
Identification of Potential Antiviral Inhibitors from Hydroxychloroquine and 1,2,4,5-Tetraoxanes Analogues and Investigation of the Mechanism of Action in SARS-CoV-2



Abstract
:1. Introduction
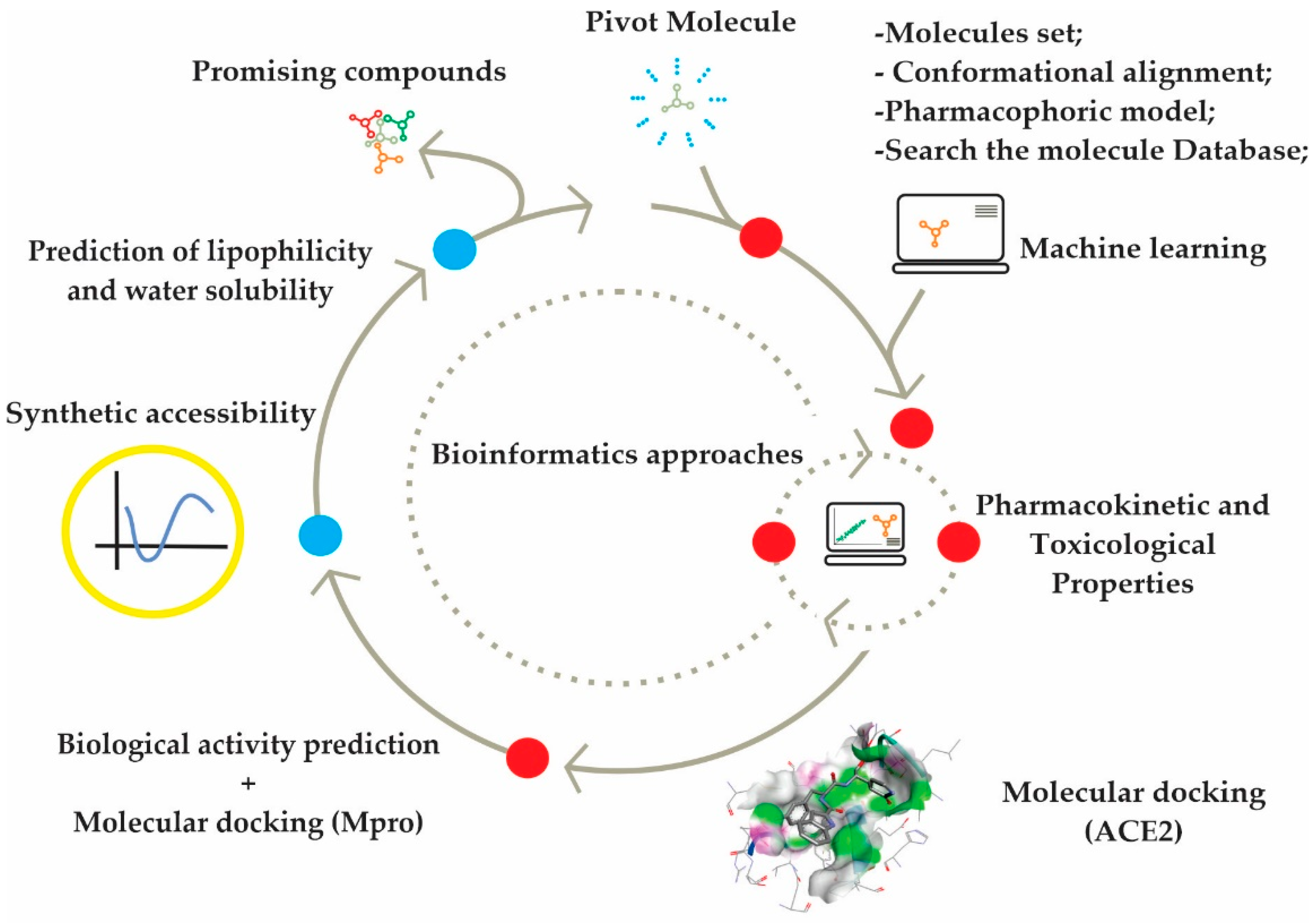
2. Results and Discussion
2.1. Molecular Docking for Obtaining and Evaluating the Pose of Selected Structures and the Pharmacophoric Model
2.2. Molecular Docking for ACE2 Receptor
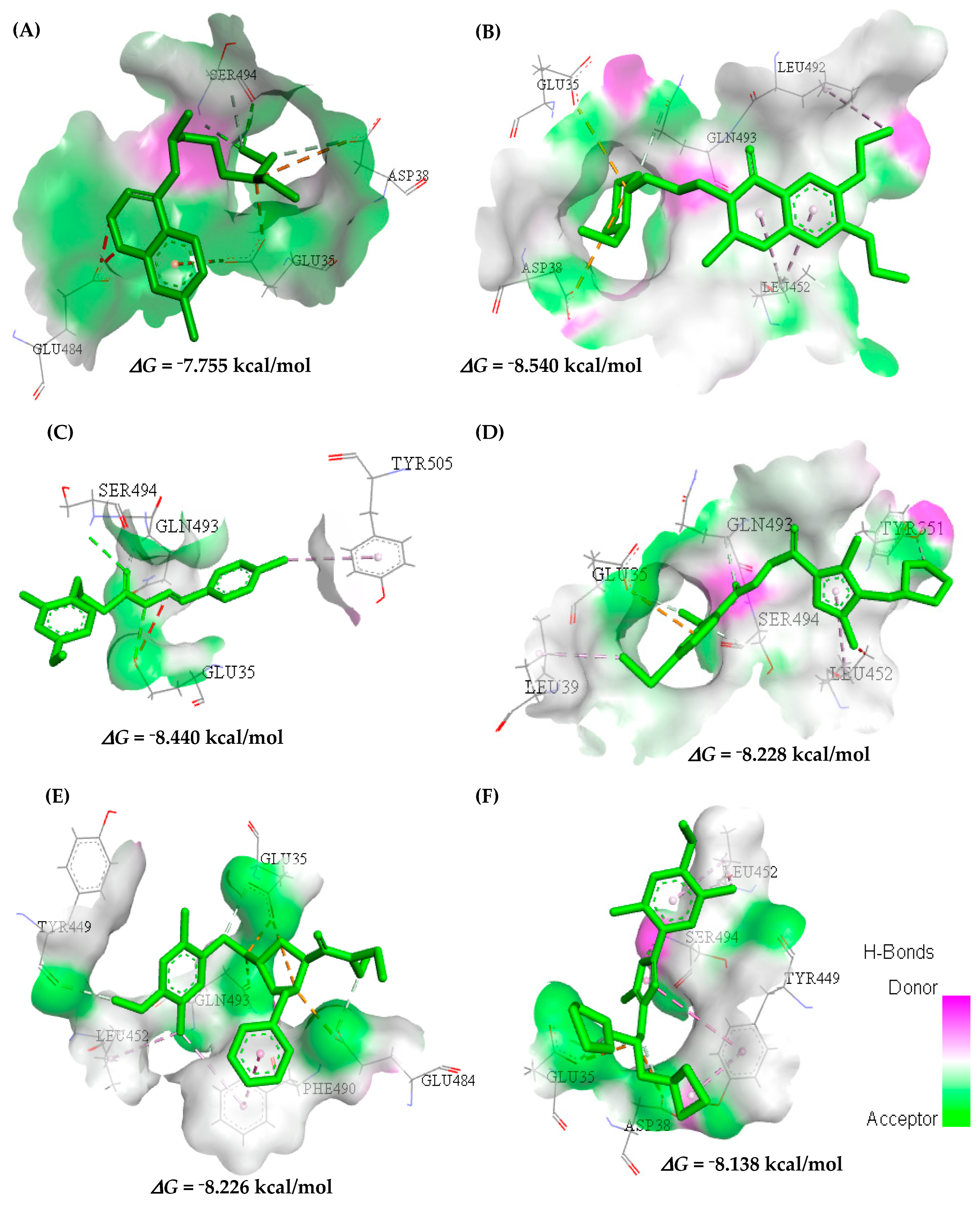
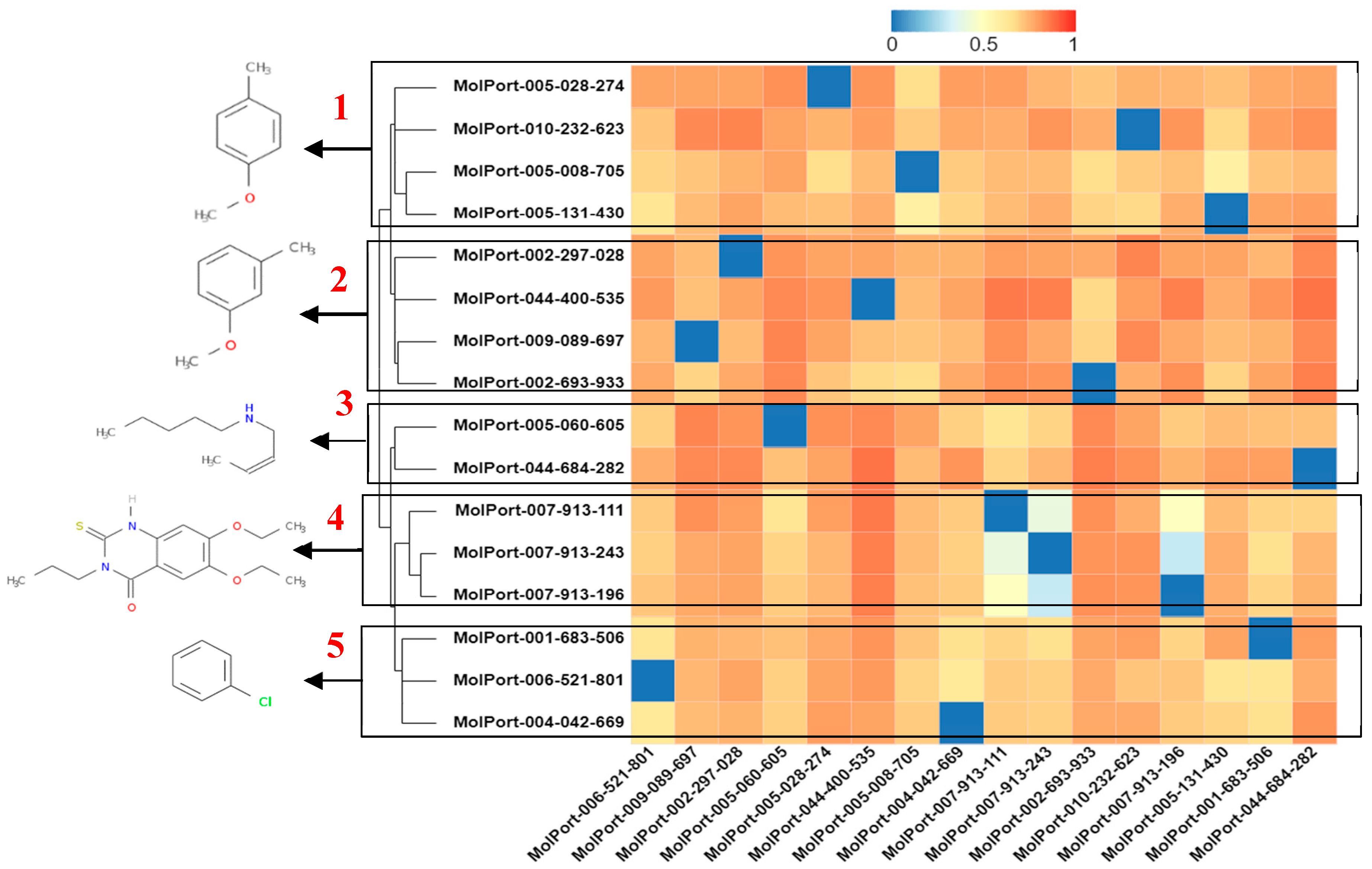
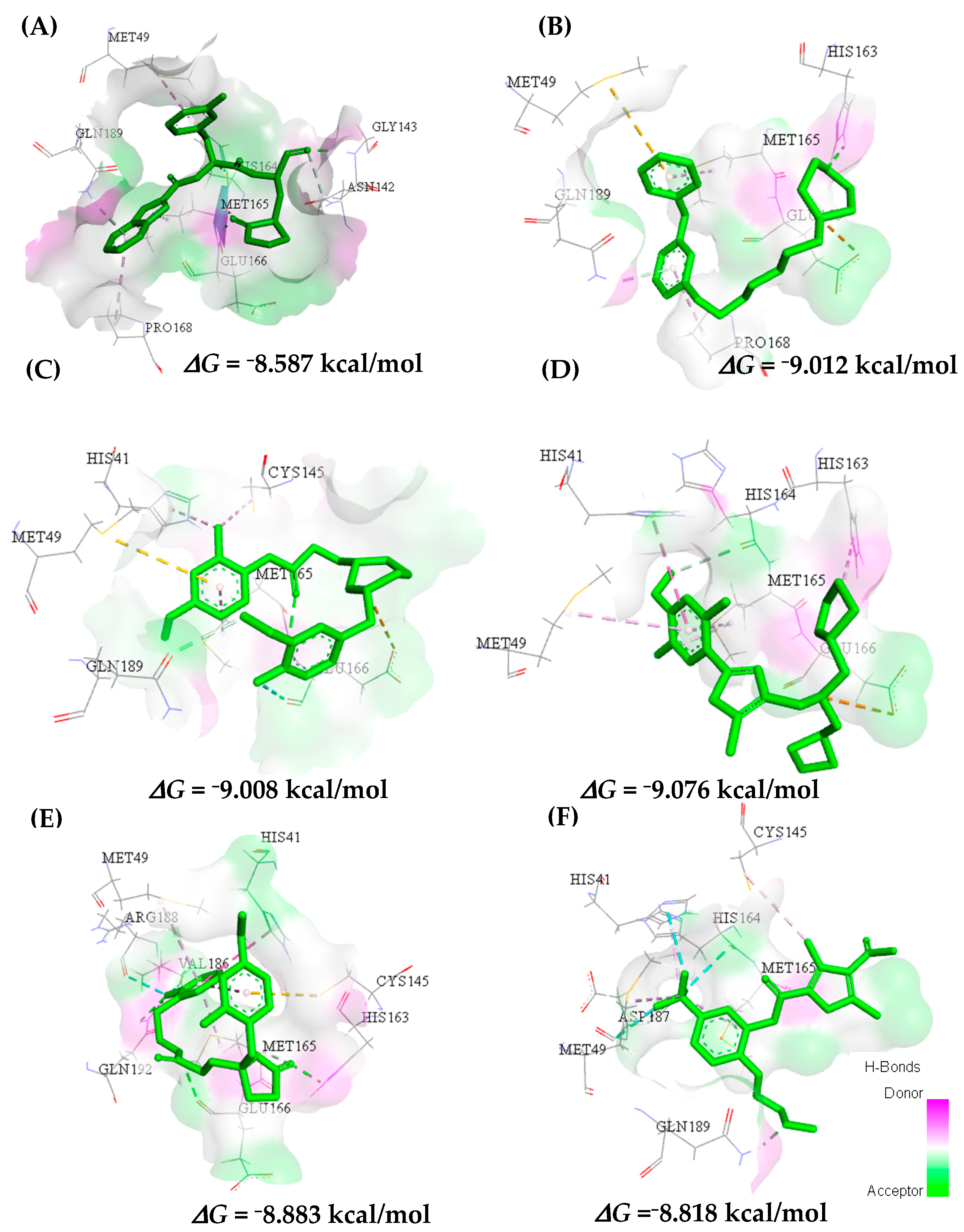
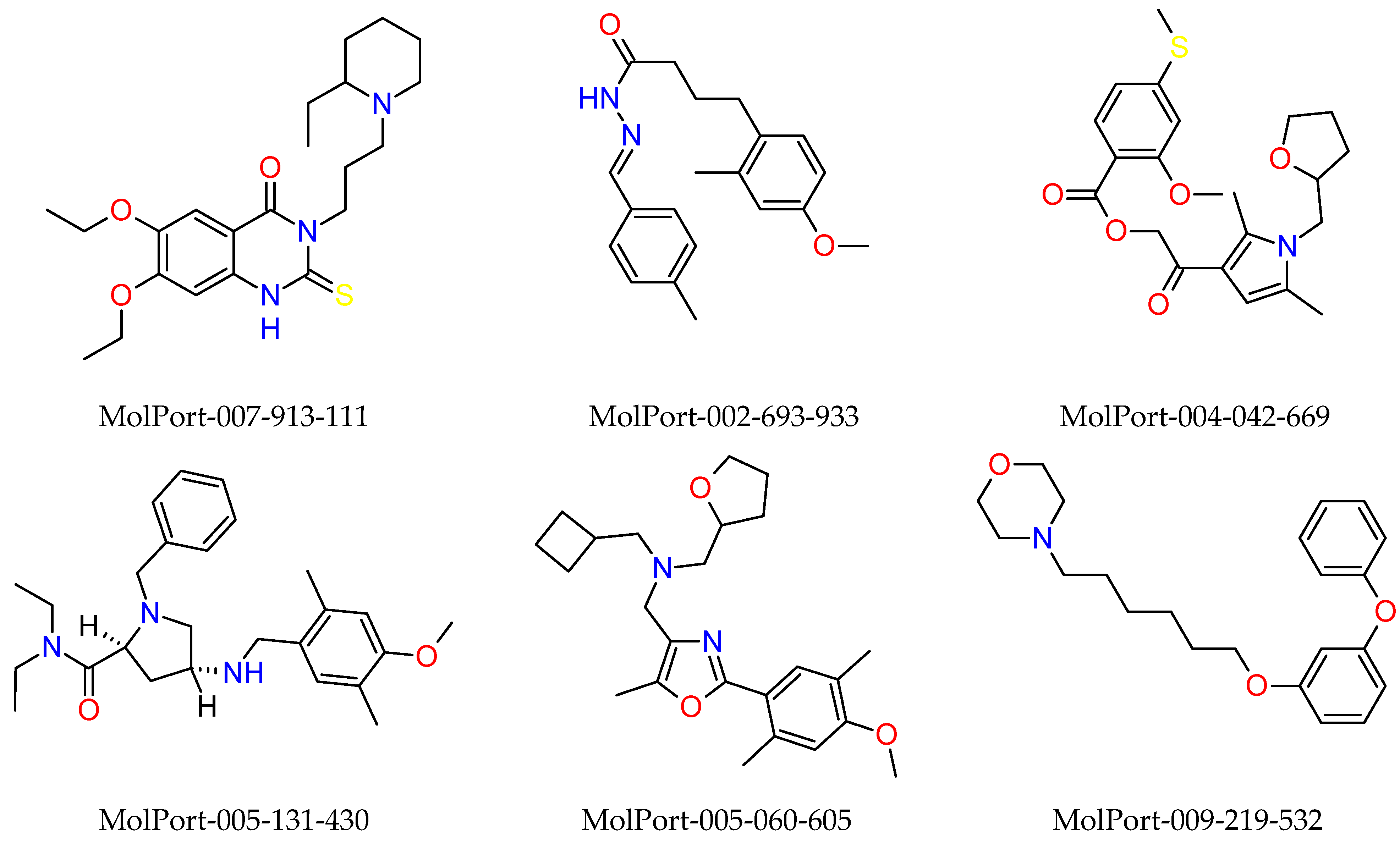
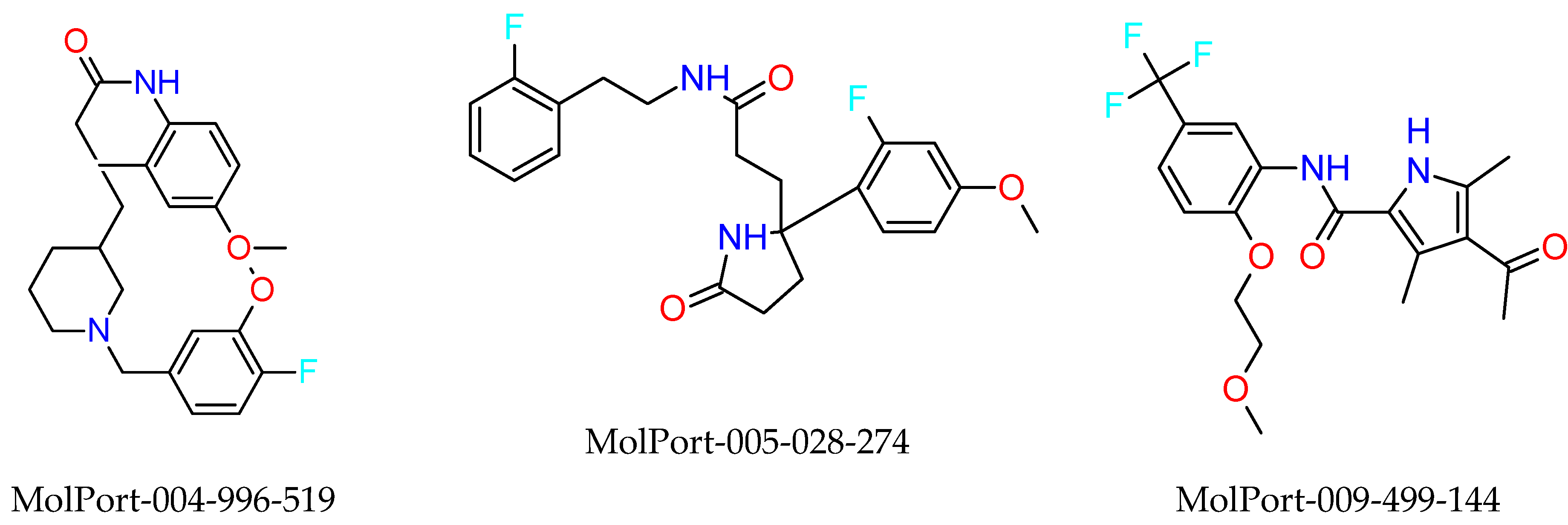
