
Metabolic Reprogramming in Response to Alterations of Mitochondrial DNA and Mitochondrial Dysfunction in Gastric Adenocarcinoma


Abstract
:1. Introduction
2. Results
2.1. Distributions of D310 Variants and mtDNA Copy Numbers of Analyzed Samples
2.2. Demographic Data of the 57 GAC Patients
2.3. Prognostic Variables and Their Hazard Ratios (HRs)
2.4. Cancer Pathological Characteristics and mtDNA Alterations
2.5. Effect of TFAM Knockdown in AGS Cells and Their Alterations
3. Discussion
4. Materials and Methods
4.1. Study Participants, Tissue Preparation, and DNA Extraction
4.2. Human GAC Cell Line, TFAM Knockdown, Stable Clone Selection, and Cellular DNA/Protein Extractions
4.3. Quantification of mtDNA and nDNA Copies and Analysis of mtDNA Copy Number
4.4. Sequencing of the D310 Region of mtDNA
4.5. Determination of Relative Protein Expression Levels
4.6. Analysis of Bioenergetic Parameters by the XFe-24 Analyzer
4.7. Wound Healing Migration Assay
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chen, Y.-J.; Yeh, S.-T.; Ou, L.-H.; Lin, C.-S.; Chien, C.-T. Impact of the extent of negative lymph nodes in gastric adenocarcinoma undergoing primary surgical resection: An institutional report. J. Chin. Med. Assoc. 2021, 84, 428–437. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; El Hajj, N.; Sittler, S.; Lammert, N.; Barnes, R.; Meloni-Ehrig, A. Gastric cancer: Classification, histology and application of molecular pathology. J. Gastrointest. Oncol. 2012, 3, 251–261. [Google Scholar] [CrossRef]
- Lee, H.-C.; Huang, K.-H.; Yeh, T.-S.; Chi, C.-W. Somatic alterations in mitochondrial DNA and mitochondrial dysfunction in gastric cancer progression. World J. Gastroenterol. 2014, 20, 3950–3959. [Google Scholar] [CrossRef]
- Wu, C.-W.; Yin, P.-H.; Hung, W.-Y.; Li, A.F.-Y.; Li, S.-H.; Chi, C.-W.; Wei, Y.-H.; Lee, H.-C. Mitochondrial DNA mutations and mitochondrial DNA depletion in gastric cancer. Genes Chromosomes Cancer 2005, 44, 19–28. [Google Scholar] [CrossRef]
- Yang, H.; Li, Y.; Hu, B. Potential role of mitochondria in gastric cancer detection: Fission and glycolysis. Oncol. Lett. 2021, 21, 439. [Google Scholar] [CrossRef]
- Lee, H.-C.; Wei, Y.-H. Mitochondrial biogenesis and mitochondrial DNA maintenance of mammalian cells under oxidative stress. Int. J. Biochem. Cell Biol. 2005, 37, 822–834. [Google Scholar] [CrossRef]
- Glancy, B.; Kane, D.A.; Kavazis, A.N.; Goodwin, M.L.; Willis, W.T.; Gladden, L.B. Mitochondrial lactate metabolism: History and implications for exercise and disease. J. Physiol. 2021, 599, 863–888. [Google Scholar] [CrossRef] [PubMed]
- Anderson, A.J.; Jackson, T.D.; Stroud, D.A.; Stojanovski, D. Mitochondria-hubs for regulating cellular biochemistry: Emerging concepts and networks. Open Biol. 2019, 9, 190126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bahat, A.; Gross, A. Mitochondrial plasticity in cell fate regulation. J. Biol. Chem. 2019, 294, 13852–13863. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.-w.; Dang, C.V. Multifaceted roles of glycolytic enzymes. Trends Biochem. Sci. 2005, 30, 142–150. [Google Scholar] [CrossRef]
- Prochownik, E.V.; Wang, H. The metabolic fates of pyruvate in normal and neoplastic cells. Cells 2021, 10, 762. [Google Scholar] [CrossRef]
