
The Multiple Functions of Fibrillin-1 Microfibrils in Organismal Physiology

{kind=link}
Abstract
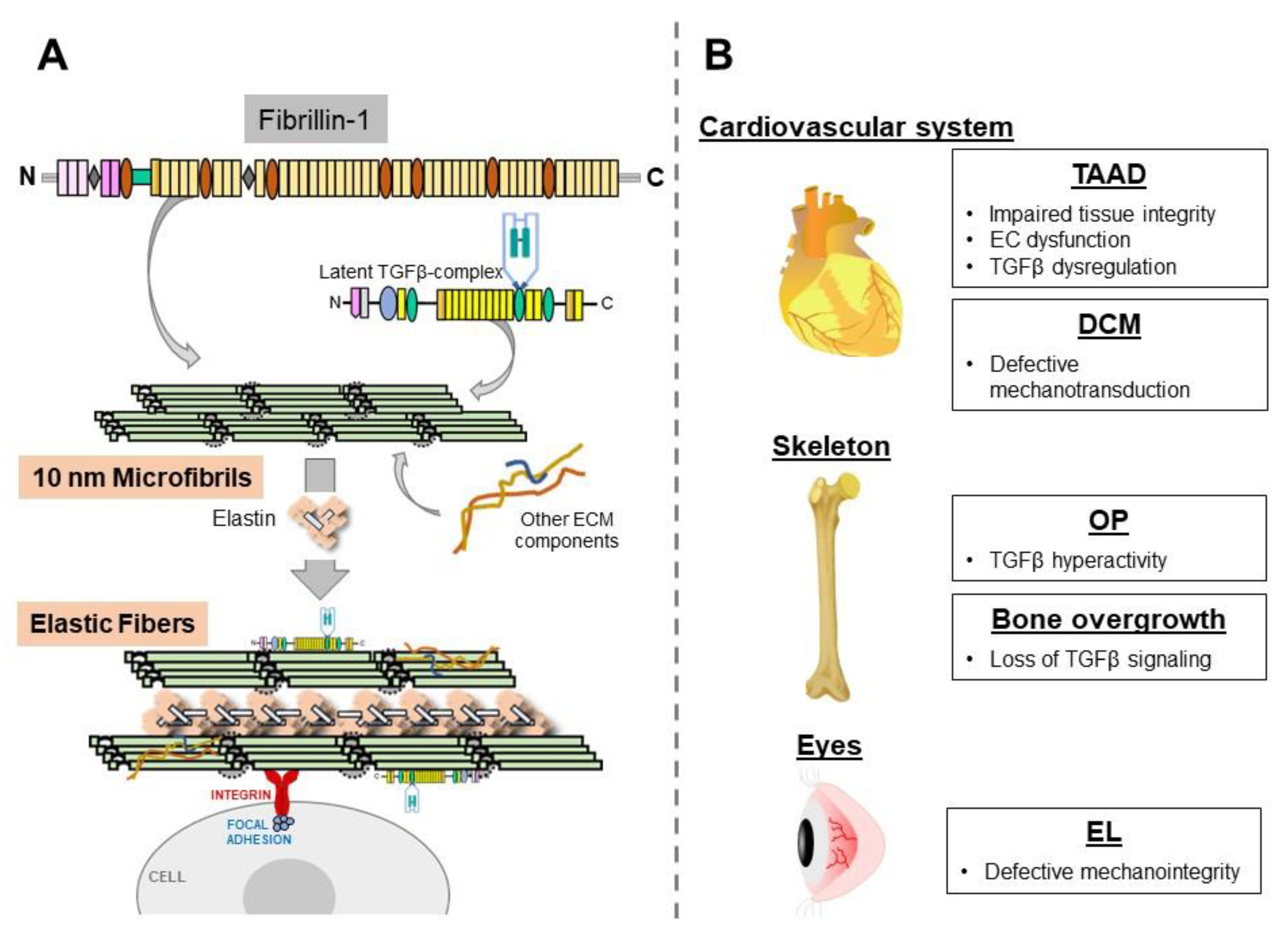
:1. Introduction
2. Thoracic Aortic Disease
3. Dilated Cardiomyopathy (DCM)
4. Osteopenia (OP)
5. Disproportionate Bone Lengthening
6. Ectopia Lentis (EL)
7. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ramirez, F.; Caescu, C.; Wondimu, E.; Galatioto, J. Marfan syndrome: A connective tissue disease at the crossroads of mechanotransduction. TGFβ signaling and cell stemness. Matrix Biol. 2018, 71–72, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, F.; Sakai, L.Y. Biogenesis and function of fibrillin assemblies. Cell Tissue Res. 2010, 339, 71–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robertson, I.B.; Rifkin, D.B. Regulation of the bioavailability of TGF-β and TGF-β-related proteins. Cold Spring Harb. Perspect. Biol. 2016, 8, a021907. [Google Scholar] [CrossRef] [PubMed]
- Hollister, D.W.; Godfrey, M.; Sakai, L.Y.; Pyeritz, R.E. Immunohistologic abnormalities of the microfibrillar-fiber system in the Marfan syndrome. N. Engl. J. Med. 1990, 323, 152–159. [Google Scholar] [CrossRef]
- Baldwin, A.K.; Simpson, A.; Steer, R.; Cain, S.A.; Kielty, C.M. Elastic fibres in health and disease. Expert Rev. Mol. Med. 2013, 15, e8. [Google Scholar] [CrossRef] [Green Version]
- Jensen, S.A.; Handford, P.A. New insights into the structure, assembly and biological roles of 10-12nm connective tissue microfibrils from fibrillin-1 studies. Biochem. J. 2016, 473, 827–838. [Google Scholar] [CrossRef]
- Godwin, A.R.F.; Singh, M.; Lockhart-Cairns, M.P.; Alanazi, Y.F.; Cain, S.A.; Baldock, C. The role of fibrillin and microfibril binding proteins in elastin and elastic fibre assembly. Matrix Biol. 2019, 84, 17–30. [Google Scholar] [CrossRef]
- Shin, S.J.; Yanagisawa, H. Recent updates on the molecular network of elastic fiber formation. Essays Biochem. 2019, 63, 365–376. [Google Scholar]
- Wagenseil, J.E.; Mecham, R.P. Vascular extracellular matrix and arterial mechanics. Physiol. Rev. 2009, 89, 957–989. [Google Scholar] [CrossRef] [Green Version]
- Milewicz, D.M.; Ramirez, F. Therapies for thoracic aortic aneurysms and acute aortic dissections. Arter. Throm. Vasc. Biol. 2019, 39, 126–136. [Google Scholar] [CrossRef] [Green Version]
- Bunton, T.E.; Jensen Biery, N.; Gayraud, B.; Ramirez, F.; Dietz, H.C. Phenotypic modulation of vascular smooth muscle cells contributes to elastolysis in a mouse model of Marfan syndrome. Circul. Res. 2001, 88, 37–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carta, L.; Pereira, L.; Arteaga-Solis, E.; Lee-Arteaga, S.Y.; Lenart, B.; Starcher, B.; Merkel, C.A.; Sukoyan, M.; Kerkis, A.; Hazeki, N.; et al. Fibrillins 1 and 2 perform partially overlapping functions during aortic development. J. Biol. Chem. 2006, 281, 8016–8023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Habashi, J.P.; Judge, D.P.; Holm, T.M.; Cohn, R.D.; Loeys, B.L.; Cooper, T.K.; Myers, L.; Klein, E.C.; Liu, G.; Calvi, C.; et al. Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome. Science 2006, 312, 117–121. [Google Scholar] [CrossRef] [Green Version]
- Cook, J.R.; Clayton, N.P.; Carta, L.; Galatioto, J.; Chiu, E.; Smaldone, S.; Nelson, C.A.; Cheng, S.H.; Wentworth, B.M.; Ramirez, F. Dimorphic effects of TGFβ signaling during aortic aneurysm progression in mice suggest a combinatorial therapy for Marfan syndrome. Arter. Thromb. Vasc. Biol. 2015, 35, 911–917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Li, Q.; Jiao, Y.; Qin, L.; Ali, R.; Zhou, J.; Ferruzzi, J.; Kim, R.W.; Geirsson, A.; Dietz, H.C.; et al. Tgfbr2 disruption in postnatal smooth muscle impairs aortic wall homeostasis. J. Clin. Invest 2014, 124, 755–767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, J.H.; Wei, H.; Jaffe, J.; Airhart, N.; Du, L.; Angelov, S.N.; Yan, J.; Allen, J.K.; Kang, I.; Wight, T.N.; et al. Postnatal deletion of the type II transforming growth factor-β receptor in smooth muscle cells causes severe aortopathy in mice. Arter. Thromb. Vasc. Biol. 2015, 35, 2647–2656. [Google Scholar] [CrossRef] [Green Version]
- Toral, M.; de la Fuente-Alonso, A.; Campanero, M.R.; Redondo, J.M. The NO signaling pathway in aortic aneurysm and dissection. Br. J. Pharmacol 2021. Epub ahead of print. [Google Scholar] [CrossRef]
- Wilson, D.G.; Bellamy, M.F.; Ramsey, M.W.; Goodfellow, J.; Brownlee, M.; Davies, S.; Wilson, J.F.; Lewis, M.J.; Stuart, A.G. Endothelial function in Marfan syndrome: Selective impairment of flow-mediated vasodilation. Circulation 1999, 99, 909–915. [Google Scholar] [CrossRef] [Green Version]
- Chung, A.W.Y.; Yeung, K.A.; Cortes, S.F.; Sandor, G.G.S.; Judge, D.P.; Dietz, H.C.; van Breemen, C. Endothelial dysfunction and compromise eNOS/Akt signaling in the thoracic aorta during the progression of Marfan syndrome. Br. J. Pharmacol. 2007, 150, 1075–1083. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.H.C.; van Breemen, C.; Chung, A.W.Y. Vasomotor dysfunction in the thoracic aorta of Marfan syndrome is associated with accumulation of oxidative stress. Vasc. Pharmacol. 2010, 52, 37–45. [Google Scholar] [CrossRef]
- Jimenez-Altayo, F.; Meirelles, T.; Crosas-Molist, E.; Sorolla, M.A.; Gorbenko del Blanco, D.; Lopez-Luque, J.; Mas-Stachurska, A.; Siegert, A.-M.; Bonorino, F.; Barbera, L.; et al. Redox stress in Marfan syndrome: Dissecting the role of the NADPH oxidase NOX4 in aortic aneurysm. Free Radic. Biol. Med. 2018, 118, 44–58. [Google Scholar] [CrossRef]