2.3. In Silico Determination of Biological Activity and Molecular Docking Simulations (Mpro)
2.4. Synthetic Accessibility (SA) Prediction
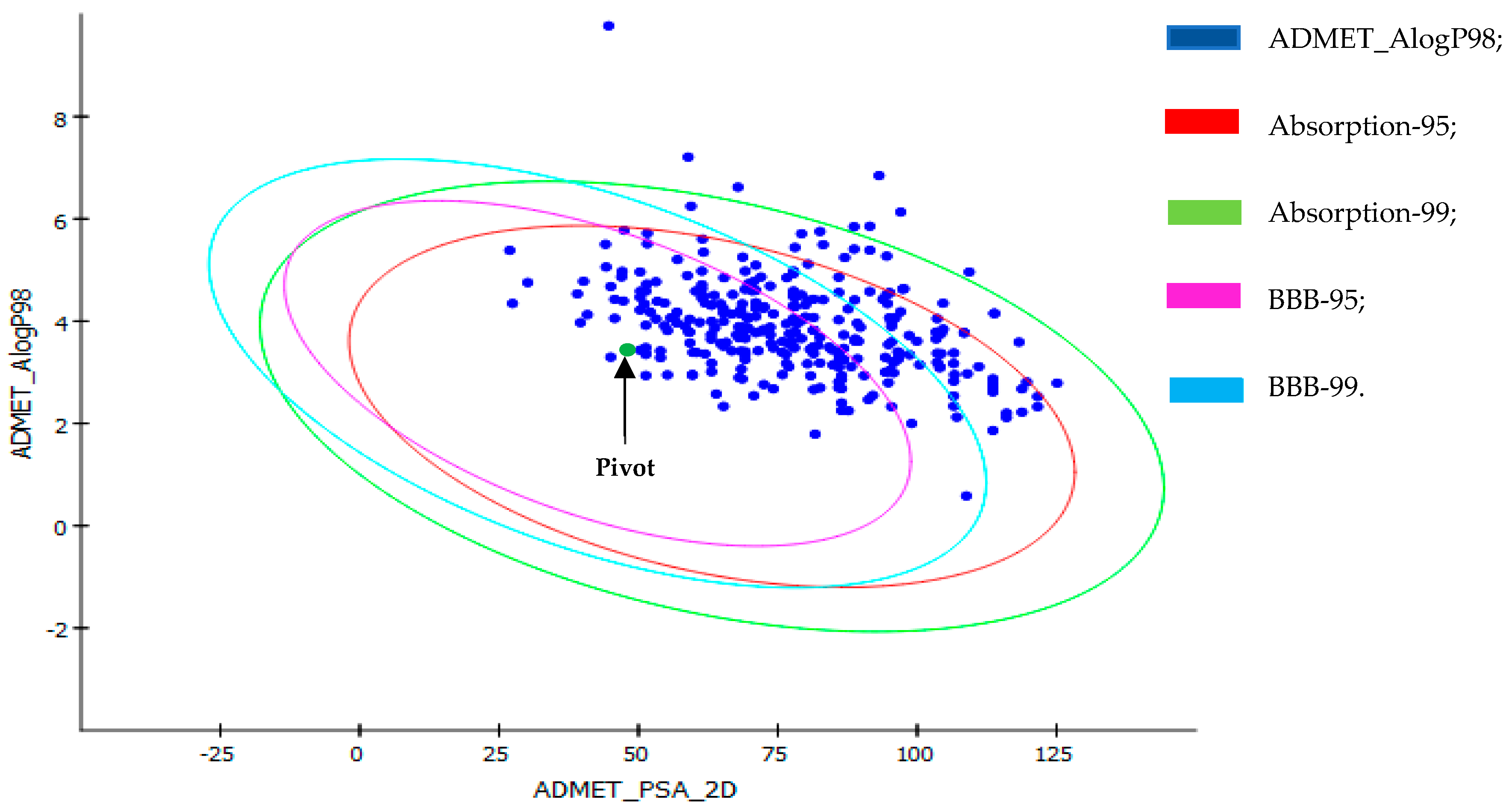
2.5. Prediction of Lipophilicity and Water Solubility for Promising Compounds
3. Materials and Methods
3.1. Obtaining, Optimizing, and Molecular Docking for Selected Structures
3.2. Generation and Evaluation of the Pharmacophoric Model
3.3. Selection of Molecules in the Database
3.4. Prediction of Pharmacokinetic and Toxicological Properties
3.5. Molecular Docking for ACE2 Receptor with DockThor
3.6. In Silico Determination of Biological Activity and Molecular Docking Simulations (Mpro)
Molecular Docking for Mpro Receptor
3.7. Structural Similarity and Synthetic Accessibility (SA) Prediction
3.8. Prediction of Lipophilicity and Water Solubility for Promising Compounds
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhu, H.; Wei, L.; Niu, P. The novel coronavirus outbreak in Wuhan, China. Glob. Health Res. Policy 2020, 5, 6. [Google Scholar] [CrossRef] [Green Version]
- Zhou, P.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [Green Version]
- Who Global Overview. Weekly Epidemiological Update on COVID-19—25 January 2022. Available online: https://www.who.int/publications/m/item/weekly-epidemiological-update-on-covid-19---25-january-2022 (accessed on 28 January 2022).