- Yan, C.; Duanmu, X.; Zeng, L.; Liu, B.; Song, Z. Mitochondrial DNA: Distribution, mutations, and elimination. Cells 2019, 8, 379. [Google Scholar] [CrossRef] [Green Version]
- Gray, M.W. Mitochondrial evolution. Cold Spring Harb. Perspect. Biol. 2012, 4, a011403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallace, D.C.; Chalkia, D. Mitochondrial DNA genetics and the heteroplasmy conundrum in evolution and disease. Cold Spring Harb. Perspect. Biol. 2013, 5, a021220. [Google Scholar] [CrossRef] [PubMed]
- Hahn, A.; Zuryn, S. Mitochondrial genome (mtDNA) mutations that generate reactive oxygen species. Antioxidants 2019, 8, 392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, A.P.; Luo, X.; Russell, W.; Yin, Y.W. Oxidative damage diminishes mitochondrial DNA polymerase replication fidelity. Nucleic Acids Res. 2020, 48, 817–829. [Google Scholar] [CrossRef]
- Mambo, E.; Gao, X.; Cohen, Y.; Guo, Z.; Talalay, P.; Sidransky, D. Electrophile and oxidant damage of mitochondrial DNA leading to rapid evolution of homoplasmic mutations. Proc. Natl. Acad. Sci. USA 2003, 100, 1838–1843. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.-S.; Chang, S.-C.; Wang, L.-S.; Chou, T.-Y.; Hsu, W.-H.; Wu, Y.-C.; Wei, Y.-H. The role of mitochondrial DNA alterations in esophageal squamous cell carcinomas. J. Thorac. Cardiovasc. Surg. 2009, 139, 189–197. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.-S.; Chang, S.-C.; Ou, L.-H.; Chen, C.-M.; Hsieh, S.S.W.; Chung, Y.-P.; King, K.-L.; Lin, S.-L.; Wei, Y.-H. Mitochondrial DNA alterations correlate with the pathological status and the immunological ER, PR, HER-2/neu, p53 and Ki-67 expression in breast invasive ductal carcinoma. Oncol. Rep. 2015, 33, 2924–2934. [Google Scholar] [CrossRef] [Green Version]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Kim, J.W.; Dang, C.V. Cancer’s molecular sweet tooth and the Warburg effect. Cancer Res. 2006, 66, 8927–8930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Hattab, A.W.; Scaglia, F. Mitochondrial DNA depletion syndromes: Review and updates of genetic basis, manifestations, and therapeutic options. Neurotherapeutics 2013, 10, 186–198. [Google Scholar] [CrossRef] [Green Version]
- Bao, X.; Zhang, J.; Huang, G.; Yan, J.; Xu, C.; Dou, Z.; Sun, C.; Zhang, H. The crosstalk between HIFs and mitochondrial dysfunctions in cancer development. Cell Death Dis. 2021, 12, 215. [Google Scholar] [CrossRef] [PubMed]
- Vaupel, P.; Multhoff, G. Revisiting the Warburg effect: Historical dogma versus current understanding. J. Physiol. 2021, 599, 1745–1757. [Google Scholar] [CrossRef]
- Lee, H.-T.; Lin, C.-S.; Pan, S.-C.; Wu, T.-H.; Lee, C.-S.; Chang, D.-M.; Tsai, C.-Y.; Wei, Y.-H. Alterations of oxygen consumption and extracellular acidification rates by glutamine in PBMCs of SLE patients. Mitochondrion 2019, 44, 65–74. [Google Scholar] [CrossRef]
- Chapman, J.; Ng, Y.S.; Nicholls, T.J. The maintenance of mitochondrial DNA integrity and dynamics by mitochondrial membranes. Life 2020, 10, 164. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-S.; Lee, H.-T.; Lee, M.-H.; Pan, S.-C.; Ke, C.-Y.; Chiu, A.W.-H.; Wei, Y.-H. Role of mitochondrial DNA copy number alteration in human renal cell carcinoma. Int. J. Mol. Sci. 2016, 17, 814. [Google Scholar] [CrossRef] [PubMed]
