byrne, F.L.; McCarroll, J.A.; Kavallaris, M. Analyses of tumor burden in vivo and metastasis ex vivo using luciferase-expressing cancer cells in an orthotopic mouse model of neuroblastoma. Methods Mol. Biol. 2016, 1372, 61–77. [Google Scholar] [PubMed] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alferiev, I.S.; Guerrero, D.T.; Soberman, D.; Guan, P.; Nguyen, F.; Kolla, V.; Fishbein, I.; Pressly, B.B.; Brodeur, G.M.; Chorny, M. Nanocarrier-Based Delivery of SN22 as a Tocopheryl Oxamate Prodrug Achieves Rapid Tumor Regression and Extends Survival in High-Risk Neuroblastoma Models. Int. J. Mol. Sci. 2022, 23, 1752. https://doi.org/10.3390/ijms23031752
Alferiev IS, Guerrero DT, Soberman D, Guan P, Nguyen F, Kolla V, Fishbein I, Pressly BB, Brodeur GM, Chorny M. Nanocarrier-Based Delivery of SN22 as a Tocopheryl Oxamate Prodrug Achieves Rapid Tumor Regression and Extends Survival in High-Risk Neuroblastoma Models. International Journal of Molecular Sciences. 2022; 23(3):1752. https://doi.org/10.3390/ijms23031752
Chicago/Turabian StyleAlferiev, Ivan S., David T. Guerrero, Danielle Soberman, Peng Guan, Ferro Nguyen, Venkatadri Kolla, Ilia Fishbein, Blake B. Pressly, Garrett M. Brodeur, and Michael Chorny. 2022. "Nanocarrier-Based Delivery of SN22 as a Tocopheryl Oxamate Prodrug Achieves Rapid Tumor Regression and Extends Survival in High-Risk Neuroblastoma Models" International Journal of Molecular Sciences 23, no. 3: 1752. https://doi.org/10.3390/ijms23031752
APA StyleAlferiev, I. S., Guerrero, D. T., Soberman, D., Guan, P., Nguyen, F., Kolla, V., Fishbein, I., Pressly, B. B., Brodeur, G. M., & Chorny, M. (2022). Nanocarrier-Based Delivery of SN22 as a Tocopheryl Oxamate Prodrug Achieves Rapid Tumor Regression and Extends Survival in High-Risk Neuroblastoma Models. International Journal of Molecular Sciences, 23(3), 1752. https://doi.org/10.3390/ijms23031752