- White, N.J.; Watson, J.A.; Hoglund, R.M.; Chan, X.H.S.; Cheah, P.Y.; Tarning, J. COVID-19 prevention and treatment: A critical analysis of chloroquine and hydroxychloroquine clinical pharmacology. PLoS Med. 2020, 17, e1003252. [Google Scholar] [CrossRef]
- Kalamatianos, K. Drug Repurposing for Coronavirus (COVID-19): In Silico Screening of Known Drugs against the SARS-CoV-2 Spike Protein Bound to Angiotensin Converting Enzyme 2 (ACE2) (6M0J). ChemRxiv 2021, 1–24. [Google Scholar] [CrossRef]
- Wu, Y.-C.; Chen, C.-S.; Chan, Y.-J. The outbreak of COVID-19: An overview. J. Chin. Med. Assoc. JCMA 2020, 83, 217–220. [Google Scholar] [CrossRef]
- Purwati; Miatmoko, A.; Nasronudin; Hendrianto, E.; Karsari, D.; Dinaryanti, A.; Ertanti, N.; Ihsan, I.S.; Purnama, D.S.; Asmarawati, T.P.; et al. An in vitro study of dual drug combinations of anti-viral agents, antibiotics, and/or hydroxychloroquine against the SARS-CoV-2 virus isolated from hospitalized patients in Surabaya, Indonesia. PLoS ONE 2021, 16, e0252302. [Google Scholar] [CrossRef]
- Wang, M.; Cao, R.; Zhang, L.; Yang, X.; Liu, J.; Xu, M.; Shi, Z.; Hu, Z.; Zhong, W.; Xiao, G. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020, 30, 269–271. [Google Scholar] [CrossRef]
- Das, S.; Sarmah, S.; Lyndem, S.; Roy, A.S. An investigation into the identification of potential inhibitors of SARS-CoV-2 main protease using molecular docking study. J. Biomol. Struct. Dyn. 2020, 39, 3347–3357. [Google Scholar] [CrossRef]
- Basu, A.; Sarkar, A.; Maulik, U. Molecular docking study of potential phytochemicals and their effects on the complex of SARS-CoV2 spike protein and human ACE2. Sci. Rep. 2020, 10, 17699. [Google Scholar] [CrossRef]
- Baysal, Ö.; Ghafoor, N.A.; Silme, R.S.; Ignatov, A.N.; Kniazeva, V. Molecular dynamics analysis of N-acetyl-D-glucosamine against specific SARS-CoV-2’s pathogenicity factors. PLoS ONE 2021, 16, e0252571. [Google Scholar] [CrossRef]
- Khan, A.A.; Baildya, N.; Dutta, T.; Ghosh, N.N. Inhibitory efficiency of potential drugs against SARS-CoV-2 by blocking human angiotensin converting enzyme-2: Virtual screening and molecular dynamics study. Microb. Pathog. 2021, 152, 104762. [Google Scholar] [CrossRef]
- Teli, D.M.; Shah, M.B.; Chhabria, M.T. In silico Screening of Natural Compounds as Potential Inhibitors of SARS-CoV-2 Main Protease and Spike RBD: Targets for COVID-19. Front. Mol. Biosci. 2021, 7, 599079. [Google Scholar] [CrossRef]
- Naqvi, A.A.T.; Fatima, K.; Mohammad, T.; Fatima, U.; Singh, I.K.; Singh, A.; Atif, S.M.; Hariprasad, G.; Hasan, G.M.; Hassan, M.I. Insights into SARS-CoV-2 Genome, Structure, Evolution, Pathogenesis and Therapies: Structural Genomics Approach. Biochim. Biophys. Acta-Mol. Basis Dis. 2020, 1866, 165878. [Google Scholar] [CrossRef]
- Law, W.Y.; Asaruddin, M.R.; Bhawani, S.A.; Mohamad, S. Pharmacophore modelling of vanillin derivatives, favipiravir, chloroquine, hydroxychloroquine, monolaurin and tetrodotoxin as MPro inhibitors of severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2). BMC Res. Notes 2020, 13, 527. [Google Scholar] [CrossRef]
- Hussien, M.A.; Abdelaziz, A.E.M. Molecular docking suggests repurposing of brincidofovir as a potential drug targeting SARS-CoV-2 ACE2 receptor and main protease. Netw. Model. Anal. Health Inform. Bioinform. 2020, 9, 56. [Google Scholar] [CrossRef]
- Avelar, L.A.A.; Camilo, C.D.; de Albuquerque, S.; Fernandes, W.B.; Gonçalez, C.; Kenny, P.W.; Leitão, A.; McKerrow, J.H.; Montanari, C.A.; Orozco, E.V.M.; et al. Molecular Design, Synthesis and Trypanocidal Activity of Dipeptidyl Nitriles as Cruzain Inhibitors. PLoS Negl. Trop. Dis. 2015, 9, e0003916. [Google Scholar] [CrossRef]
- Gomes, J.C.; Cianni, L.; Ribeiro, J.; dos Reis Rocho, F.; da Costa Martins Silva, S.; Batista, P.H.J.; Moraes, C.B.; Franco, C.H.; Freitas-Junior, L.H.G.; Kenny, P.W.; et al. Synthesis and structure-activity relationship of nitrile-based cruzain inhibitors incorporating a trifluoroethylamine-based P2 amide replacement. Bioorg. Med. Chem. 2019, 27, 115083. [Google Scholar] [CrossRef]
- Wang, N.; Han, S.; Liu, R.; Meng, L.; He, H.; Zhang, Y.; Wang, C.; Lv, Y.; Wang, J.; Li, X.; et al. Chloroquine and hydroxychloroquine as ACE2 blockers to inhibit viropexis of 2019-nCoV Spike pseudotyped virus. Phytomedicine 2020, 79, 153333. [Google Scholar] [CrossRef]
- Yavuz, S.; Ünal, S. Antiviral treatment of COVID-19. Turk. J. Med. Sci. 2020, 50, 611–619. [Google Scholar] [CrossRef]
- Alamri, M.A.; Qamar, M.T.U.; Mirza, M.U.; Bhadane, R.; Alqahtani, S.M.; Muneer, I.; Froeyen, M.; Salo-Ahen, O.M.H. Pharmacoinformatics and molecular dynamics simulation studies reveal potential covalent and FDA-approved inhibitors of SARS-CoV-2 main protease 3CLpro. J. Biomol. Struct. Dyn. 2021, 39, 4936–4948. [Google Scholar] [CrossRef]
- Ferreira, E.F.B.; Silva, L.B.; Costa, G.V.; Costa, J.S.; Fujishima, M.A.T.; Leão, R.P.; Ferreira, A.L.S.; Federico, L.B.; Silva, C.H.T.P.; Rosa, J.M.C.; et al. Identification of New Inhibitors with Potential Antitumor Activity from Polypeptide Structures via Hierarchical Virtual Screening. Molecules 2019, 24, 2943. [Google Scholar] [CrossRef] [Green Version]
- Koes, D.R.; Camacho, C.J. ZINCPharmer: Pharmacophore search of the ZINC database. Nucleic Acids Res. 2012, 40, W409–W414. [Google Scholar] [CrossRef]
- QSAR, ADMET and Predictive Toxicology with Biovia Discovery Studio Datasheet Predicting Development Risks; DS No. 3057-1014; Dassault Systèmes Corporate: Waltham, MA, USA; BIOVIA Corporate Americas: San Diego, CA, USA; BIOVIA Corporate Europe: Cambridge, UK, 2016.