- Yin, P.H.; Lee, H.C.; Chau, G.Y.; Wu, Y.T.; Li, S.H.; Lui, W.Y.; Wei, Y.H.; Liu, T.Y.; Chi, C.W. Alteration of the copy number and deletion of mitochondrial DNA in human hepatocellular carcinoma. Br. J. Cancer 2004, 90, 2390–2396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tseng, L.M.; Yin, P.H.; Chi, C.W.; Hsu, C.Y.; Wu, C.W.; Lee, L.M.; Wei, Y.H.; Lee, H.C. Mitochondrial DNA mutations and mitochondrial DNA depletion in breast cancer. Genes Chromosomes Cancer 2006, 45, 629–638. [Google Scholar] [CrossRef]
- Wen, S.-L.; Zhang, F.; Feng, S. Decreased copy number of mitochondrial DNA: A potential diagnostic criterion for gastric cancer. Oncol. Lett. 2013, 6, 1098–1102. [Google Scholar] [CrossRef] [Green Version]
- Moindjie, H.; Rodrigues-Ferreira, S.; Nahmias, C. Mitochondrial metabolism in carcinogenesis and cancer therapy. Cancers 2021, 13, 3311. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-S.; Huang, Y.-Y.; Pan, S.-C.; Cheng, C.-T.; Liu, C.-C.; Shih, C.-H.; Ho, H.-L.; Yeh, Y.-C.; Chou, T.-Y.; Lee, M.-Y.; et al. Involvement of increased p53 expression in the decrease of mitochondrial DNA copy number and increase of SUVmax of FDG-PET scan in esophageal squamous cell carcinoma. Mitochondrion 2019, 47, 54–63. [Google Scholar] [CrossRef]
- Lin, C.-S.; Yeh, Y.-C.; Pan, S.-C.; Lu, S.-Y.; Chen, Y.-J.; Chueh, W.-Y.; Wei, Y.-H. Role of mitochondrial DNA copy number alteration in non-small cell lung cancer. Formos. J. Surg. 2020, 53, 165–176. [Google Scholar] [CrossRef]
- Lin, C.-S.; Wang, L.-S.; Tsai, C.-M.; Wei, Y.-H. Low copy number and low oxidative damage of mitochondrial DNA are associated with tumor progression in lung cancer tissues after neoadjuvant chemotherapy. Interact. Cardiovasc. Thorac. Surg. 2008, 7, 954–958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, M.; Zhou, Y.; Shi, Y.; Ning, L.; Yang, Y.; Wei, X.; Zhang, N.; Hao, X.; Niu, R. Reduced mitochondrial DNA copy number is correlated with tumor progression and prognosis in Chinese breast cancer patients. IUBMB Life 2007, 59, 450–457. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.-Z.; Fang, Y.; Shi, Y.-H.; Cui, J.-H.; Li, L.-Y.; Xu, Y.-C.; Ling, B. Mitochondrial D310 instability in Chinese lung cancer patients. Mitochondrial DNA Part A DNA Mapp. Seq. Anal. 2016, 27, 1177–1180. [Google Scholar] [CrossRef] [PubMed]
- Matsuyama, W.; Nakagawa, M.; Wakimoto, J.; Hirotsu, Y.; Kawabata, M.; Osame, M. Mitochondrial DNA mutation correlates with stage progression and prognosis in non-small cell lung cancer. Hum. Mutat. 2003, 21, 441–443. [Google Scholar] [CrossRef]
- Lièvre, A.; Chapusot, C.; Bouvier, A.-M.; Zinzindohoué, F.; Piard, F.; Roignot, P.; Arnould, L.; Beaune, P.; Faivre, J.; Laurent-Puig, P. Clinical value of mitochondrial mutations in colorectal cancer. J. Clin. Oncol. 2005, 23, 3517–3525. [Google Scholar] [CrossRef]
- Sharma, P.; Sampath, H. Mitochondrial DNA integrity: Role in health and disease. Cells 2019, 8, 100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wai, T.; Langer, T. Mitochondrial Dynamics and Metabolic Regulation. Trends Endocrinol. Metab. 2016, 27, 105–117. [Google Scholar] [CrossRef]