- Zhu, Y.; Han, Y.; Ma, Y.; Yang, P. ADME/toxicity prediction and antitumor activity of novel nitrogenous heterocyclic compounds designed by computer targeting of alkylglycerone phosphate synthase. Oncol. Lett. 2018, 16, 1431–1438. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Gonzalez, E.; Jain, S.; Shah, P.; Torimoto-Katori, N.; Zakharov, A.; Nguyễn, Ð.-T.; Sakamuru, S.; Huang, R.; Xia, M.; Obach, R.S.; et al. Development of Robust QSAR Models for CYP2C9, CYP2D6, and CYP3A4 Catalysis and Inhibition. Drug Metab. Dispos. 2021, 49, 822–832. [Google Scholar] [CrossRef] [PubMed]
- Leão, R.P.; Cruz, J.V.; da Costa, G.V.; Cruz, J.N.; Ferreira, E.F.B.; Silva, R.C.; de Lima, L.R.; Borges, R.S.; dos Santos, G.B.; Santos, C.B.R. Identification of New Rofecoxib-Based Cyclooxygenase-2 Inhibitors: A Bioinformatics Approach. Pharmaceuticals 2020, 13, 209. [Google Scholar] [CrossRef] [PubMed]
- Ohtsu, Y.; Susaki, Y.; Noguchi, K. Absorption, Distribution, Metabolism, and Excretion of the Novel Helicase-Primase Inhibitor, Amenamevir (ASP2151), in Rodents. Eur. J. Drug Metab. Pharmacokinet. 2018, 43, 693–706. [Google Scholar] [CrossRef] [Green Version]
- Yong, T.; Chen, S.; Xie, Y.; Shuai, O.; Li, X.; Chen, D.; Su, J.; Jiao, C.; Liang, Y. Hypouricemic Effects of Extracts from Agrocybe aegerita on Hyperuricemia Mice and Virtual Prediction of Bioactives by Molecular Docking. Front. Pharmacol. 2018, 9, 498. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Yu, S.; Su, J.; Sun, L. Computational Study on New Natural Compound Inhibitors of Pyruvate Dehydrogenase Kinases. Int. J. Mol. Sci. 2016, 17, 340. [Google Scholar] [CrossRef] [Green Version]
- Brown, E.; Yedjou, C.G.; Tchounwou, P.B. Cytotoxicity and oxidative stress in human liver carcinoma cells exposed to arsenic trioxide (HepG2). Met. Ions Biol. Med. 2008, 10, 583–587. [Google Scholar]
- Pajouhesh, H.; Lenz, G.R. Medicinal chemical properties of successful central nervous system drugs. NeuroRX 2005, 2, 541–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.-L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sternberg, A.; Naujokat, C. Structural Features of Coronavirus SARS-CoV-2 Spike Protein: Targets for Vaccination. Life Sci. 2020, 257, 118056. [Google Scholar] [CrossRef] [PubMed]
- Gorkhali, R.; Koirala, P.; Rijal, S.; Mainali, A.; Baral, A.; Bhattarai, H.K. Structure and Function of Major SARS-CoV-2 and SARS-CoV Proteins. Bioinform. Biol. Insights 2021, 15, 11779322211025876. [Google Scholar] [CrossRef] [PubMed]
- Hatmal, M.M.; Alshaer, W.; Al-Hatamleh, M.A.I.; Hatmal, M.; Smadi, O.; Taha, M.O.; Oweida, A.J.; Boer, J.C.; Mohamud, R.; Plebanski, M. Comprehensive Structural and Molecular Comparison of Spike Proteins of SARS-CoV-2, SARS-CoV and MERS-CoV, and Their Interactions with ACE2. Cells 2020, 9, 2638. [Google Scholar] [CrossRef]
- Khelfaoui, H.; Harkati, D.; Saleh, B.A. Molecular docking, molecular dynamics simulations and reactivity, studies on approved drugs library targeting ACE2 and SARS-CoV-2 binding with ACE2. J. Biomol. Struct. Dyn. 2021, 39, 7246–7262. [Google Scholar] [CrossRef]
- Liu, Q.; Wang, Y.; Leung, E.L.-H.; Yao, X. In silico study of intrinsic dynamics of full-length apo-ACE2 and RBD-ACE2 complex. Comput. Struct. Biotechnol. J. 2021, 19, 5455–5465. [Google Scholar] [CrossRef] [PubMed]
- Faisal, H.M.N.; Katti, K.S.; Katti, D.R. Binding of SARS-COV-2 (COVID-19) and SARS-COV to human ACE2: Identifying binding sites and consequences on ACE2 stiffness. Chem. Phys. 2021, 551, 111353. [Google Scholar] [CrossRef]
- Chowdhury, R.; Boorla, V.S.; Maranas, C.D. Computational biophysical characterization of the SARS-CoV-2 spike protein binding with the ACE2 receptor and implications for infectivity. Comput. Struct. Biotechnol. J. 2020, 18, 2573–2582. [Google Scholar] [CrossRef]
- Borges, R.S.; Palheta, I.C.; Ota, S.S.B.; Morais, R.B.; Barros, V.A.; Ramos, R.S.; Silva, R.C.; Costa, J.D.S.; Silva, C.H.T.P.; Campos, J.M.; et al. Toward of Safer Phenylbutazone Derivatives by Exploration of Toxicity Mechanism. Molecules 2019, 24, 143. [Google Scholar] [CrossRef] [Green Version]
- Refaey, R.H.; El-Ashrey, M.K.; Nissan, Y.M. Repurposing of renin inhibitors as SARS-COV-2 main protease inhibitors: A computational study. Virology 2021, 554, 48–54. [Google Scholar] [CrossRef]
- Gowthaman, U.; Jayakanthan, M.; Sundar, D. Molecular docking studies of dithionitrobenzoic acid and its related compounds to protein disulfide isomerase: Computational screening of inhibitors to HIV-1 entry. BMC Bioinform. 2008, 9, S14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hevener, K.E.; Zhao, W.; Ball, D.M.; Babaoglu, K.; Qi, J.; White, S.W.; Lee, R.E. Validation of Molecular Docking Programs for Virtual Screening against Dihydropteroate Synthase. J. Chem. Inf. Model. 2009, 49, 444–460. [Google Scholar] [CrossRef]
- Ramos, R.S.; Macêdo, W.J.C.; Costa, J.S.; da Silva, C.H.T.D.P.; Rosa, J.M.C.; da Cruz, J.N.; de Oliveira, M.S.; de Aguiar Andrade, E.H.; e Silva, R.B.L.; Souto, R.N.P.; et al. Potential inhibitors of the enzyme acetylcholinesterase and juvenile hormone with insecticidal activity: Study of the binding mode via docking and molecular dynamics simulations. J. Biomol. Struct. Dyn. 2020, 38, 4687–4709. [Google Scholar] [CrossRef] [PubMed]