- Guido, C.; Whitaker-Menezes, D.; Lin, Z.; Pestell, R.G.; Howell, A.; Zimmers, T.A.; Casimiro, M.C.; Aquila, S.; Ando, S.; Martinez-Outschoorn, U.E.; et al. Mitochondrial fission induces glycolytic reprogramming in cancer-associated myofibroblasts, driving stromal lactate production, and early tumor growth. Oncotarget 2012, 3, 798–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, C.S.; Lee, H.T.; Lee, S.Y.; Shen, Y.A.; Wang, L.S.; Chen, Y.J.; Wei, Y.H. High mitochondrial DNA copy number and bioenergetic function are associated with tumor invasion of esophageal squamous cell carcinoma cell lines. Int. J. Mol. Sci. 2012, 13, 11228–11246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, C.S.; Liu, L.T.; Ou, L.H.; Pan, S.C.; Lin, C.I.; Wei, Y.H. Role of mitochondrial function in the invasiveness of human colon cancer cells. Oncol. Rep. 2018, 39, 316–330. [Google Scholar] [CrossRef] [Green Version]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [Green Version]
- Moro, L.; Arbini, A.A.; Yao, J.L.; di Sant’Agnese, P.A.; Marra, E.; Greco, M. Mitochondrial DNA depletion in prostate epithelial cells promotes anoikis resistance and invasion through activation of PI3K/Akt2. Cell Death Differ. 2009, 16, 571–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, M.; Marayati, R.; Moffitt, R.; Yeh, J.J. Hexokinase 2 promotes tumor growth and metastasis by regulating lactate production in pancreatic cancer. Oncotarget 2017, 8, 56081–56094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, D.; Park, J.H.; Kaur, H.; Jung, K.H.; Yang, S.; Tripathi, S.; Galbraith, M.; Deng, Y.; Jolly, M.K.; Kaipparettu, B.A.; et al. Towards decoding the coupled decision-making of metabolism and epithelial-to-mesenchymal transition in cancer. Br. J. Cancer 2021, 124, 1902–1911. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, P.L. Warburg, me and hexokinase 2: Multiple discoveries of key molecular events underlying one of cancers’ most common phenotypes, the “Warburg Effect”, i.e., elevated glycolysis in the presence of oxygen. J. Bioenerg. Biomembr. 2007, 39, 211. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mo, M.; Peng, F.; Wang, L.; Peng, L.; Lan, G.; Yu, S. Roles of mitochondrial transcription factor A and microRNA-590-3p in the development of bladder cancer. Oncol Lett 2013, 6, 617–623. [Google Scholar] [CrossRef] [Green Version]
- McCann, C.; Kerr, E.M. Metabolic reprogramming: A friend or foe to cancer therapy? Cancers 2021, 13, 3351. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Schiliro, C.; Firestein, B.L. Mechanisms of metabolic reprogramming in cancer cells supporting enhanced growth and proliferation. Cells 2021, 10, 1056. [Google Scholar] [CrossRef] [PubMed]
- Ward, P.S.; Thompson, C.B. Metabolic reprogramming: A cancer hallmark even warburg did not anticipate. Cancer Cell 2012, 21, 297–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rehman, J.; Zhang, H.J.; Toth, P.T.; Zhang, Y.; Marsboom, G.; Hong, Z.; Salgia, R.; Husain, A.N.; Wietholt, C.; Archer, S.L. Inhibition of mitochondrial fission prevents cell cycle progression in lung cancer. FASEB J. 2012, 26, 2175–2186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, J.; Zhang, J.; Yu, M.; Xie, Y.; Huang, Y.; Wolff, D.W.; Abel, P.W.; Tu, Y. Mitochondrial dynamics regulates migration and invasion of breast cancer cells. Oncogene 2013, 32, 4814–4824. [Google Scholar] [CrossRef] [PubMed]
- Saunier, E.; Benelli, C.; Bortoli, S. The pyruvate dehydrogenase complex in cancer: An old metabolic gatekeeper regulated by new pathways and pharmacological agents. Int. J. Cancer 2016, 138, 809–817. [Google Scholar] [CrossRef]
- Park, S.; Jeon, J.-H.; Min, B.-K.; Ha, C.-M.; Thoudam, T.; Park, B.-Y.; Lee, I.-K. Role of the Pyruvate Dehydrogenase Complex in Metabolic Remodeling: Differential Pyruvate Dehydrogenase Complex Functions in Metabolism. DMJ 2018, 42, 270–281. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Iglesias, A.; Mañes, S. The importance of mitochondrial pyruvate carrier in cancer cell metabolism and tumorigenesis. Cancers 2021, 13, 1488. [Google Scholar] [CrossRef] [PubMed]
- Hoxhaj, G.; Manning, B.D. The PI3K-AKT network at the interface of oncogenic signalling and cancer metabolism. Nat. Rev. Cancer 2020, 20, 74–88. [Google Scholar] [CrossRef]
- Hsieh, Y.-T.; Tu, H.-F.; Yang, M.-H.; Chen, Y.-F.; Lan, X.-Y.; Huang, C.-L.; Chen, H.-M.; Li, W.-C. Mitochondrial genome and its regulator TFAM modulates head and neck tumourigenesis through intracellular metabolic reprogramming and activation of oncogenic effectors. Cell Death Dis. 2021, 12, 961. [Google Scholar] [CrossRef] [PubMed]
- Nunes, J.B.; Peixoto, J.; Soares, P.; Maximo, V.; Carvalho, S.; Pinho, S.S.; Vieira, A.F.; Paredes, J.; Rego, A.C.; Ferreira, I.L.; et al. OXPHOS dysfunction regulates integrin-β1 modifications and enhances cell motility and migration. Hum. Mol. Genet. 2014, 24, 1977–1990. [Google Scholar] [CrossRef] [Green Version]
- Marano, L.; D’Ignazio, A.; Cammillini, F.; Angotti, R.; Messina, M.; Marrelli, D.; Roviello, F. Comparison between 7th and 8th edition of AJCC TNM staging system for gastric cancer: Old problems and new perspectives. Transl. Gastroenterol. Hepatol. 2019, 4, 22. [Google Scholar] [CrossRef]
- Wang, S.-F.; Huang, K.-H.; Tseng, W.-C.; Lo, J.-F.; Li, A.F.-Y.; Fang, W.-L.; Chen, C.-F.; Yeh, T.-S.; Chang, Y.-L.; Chou, Y.-C.; et al. DNAJA3/Tid1 is required for mitochondrial DNA maintenance and regulates migration and invasion of human gastric cancer cells. Cancers 2020, 12, 3463. [Google Scholar] [CrossRef] [PubMed]
- Sung, M.-T.; Hsu, H.-T.; Lee, C.-C.; Lee, H.-C.; Kuo, Y.J.; Hua, K.; Hsia, C.-Y.; Chi, C.-W. Krüppel-like factor 4 modulates the migration and invasion of hepatoma cells by suppressing TIMP-1 and TIMP-2. Oncol. Rep. 2015, 34, 439–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Unal, I. Defining an optimal cut-point value in ROC analysis: An alternative approach. Comput. Math. Methods Med. 2017, 2017, 3762651. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| DNA Variables | Subjects/Analyzed Tissue Samples | |||||
|---|---|---|---|---|---|---|
| Overall (n = 131, 100.0%) | Controls (n = 17) | GAC Patients (n = 57) | p-Value | |||
| N-GM (n = 17, 100.0%) | NC-GM (n = 57, 100.0%) | GAC (n = 57, 100.0%) | a | b | ||
| D310 pattern | ||||||
| Homoplasmy | 45 (34.4%) | 6 (35.3%) | 17 (29.8%) | 22 (38.6%) | 0.324 * | 0.547 **** |