- Ramos, R.D.S.; Costa, J.D.S.; Silva, R.C.; da Costa, G.V.; Rodrigues, A.B.L.; de Menezes Rabelo, É.; Souto, R.N.P.; Taft, C.A.; da Silva, C.H.T.D.P.; Rosa, J.M.C.; et al. Identification of Potential Inhibitors from Pyriproxyfen with Insecticidal Activity by Virtual Screening. Pharmaceuticals 2019, 12, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murthy, T.P.K.; Joshi, T.; Gunnan, S.; Kulkarni, N.; Priyanka, V.; Kumar, S.B.; Gowrishankar, B.S. In silico analysis of Phyllanthus amarus phytochemicals as potent drugs against SARS-CoV-2 main protease. Curr. Res. Green Sustain. Chem. 2021, 4, 100159. [Google Scholar] [CrossRef]
- Yan, F.; Gao, F. An overview of potential inhibitors targeting non-structural proteins 3 (PLpro and Mac1) and 5 (3CLpro/Mpro) of SARS-CoV-2. Comput. Struct. Biotechnol. J. 2021, 19, 4868–4883. [Google Scholar] [CrossRef]
- García-Gutiérrez, P.; Zubillaga, R.A.; Ibarra, I.A.; Martínez, A.; Vargas, R.; Garza, J. Non-conventional interactions of N3 inhibitor with the main protease of SARS-CoV and SARS-CoV-2. Comput. Struct. Biotechnol. J. 2021, 19, 4669–4675. [Google Scholar] [CrossRef] [PubMed]
- Kochev, N.T.; Paskaleva, V.H.; Jeliazkova, N. Ambit-Tautomer: An Open Source Tool for Tautomer Generation. Mol. Inform. 2013, 32, 481–504. [Google Scholar] [CrossRef] [PubMed]
- Jeliazkova, N.; Chomenidis, C.; Doganis, P.; Fadeel, B.; Grafström, R.; Hardy, B.; Hastings, J.; Hegi, M.; Jeliazkov, V.; Kochev, N.; et al. The eNanoMapper database for nanomaterial safety information. Beilstein J. Nanotechnol. 2015, 6, 1609–1634. [Google Scholar] [CrossRef] [Green Version]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]
- Daina, A.; Zoete, V. A BOILED-Egg to Predict Gastrointestinal Absorption and Brain Penetration of Small Molecules. ChemMedChem 2016, 11, 1117–1121. [Google Scholar] [CrossRef] [Green Version]
- Daina, A.; Michielin, O.; Zoete, V. iLOGP: A Simple, Robust, and Efficient Description of n-Octanol/Water Partition Coefficient for Drug Design Using the GB/SA Approach. J. Chem. Inf. Model. 2014, 54, 3284–3301. [Google Scholar] [CrossRef]
- Costa, E.B.; Silva, R.C.; Espejo-Román, J.M.; Neto, M.F.D.A.; Cruz, J.N.; Leite, F.H.A.; Silva, C.H.T.P.; Pinheiro, J.C.; Macêdo, W.J.C.; Santos, C.B.R. Chemometric methods in antimalarial drug design from 1,2,4,5-tetraoxanes analogues. SAR QSAR Environ. Res. 2020, 31, 677–695. [Google Scholar] [CrossRef]
- Schneidman-Duhovny, D.; Dror, O.; Inbar, Y.; Nussinov, R.; Wolfson, H.J. PharmaGist: A webserver for ligand-based pharmacophore detection. Nucleic Acids Res. 2008, 36, W223–W228. [Google Scholar] [CrossRef] [Green Version]
- Araújo, P.H.F.; Ramos, R.S.; da Cruz, J.N.; Silva, S.G.; Ferreira, E.F.B.; de Lima, L.R.; Macêdo, W.J.C.; Espejo-Román, J.M.; Campos, J.M.; Santos, C.B.R. Identification of Potential COX-2 Inhibitors for the Treatment of Inflammatory Diseases Using Molecular Modeling Approaches. Molecules 2020, 25, 4183. [Google Scholar] [CrossRef]
- Nazemi, H.; Mirzaei, M.; Jafari, E. Antidepressant Activity of Curcumin by Monoamine Oxidase–A Inhibition. Adv. J. Chem. Sect. B 2019, 1, 3–9. [Google Scholar] [CrossRef]
- Shukla, A.; Sharma, P.; Prakash, O.; Singh, M.; Kalani, K.; Khan, F.; Bawankule, D.U.; Luqman, S.; Srivastava, S.K. QSAR and Docking Studies on Capsazepine Derivatives for Immunomodulatory and Anti-Inflammatory Activity. PLoS ONE 2014, 9, e100797. [Google Scholar] [CrossRef] [Green Version]
- Jurrus, E.; Engel, D.; Star, K.; Monson, K.; Brandi, J.; Felberg, L.E.; Brookes, D.H.; Wilson, L.; Chen, J.; Liles, K.; et al. Improvements to the APBS biomolecular solvation software suite. Protein Sci. 2018, 27, 112–128. [Google Scholar] [CrossRef] [Green Version]
- Dolinsky, T.J.; Nielsen, J.E.; McCammon, J.A.; Baker, N.A. PDB2PQR: An automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res. 2004, 32, W665–W667. [Google Scholar] [CrossRef]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef] [Green Version]
- dos Santos, K.B.; Guedes, I.A.; Karl, A.L.M.; Dardenne, L.E. Highly Flexible Ligand Docking: Benchmarking of the DockThor Program on the LEADS-PEP Protein–Peptide Data Set. J. Chem. Inf. Model. 2020, 60, 667–683. [Google Scholar] [CrossRef]
- Guedes, I.A.; Barreto, A.M.S.; Marinho, D.; Krempser, E.; Kuenemann, M.A.; Sperandio, O.; Dardenne, L.E.; Miteva, M.A. New machine learning and physics-based scoring functions for drug discovery. Sci. Rep. 2021, 11, 3198. [Google Scholar] [CrossRef]
- Goel, R.K.; Singh, D.; Lagunin, A.; Poroikov, V. PASS-assisted exploration of new therapeutic potential of natural products. Med. Chem. Res. 2011, 20, 1509–1514. [Google Scholar] [CrossRef]
- Kirchmair, J.; Göller, A.H.; Lang, D.; Kunze, J.; Testa, B.; Wilson, I.D.; Glen, R.C.; Schneider, G. Predicting drug metabolism: Experiment and/or computation? Nat. Rev. Drug Discov. 2015, 14, 387–404. [Google Scholar] [CrossRef] [Green Version]
- Dai, W.; Zhang, B.; Jiang, X.-M.; Su, H.; Li, J.; Zhao, Y.; Xie, X.; Jin, Z.; Peng, J.; Liu, F.; et al. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science 2020, 368, 1331–1335. [Google Scholar] [CrossRef] [Green Version]
- Backman, T.W.H.; Cao, Y.; Girke, T. ChemMine tools: An online service for analyzing and clustering small molecules. Nucleic Acids Res. 2011, 39, W486–W491. [Google Scholar] [CrossRef]
- Sun, J.; Jeliazkova, N.; Chupakhin, V.I.; Golib-Dzib, J.-F.; Engkvist, O.; Carlsson, L.; Wegner, J.K.; Ceulemans, H.; Georgiev, I.; Jeliazkov, V.; et al. ExCAPE-DB: An integrated large scale dataset facilitating Big Data analysis in chemogenomics. J. Cheminform. 2017, 9, 17. [Google Scholar] [CrossRef] [Green Version]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
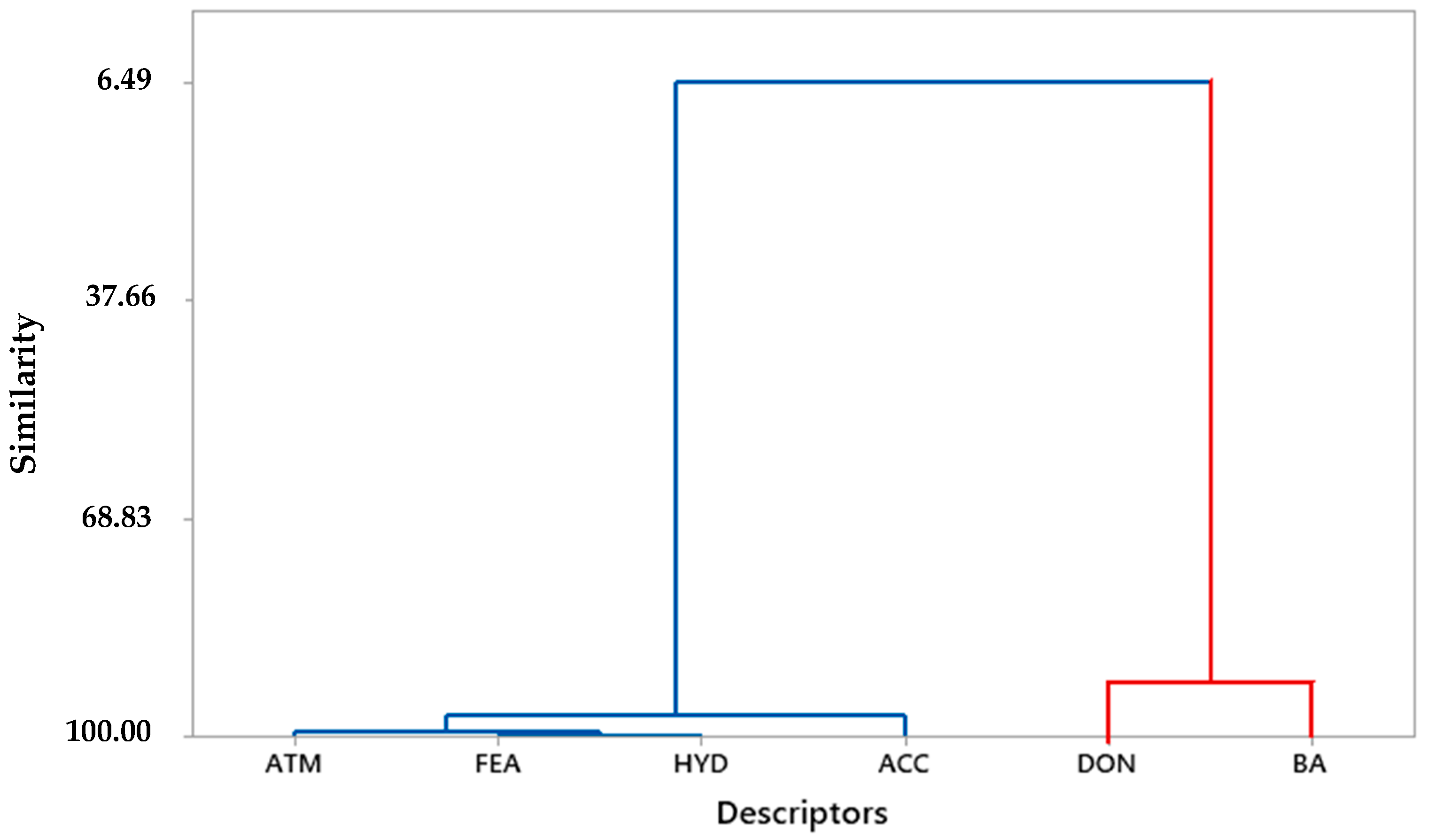
| Molecules | ATM | FEA | HYD | DON | ACC | BA | pKi (µM) |
|---|---|---|---|---|---|---|---|
| 1 * | 49 | 10 | 3 | 2 | 3 | −7.755 | 2.103 |
| 2 | 48 | 9 | 4 | 1 | 2 | −7.709 | 2.273 |
| 3 | 96 | 34 | 24 | 0 | 10 | −9.413 | 0.128 |
| 4 | 99 | 35 | 25 | 0 | 10 | −9.230 | 0.175 |
| 5 | 105 | 37 | 27 | 0 | 10 | −9.216 | 0.179 |
| 6 | 114 | 43 | 33 | 0 | 10 | −9.214 | 0.180 |
| 7 | 102 | 37 | 27 | 0 | 10 | −9.031 | 0.245 |
| 8 | 98 | 37 | 26 | 0 | 10 | −9.009 | 0.254 |
| 9 | 101 | 38 | 27 | 0 | 10 | −8.681 | 0.442 |
| 10 | 106 | 36 | 26 | 1 | 9 | −8.655 | 0.461 |
| 11 | 103 | 37 | 27 | 1 | 9 | −8.600 | 0.506 |
| 12 | 103 | 36 | 26 | 1 | 9 | −8.589 | 0.516 |
| 13 | 106 | 36 | 26 | 1 | 9 | −8.533 | 0.567 |
| 14 | 109 | 37 | 27 | 1 | 9 | −8.533 | 0.570 |
| 15 | 103 | 37 | 27 | 1 | 9 | −8.529 | 0.573 |
| 16 | 106 | 37 | 27 | 1 | 9 | −8.526 | 0.607 |
| ATM | 1.000 | - | - | - | - | - | - |
| FEA | 0.988 | 1.000 | - | - | - | - | - |
| HYD | 0.989 | 0.998 | 1.000 | - | - | - | - |
| DON | −0.477 | −0.563 | −0.563 | 1.000 | - | - | - |
| ACC | 0.939 | 0.967 | 0.955 | −0.671 | 1.000 | - | - |
| BA | −0.719 | −0.769 | −0.763 | 0.849 | −0.870 | 1.000 | - |
| Pharmacophoric Characteristics | Coordinates | ||||
|---|---|---|---|---|---|
| X | Y | Z | Radius (Å) | ||
 | Hydrogen bond acceptor (ACC 1) | 30.171 | −13.304 | −1.102 | 0.5 |
| Hydrogen bond acceptor (ACC 2) | 26.428 | −22.656 | −0.807 | 0.5 | |
| Hydrophobic (HYD 1) | 32.525 | −13.999 | −1.149 | 1.0 | |
| Hydrophobic (HYD 2) | 28.372 | −15.871 | −0.992 | 1.0 | |
| Hydrophobic (HYD 3) | 28.789 | −18.153 | −2.161 | 1.0 | |
| Molecules | MW | RotBonds | LogP | TPSA | ARO | HBA | HBD |
|---|---|---|---|---|---|---|---|
| 1 * | 335.88 | 9 | 4.00 | 48.38 | 2 | 4 | 2 |
| 2 | 319.87 | 8 | 5.00 | 28.16 | 2 | 2 | 1 |
| 3 | 620.36 | 9 | 7.45 | 115.85 | 0 | 10 | 0 |
| 4 | 634.37 | 9 | 7.69 | 115.85 | 0 | 10 | 0 |
| 5 | 662.40 | 9 | 7.93 | 115,85 | 0 | 10 | 0 |
| 6 | 704.45 | 8 | 7.38 | 126.84 | 0 | 10 | 0 |
| 7 | 648.39 | 9 | 7.63 | 126.84 | 0 | 10 | 0 |
| 8 | 633.36 | 9 | 6.91 | 126.84 | 0 | 10 | 0 |
| 9 | 647.38 | 8 | 6.63 | 132.64 | 0 | 9 | 1 |
| 10 | 661.42 | 10 | 7.88 | 118.64 | 0 | 9 | 1 |
| 11 | 647.40 | 8 | 6.77 | 118.64 | 0 | 9 | 1 |
| 12 | 647.40 | 9 | 7.15 | 118.64 | 0 | 9 | 1 |
| 13 | 661.42 | 10 | 7.65 | 118.64 | 0 | 9 | 1 |
| 14 | 675.44 | 8 | 6.40 | 132.64 | 0 | 9 | 1 |
| 15 | 647.40 | 8 | 6.77 | 118.64 | 0 | 9 | 1 |
| 16 | 661.42 | 9 | 7.15 | 118.64 | 0 | 9 | 1 |
| Min. | 319.872 | 8 | 4.00 | 28.16 | 0 | 2 | 0 |
| Max. | 704.450 | 10 | 7.93 | 132.64 | 2 | 10 | 2 |
| Molecules | Oral Bioavailability | MW | AlogP | HBD | HBA | R5 |
|---|---|---|---|---|---|---|
| Normal range | (<140 A°2) | (<500) | (≤5) | (≤5) | (≤10) | Max 4 |
| Hydroxychloroquine | 48.239 | 335.872 | 3.457 | 2 | 4 | 0 |
| MolPort-009-219-532 | 30.142 | 355.471 | 4.755 | 0 | 4 | 0 |
| MolPort-004-996-519 | 51.323 | 414.513 | 4.588 | 1 | 4 | 0 |
| MolPort-005-060-605 | 45.027 | 398.538 | 4.677 | 0 | 4 | 0 |
| MolPort-005-028-274 | 69.152 | 416.461 | 3.347 | 2 | 3 | 0 |
| MolPort-004-042-669 | 66.740 | 417.518 | 3.416 | 0 | 6 | 0 |
| MolPort-007-913-111 | 54.676 | 419.581 | 4.826 | 1 | 5 | 0 |
| MolPort-002-693-933 | 50.364 | 324.417 | 4.586 | 1 | 3 | 0 |
| MolPort-005-083-430 | 40.152 | 426.618 | 4.778 | 0 | 5 | 0 |
| MolPort-009-499-144 | 80.327 | 398.376 | 3.166 | 2 | 4 | 0 |
| Molecules | PPB | Hepatotoxic | CYP2D6 | Solubility | BBB | IA |
|---|---|---|---|---|---|---|
| Hydroxychloroquine | false | true | true | 3 | 1 | 0 |
| MolPort-009-219-532 | true | false | true | 2 | 0 | 0 |
| MolPort-004-996-519 | true | false | false | 2 | 1 | 0 |
| MolPort-005-060-605 | true | false | false | 2 | 1 | 0 |
| MolPort-005-028-274 | true | false | false | 2 | 2 | 0 |
| MolPort-004-042-669 | true | false | false | 2 | 2 | 0 |
| MolPort-007-913-111 | false | false | false | 2 | 1 | 0 |
| MolPort-002-693-933 | true | false | false | 2 | 1 | 0 |
| MolPort-005-083-430 | true | false | true | 2 | 1 | 0 |
| MolPort-009-499-144 | true | false | false | 2 | 2 | 0 |
| Molecules | Mouse Female | Rat Female | Ames Mutagenicity | Skin Irritancy |
|---|---|---|---|---|
| Hydroxychloroquine | Non-Carcinogen | Non-Carcinogen | Mutagen | None |
| MolPort-009-219-532 | Multi-Carcinogen | Non-Carcinogen | Non-Mutagen | None |
| MolPort-004-996-519 | Non-Carcinogen | Single-Carcinogen | Non-Mutagen | None |
| MolPort-005-060-605 | Non-Carcinogen | Non-Carcinogen | Non-Mutagen | None |
| MolPort-005-028-274 | Non-Carcinogen | Multi-Carcinogen | Non-Mutagen | Mild |
| MolPort-004-042-669 | Non-Carcinogen | Non-Carcinogen | Non-Mutagen | None |
| MolPort-007-913-111 | Multi-Carcinogen | Single-Carcinogen | Non-Mutagen | Mild |
| MolPort-002-693-933 | Multi-Carcinogen | Single-Carcinogen | Non-Mutagen | Mild |
| MolPort-005-083-430 | Non-Carcinogen | Non-Carcinogen | Non-Mutagen | None |