| Heteroplasmy | 86 (65.6%) | 11 (64.7%) | 40 (70.2%) | 35 (61.4%) | ||
| Major D310 variant | ||||||
| C7TC6 | 47 (35.9%) | 4 (23.5%) | 20 (35.1%) | 23 (40.4%) | 0.578 * | 0.105 **** |
| C8TC6 | 64 (48.9%) | 8 (47.1%) | 29 (50.9%) | 27 (47.4%) | ||
| Others | 20 (15.3%) | 5 (29.4%) | 8 (14.0%) | 7 (12.3%) | ||
| Number of D310 variants | 1.9 ± 1.0 | 2.4 ± 1.9 | 1.9 ± 0.7 | 1.9 ± 0.8 | 0.829 ** | 0.179 *** |
| D310 mutation | ||||||
| No | 30 (52.6%) | |||||
| Yes | 27 (47.4%) | |||||
| mtDNA copy number | ||||||
| Mean ± SD | 0.066 ± 0.060 | 0.060 ± 0.030 | 0.078 ± 0.076 | 0.055 ± 0.045 | <0.001 ** | 0.071 *** |
| Median | 0.050 | 0.058 | 0.055 | 0.045 | 0.030 *** | |
| ≤0.050 | 66 (50.4%) | 6 (35.3%) | 24 (42.1%) | 36 (63.2%) | 0.013 **** | |
| >0.050 | 65 (49.6%) | 11(64.7%) | 33 (57.9%) | 21 (36.8%) | ||
| mtDNA copy number ratio | Mean ± SD | p-value ***** | ||||
| Overall (n = 57, 100.0%) | 0.891 ± 0.968 | |||||
| Survival status | ||||||
| Alive (n = 29, 50.9%) | 1.081 ± 1.314 | 0.032 | ||||
| Dead (n = 28, 49.1%) | 0.694 ± 0.273 | |||||
| Subgroups | ||||||
| Low (≤0.804, n = 35, 61.4%) | 0.579 ± 0.138 | <0.001 | ||||
| High (>0.804, n = 22, 38.6%) | 1.387 ± 1.431 | |||||
| Variables | Case Number (%)/Mean ± SD |
|---|---|
| Sex | |
| Male/Female | 37 (64.9)/20 (35.1) |
| Age (years) | 67.1 ± 13.4 |
| Type of resection | |
| Partial/subtotal/total gastrectomy | 1 (1.8)/4 (77.2)/12 (21.1) |
| Pathological finding | |
| T-status * | |
| T1/T2/T3/T4 | 12 (21.1)/6 (10.5)/27 (47.4)/12 (21.1) |
| N-status * | |
| N0/N1/N2/N3 | 19 (33.3)/3 (5.3)/13 (22.8)/22 (38.6) |
| M-status * | |
| M0/M1 | 44 (77.2)/13 (22.8) |
| Differentiation | |
| Well/moderate/poor | 8 (14.0)/23 (40.4)/26 (45.6) |
| Maximal tumor diameter (cm) | 4.4 ± 2.5 |
| ≤4 cm/>4 cm | 30 (52.6)/27 (47.4) |
| Resection margin invasion | |
| No/Yes | 53 (93.0)/4 (7.0) |
| Lymphovascular invasion | |
| No/Yes | 22 (38.6)/35 (61.4) |
| Perineural invasion | |
| No/Yes | 22 (38.6)/35 (61.4) |
| mtDNA copy number ratio | 0.891 ± 0.968 |
| High (>0.804)/Low (≤0.804) | 22 (38.6%)/35 (61.4%) |
| D310 mutation | |
| No/Yes | 30 (52.6%)/27 (47.4%) |
| Survivals (months) | 44.2 ± 5.7 |
| Follow-up periods (months) | 22.5 ± 21.9 |
| Survival Differences | Log-Rank | Univariate Cox’s Regression | Multivariate Cox’s Regression | |||
|---|---|---|---|---|---|---|
| Variables/Case Number | Survival (95% CI), Months | p-Value | HRs (95% CI) | p-Value | HRs (95% CI) | p-Value |
| Sex | 0.584 | |||||
| Male (n = 37) | 42.2 (29.1−55.3) | 1.000 | ||||
| Female (n = 20) | 36.0 (24.4−47.7) | 0.794 (0.347−1.819) | 0.585 | |||
| Age | 0.410 | |||||
| ≤65 (n = 27) | 46.8 (31.5−62.1) | 1.000 | ||||
| >65 (n = 30) | 40.0 (25.6−54.5) | 1.375 (0.642−2.946) | 0.412 | |||
| Type of resection | 0.400 | |||||
| Partial gastrectomy (n = 1) | 7.9 (7.9−7.9) | 3.649 (0.473−28.126) | 0.214 | |||
| Subtotal gastrectomy (n = 44) | 44.1 (32.3−55.8) | 1.000 | ||||
| Total gastrectomy (n = 12) | 37.9 (15.9−59.9) | 1.209 (0.511−2.862) | 0.666 | |||
| Pathological finding | ||||||
| T-status * | 0.008 | |||||
| T1 (n = 12) | 66.7 (54.4−79.0) | 1.000 | 1.000 | |||