| MolPort-009-499-144 | Non-Carcinogen | Non-Carcinogen | Mutagen | None |
| Molecules | Rate Oral LD50 (g/kg Body Weight) | Daphnia EC50 (mg/L) * | Rat Chronic LOAEL (g/kg Body Weight) | Fathead Minnow LC50 (g/L) |
|---|---|---|---|---|
| Hydroxychloroquine | 0.207 | 34.619 | 0.033 | 0.0240 |
| MolPort-009-219-532 | 0.520 | 0.011 | 0.014 | 0.0006 |
| MolPort-004-996-519 | 0.867 | 0.394 | 0.005 | 0.0010 |
| MolPort-005-060-605 | 4.923 | 0.104 | 0.005 | 0.0004 |
| MolPort-005-028-274 | 5.528 | 0.370 | 0.021 | 0.0010 |
| MolPort-004-042-669 | 0.819 | 1.157 | 0.024 | 0.0004 |
| MolPort-007-913-111 | 1.803 | 0.022 | 0.051 | 0.0003 |
| MolPort-002-693-933 | 1.560 | 0.442 | 0.066 | 0.0002 |
| MolPort-005-083-430 | 0.063 | 0.720 | 0.014 | 0.0001 |
| MolPort-009-499-144 | 1.065 | 2.801 | 0.016 | 0.0020 |
| Molecules | Mouse | Rat | RMTD * |
|---|---|---|---|
| Hydroxychloroquine | 13.868 | 1.305 | 357 |
| MolPort-009-219-532 | 147.089 | 51.500 | 90 |
| MolPort-004-996-519 | 43.816 | 1.234 | 83 |
| MolPort-005-060-605 | 3.365 | 0.445 | 26 |
| MolPort-005-028-274 | 329.611 | 25.088 | 89 |
| MolPort-004-042-669 | 178.986 | 9.745 | 26 |
| MolPort-007-913-111 | 116.065 | 11.490 | 76 |
| MolPort-002-693-933 | 80.973 | 10.346 | 91 |
| MolPort-005-083-430 | 8.858 | 56.407 | 44 |
| MolPort-009-499-144 | 496.259 | 15.599 | 42 |
| Molecule | Molinspiration | PASS | |||
|---|---|---|---|---|---|
| Score | Bioactivity | Pa [a] | Pi [b] | Biological Activity | |
| 11b | 0.65 0.28 | Protease inhibitor Enzyme inhibitor | 0.265 | 0.016 | Protease inhibitor |
| MolPort-009-219-532 | 0.11 0.04 | Protease inhibitor Enzyme inhibitor | - | - | - |
| MolPort-004-996-519 | −0.08 −0.17 | Protease inhibitor Enzyme inhibitor | - | - | - |
| MolPort-005-060-605 | −0.48 −0.35 | Protease inhibitor Enzyme inhibitor | - | - | - |
| MolPort-005-028-274 | −0.36 −0.47 | Protease inhibitor Enzyme inhibitor | 0.134 | 0.059 | Protease inhibitor |
| MolPort-009-499-144 | −0.52 −0.47 | Protease inhibitor Enzyme inhibitor | - | - | - |
| Molecules | SA | Target |
|---|---|---|
| MolPort-007-913-111 | 65.579 | ACE2 |
| MolPort-002-693-933 | 79.254 | |
| MolPort-004-042-669 | 67.940 | |
| MolPort-005-131-430 | 61.351 | |
| MolPort-005-060-605 | 67.338 | |
| MolPort-009-219-532 | 81.768 | Mpro |
| MolPort-004-996-519 | 68.009 | |
| MolPort-005-028-274 | 67.051 | |
| MolPort-009-499-144 | 76.392 |
| Moleclues | iLOGP | XLOGP | WLOGP | MLOGP | SILICOS-IT | Consensus LogP |
|---|---|---|---|---|---|---|
| Pivot | 3.58 | 3.58 | 3.59 | 2.35 | 3.73 | 3.37 |
| MolPort-007-913-111 | 4.50 | 4.10 | 4.13 | 2.71 | 5.60 | 4.21 |
| MolPort-002-693-933 | 3.33 | 4.12 | 3.79 | 3.46 | 5.20 | 3.98 |
| MolPort-004-042-669 | 3.99 | 3.55 | 4.05 | 2.03 | 4.62 | 3.65 |
| MolPort-005-131-430 | 4.17 | 4.02 | 3.23 | 2.78 | 4.48 | 3.74 |
| MolPort-005-060-605 | 4.61 | 4.67 | 4.90 | 2.76 | 5.71 | 4.53 |
| MolPort-009-219-532 | 4.58 | 4.51 | 4.37 | 2.98 | 4.78 | 4.24 |
| MolPort-004-996-519 | 4.47 | 4.07 | 4.48 | 3.28 | 4.99 | 4.26 |
| MolPort-005-028-274 | 3.46 | 3.18 | 3.76 | 3.23 | 5.62 | 3.85 |
| MolPort-009-499-144 | 2.95 | 3.04 | 5.09 | 1.56 | 5.05 | 3.54 |
| Moleclues | ESOL | Ali | SILICOS-IT | Consensus LogS |
|---|---|---|---|---|
| Pivot | −3.91 | −4.28 | −6.35 | −5.81 |
| MolPort-007-913-111 | −4.69 | −5.73 | −5.99 | −5.95 |
| MolPort-002-693-933 | −4.29 | −4.89 | −7.10 | −5.35 |
| MolPort-004-042-669 | −4.35 | −5.17 | −5.67 | −6.33 |
| MolPort-005-131-430 | −4.63 | −4.66 | −7.43 | −6.03 |
| MolPort-005-060-605 | −5.01 | −5.40 | −7.02 | −5.42 |
| MolPort-009-219-532 | −4.57 | −4.88 | −6.97 | −5.27 |
| MolPort-004-996-519 | −4.68 | −4.84 | −7.45 | −5.94 |
| MolPort-005-028-274 | −4.06 | −4.27 | −8.57 | −6.58 |
| MolPort-009-499-144 | −3.92 | −4.39 | −6.72 | −6.57 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramos, R.S.; Borges, R.S.; de Souza, J.S.N.; Araujo, I.F.; Chaves, M.H.; Santos, C.B.R. Identification of Potential Antiviral Inhibitors from Hydroxychloroquine and 1,2,4,5-Tetraoxanes Analogues and Investigation of the Mechanism of Action in SARS-CoV-2. Int. J. Mol. Sci. 2022, 23, 1781. https://doi.org/10.3390/ijms23031781
Ramos RS, Borges RS, de Souza JSN, Araujo IF, Chaves MH, Santos CBR. Identification of Potential Antiviral Inhibitors from Hydroxychloroquine and 1,2,4,5-Tetraoxanes Analogues and Investigation of the Mechanism of Action in SARS-CoV-2. International Journal of Molecular Sciences. 2022; 23(3):1781. https://doi.org/10.3390/ijms23031781
Chicago/Turabian StyleRamos, Ryan S., Rosivaldo S. Borges, João S. N. de Souza, Inana F. Araujo, Mariana H. Chaves, and Cleydson B. R. Santos. 2022. "Identification of Potential Antiviral Inhibitors from Hydroxychloroquine and 1,2,4,5-Tetraoxanes Analogues and Investigation of the Mechanism of Action in SARS-CoV-2" International Journal of Molecular Sciences 23, no. 3: 1781. https://doi.org/10.3390/ijms23031781
APA StyleRamos, R. S., Borges, R. S., de Souza, J. S. N., Araujo, I. F., Chaves, M. H., & Santos, C. B. R. (2022). Identification of Potential Antiviral Inhibitors from Hydroxychloroquine and 1,2,4,5-Tetraoxanes Analogues and Investigation of the Mechanism of Action in SARS-CoV-2. International Journal of Molecular Sciences, 23(3), 1781. https://doi.org/10.3390/ijms23031781