| T2 (n = 6) | 43.3 (21.4−65.1) | 4.490 (0.407−49.541) | 0.220 | 2.112 (0.183−27.412) | 0.567 | |
| T3 (n = 27) | 23.9 (13.1−34.7) | 13.468 (1.789−101.421) | 0.012 | 2.554 (0.180−36.301) | 0.489 | |
| T4 (n = 12) | 36.7 (13.5−59.9) | 10.679 (1.310−87.034) | 0.027 | 1.572 (0.092−26.766) | 0.755 | |
| N-status * | 0.049 | |||||
| N0 (n = 19) | 54.7 (40.7−68.6) | 1.000 | 1.000 | |||
| N1 (n = 3) | 58.7 (11.8−105.6) | 1.804 (0.210−15.480) | 0.591 | 1.306 (0.080−21.287) | 0.852 | |
| N2 (n = 13) | 38.1 (17.5−58.8) | 2.813 (0.890-8.887) | 0.078 | 1.370 (0.178-10.548) | 0.762 | |
| N3 (n = 22) | 19.9 (12.5−27.2) | 3.925 (1.407−10.944) | 0.009 | 0.784 (0.103−5.975) | 0.815 | |
| M-status * | <0.001 | |||||
| M0 (n = 44) | 53.0 (40.4−65.6) | 1.000 | 1.000 | |||
| M1 (n = 13) | 8.8 (4.7−12.9) | 4.648 (2.074−10.418) | <0.001 | 2.902 (0.991−8.496) | 0.052 | |
| Cancer cell differentiation | 0.091 | |||||
| Well (n = 8) | 57.0 (36.1−77.9) | 1.000 | 1.000 | |||
| Moderate/Poor (n = 49) | 40.2 (28.1−52.3) | 3.236 (0.765−13.693) | 0.111 | 2.300 (0.356−14.850) | 0.382 | |
| Maximal tumor diameter | 0.002 | |||||
| ≤4 cm (n = 30) | 56.6 (43.3−70.0) | 1.000 | 1.000 | |||
| >4 cm (n = 27) | 26.9 (13.3−40.6) | 3.273 (1.474−7.270) | 0.004 | 2.185 (0.721−6.625) | 0.167 | |
| Resection margin invasion | 0.345 | |||||
| No (n = 53) | 46.4 (34.8−58.0) | 1.000 | ||||
| Yes (n = 4) | 23.5 (1.1−45.8) | 1.771 (0.532−5.895) | 0.352 | |||
| Lymphovascular invasion | 0.015 | |||||
| No (n = 22) | 62.5 (45.3−79.7) | 1.000 | 1.000 | |||
| Yes (n = 35) | 30.9 (19.1−42.7) | 2.922 (1.180−7.236) | 0.020 | 0.671 (0.104−4.313) | 0.674 | |
| Perineural invasion | 0.002 | |||||
| No (n = 22) | 68.4 (53.4−83.4) | 1.000 | 1.000 | |||
| Yes (n = 35) | 25.0 (14.3−35.7) | 4.184 (1.558−11.239) | 0.005 | 2.708 (0.690−10.626) | 0.153 | |
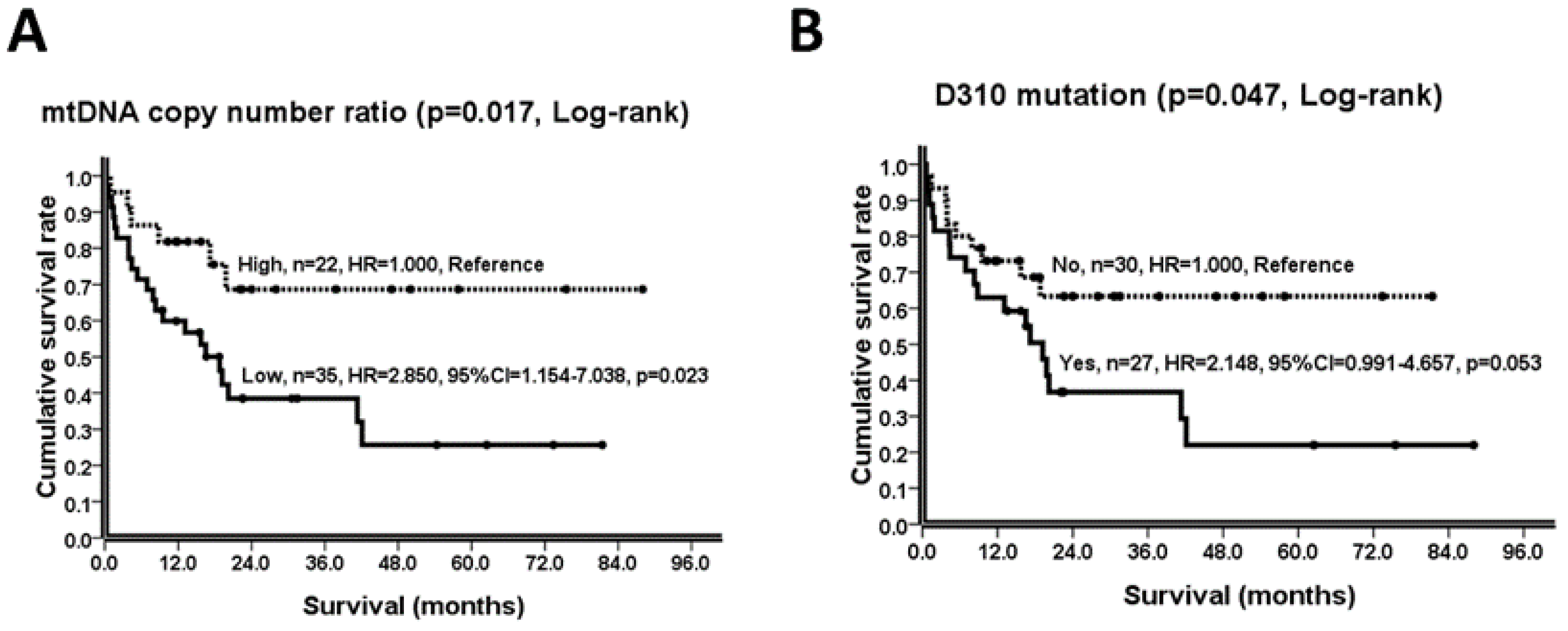
| mtDNA copy number ratio | 0.017 | |||||
| High (n = 22) | 63.7 (47.2−80.1) | 1.000 | 1.000 | |||
| Low (n = 35) | 31.6 (19.8−43.4) | 2.850 (1.154−7.038) | 0.023 | 1.597 (0.530−4.812) | 0.406 | |
| D310 mutation | 0.047 | |||||
| No (n = 30) | 54.5 (40.9−68.1) | 1.000 | 1.000 | |||
| Yes (n = 27) | 31.6 (17.9−45.3) | 2.148 (0.991−4.657) | 0.053 | 1.825 (0.724−4.601) | 0.202 | |
| mtDNA Copy Number Ratio | D310 Mutation | |||||
|---|---|---|---|---|---|---|
| Variables (Case Number, %) | Low (n = 35) | High (n = 22) | p-Value * | Yes (n = 27) | No (n = 30) | p-Value * |
| T-status | 0.064 | 0.007 | ||||
| T1 (n = 12, 100.0%) | 4 (33.3%) | 8 (66.7%) | 2 (16.7%) | 10 (83.3%) | ||
| T2 (n = 6, 100.0%) | 4 (66.7%) | 2 (33.3%) | 2 (33.3%) | 4 (66.7%) | ||
| T3 (n = 27, 100.0%) | 19 (70.4%) | 8 (29.6%) | 15 (55.6%) | 12 (44.4%) | ||
| T4 (n = 12,100.0%) | 8 (66.7%) | 4 (33.3%) | 8 (66.7%) | 4 (33.3%) | ||
| N-status | 0.070 | 0.153 | ||||
| N0 (n = 19, 100.0%) | 9 (47.4%) | 10 (52.6%) | 7 (36.8%) | 12 (63.2%) | ||
| N1 (n = 3, 100.0%) | 2 (66.7%) | 1 (33.3%) | 1 (33.3%) | 2 (66.7%) | ||
| N2 (n = 13, 100.0%) | 7 (53.8%) | 6 (46.2%) | 6 (46.2%) | 7 (53.8%) | ||
| N3 (n = 22, 100.0%) | 17 (77.3%) | 5 (22.7%) | 13 (59.1%) | 9 (40.9%) | ||
| M-status | 0.009 | 0.464 | ||||
| M0 (n = 44, 100.0%) | 23 (52.3%) | 21 (47.7%) | 22 (50.0%) | 22 (50.0%) | ||
| M1 (n = 13, 100.0%) | 12 (92.3%) | 1 (7.7%) | 5 (38.5%) | 8 (61.5%) | ||
| Differentiation | 0.002 | 0.547 | ||||
| Well (n = 8, 100.0%) | 1 (12.5%) | 7 (87.5%) | 3 (37.5%) | 5 (52.5%) | ||
| Moderate/Poor (n = 49, 100.0%) | 34 (69.4%) | 15 (30.6%) | 24 (49.0%) | 25 (51.0%) | ||
| Maximal tumor diameter | 0.752 | 0.006 | ||||
| ≤4 cm (n = 30, 100.0%) | 19 (63.3%) | 11 (36.7%) | 9 (30.0%) | 21 (70.0%) | ||
| >4 cm (n = 27, 100.0%) | 16 (59.3%) | 11 (40.7%) | 18 (66.7%) | 9 (33.3%) | ||
| Lymphovascular invasion | 0.161 | 0.439 | ||||
| No (n = 22, 100.0%) | 11 (50.0%) | 11 (50.0%) | 9 (40.9%) | 13 (59.1%) | ||
| Yes (n = 35, 100.0%) | 24 (68.6%) | 11 (31.4%) | 18 (51.4%) | 17 (48.6%) | ||
| Perineural invasion | 0.161 | 0.062 | ||||
| No (n = 22, 100.0%) | 11 (50.0%) | 11 (50.0%) | 7 (31.8%) | 15 (68.2%) | ||
| Yes (n = 35, 100.0%) | 24 (68.6%) | 11 (31.4%) | 20 (57.1%) | 15 (42.9%) | ||
| AGS Cell Lines | |||||
|---|---|---|---|---|---|
| Variables (Mean ± SD) | PT (n = 3) | NT (n = 3) | TFAM-KD-C (n = 3) | pa | pb |
| mtDNA copy number | 1.000 ± 0.000 | 0.958 ± 0.168 | 0.610 ± 0.154 | 0.700 | 0.050 |
| Oxygen consumption rate (OCR, pmol/min/106 cells) | |||||
| Basal respiration (OCRBR) | 2618.8 ± 410.9 | 945.4 ± 67.5 | 0.050 | ||
| ATP-coupled (OCRATP) | 1257.1 ± 243.8 | 534.0 ± 139.3 | 0.050 | ||
| Reserve capacity (OCRRC) | 2205.7 ± 397.9 | 586.5 ± 309.8 | 0.050 | ||
| Proton leak (OCRPL) | 1361.8 ± 374.8 | 411.4 ± 134.5 | 0.050 | ||
| Extracellular acidification rate (ECAR, mpH/min/106 cells) | 1884.7 ± 80.2 | 1183.3 ± 153.5 | 0.050 | ||
| ECAR/OCRBR ratio | 0.730 ± 0.098 | 1.248 ± 0.071 | 0.050 | ||
| Wound width (μm) | |||||
| 0 h | 493.1 ± 8.8 | 491.4 ± 14.3 | 1.000 | ||
| 6 h | 459.0 ± 13.6 | 333.8 ± 19.7 | 0.050 | ||
| 12 h | 361.7 ± 3.1 | 251.9 ± 2.0 | 0.050 | ||
| 18 h | 330.0 ± 22.0 | 115.7 ± 11.1 | 0.050 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chang, T.-C.; Lee, H.-T.; Pan, S.-C.; Cho, S.-H.; Cheng, C.; Ou, L.-H.; Lin, C.-I.; Lin, C.-S.; Wei, Y.-H. Metabolic Reprogramming in Response to Alterations of Mitochondrial DNA and Mitochondrial Dysfunction in Gastric Adenocarcinoma. Int. J. Mol. Sci. 2022, 23, 1857. https://doi.org/10.3390/ijms23031857
Chang T-C, Lee H-T, Pan S-C, Cho S-H, Cheng C, Ou L-H, Lin C-I, Lin C-S, Wei Y-H. Metabolic Reprogramming in Response to Alterations of Mitochondrial DNA and Mitochondrial Dysfunction in Gastric Adenocarcinoma. International Journal of Molecular Sciences. 2022; 23(3):1857. https://doi.org/10.3390/ijms23031857
Chicago/Turabian StyleChang, Tzu-Ching, Hui-Ting Lee, Siao-Cian Pan, Shih-Han Cho, Chieh Cheng, Liang-Hung Ou, Chia-I Lin, Chen-Sung Lin, and Yau-Huei Wei. 2022. "Metabolic Reprogramming in Response to Alterations of Mitochondrial DNA and Mitochondrial Dysfunction in Gastric Adenocarcinoma" International Journal of Molecular Sciences 23, no. 3: 1857. https://doi.org/10.3390/ijms23031857
APA StyleChang, T. -C., Lee, H. -T., Pan, S. -C., Cho, S. -H., Cheng, C., Ou, L. -H., Lin, C. -I., Lin, C. -S., & Wei, Y. -H. (2022). Metabolic Reprogramming in Response to Alterations of Mitochondrial DNA and Mitochondrial Dysfunction in Gastric Adenocarcinoma. International Journal of Molecular Sciences, 23(3), 1857. https://doi.org/10.3390/ijms23031857