- Huang, K.; Wang, Y.; Siu, K.L.; Zhang, Y.; Cai, H. Targeting feed-forward signaling of TGFβ/NOX4/DHFR/eNOS uncoupling/TGFβ axis with anti-TGFβ and folic acid attenuates formation of aortic aneurysms: Novel mechanisms and therapeutics. Redox Biol. 2021, 38, 101757. [Google Scholar] [CrossRef] [PubMed]
- Oller, J.; Mendez-Barbero, N.; Ruiz, E.J.; Villahoz, S.; Renard, M.; Canelas, L.I.; Briones, A.M.; Alberca, R.; Lozano-Vidal, N.; Hurle, M.A.; et al. Nitric oxide mediates aortic disease in mice deficient in the metalloprotease Admats1 and in a mouse model of Marfan syndrome. Nat. Med. 2017, 23, 200–212. [Google Scholar] [CrossRef]
- De la Fuente-Alonso, A.; Toral, M.; Alfayate, A.; Ruiz-Rodriguez, M.J.; Bonzon-Kulichenko, E.; Teixdo-Tura, G.; Martinez-Martinez, S.; Mendez-Olivares, M.J.; Lopez-Maderuelo, D.; Gonzalez-Validez, I.; et al. Aortic disease in Marfan syndrome is caused by overactivation of sGC-PRKG signaling by NO. Nat. Commun. 2021, 12, 2628. [Google Scholar] [CrossRef] [PubMed]
- De Backer, J.F.; Devos, D.; Segers, P.; Matthys, D.; Francois, K.; Gillebert, T.C.; De Paepe, A.M.; De Sutter, J. Primary impairment of left ventricular function in Marfan syndrome. Int. J. Cardiol. 2006, 112, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Alpendurada, F.; Wong, J.; Kiotsekoglou, A.; Banya, W.; Child, A.; Prasad, S.K.; Pennell, D.J.; Mohiaddin, R.H. Evidence for Marfan cardiomyopathy. Eur. J. Heart Fail. 2010, 12, 1085–1091. [Google Scholar] [CrossRef]
- De Witte, P.; Aalberts, J.J.J.; Radonic, T.; Timmermans, J.; Scholte, A.J.; Zwinderman, A.H.; Mulder, B.J.M.; Groenink, M.; van den Berg, M.P. Intrinsic biventricular dysfunction in Marfan syndrome. Heart 2011, 97, 2063–2068. [Google Scholar] [CrossRef] [Green Version]
- Cook, J.R.; Carta, L.; Benard, L.; Chemaly, E.R.; Chiu, E.; Rao, S.K.; Hampton, T.G.; Yurchenco, P.; Costa, K.D.; Hajjar, R.J.; et al. Abnormal muscle mechanosignaling triggers cardiomyopathy in mice with Marfan syndrome. J. Clin. Invest 2014, 124, 1329–1339. [Google Scholar] [CrossRef] [Green Version]
- Campens, L.; Renard, M.; Trachet, B.; Segers, P.; Mosquera, L.M.; De Sutter, J.; Sakai, L.; De Paepe, A.; De Backer, J. Intrinsic cardiomyopathy in Marfan syndrome: Results from in-vivo and ex-vivo studies of the Fbn1C1039G/+ model and longitudinal findings in humans. Pediatr Res. 2015, 78, 256–263. [Google Scholar] [CrossRef] [Green Version]
- Seidman, J.G.; Seidman, C. The genetic basis for cardiomyopathy: From mutation identification to mechanistic paradigms. Cell 2001, 104, 557–567. [Google Scholar] [CrossRef] [Green Version]
- Carter, N.; Duncan, E.; Wordsworth, P. Bone mineral density in adults with Marfan syndrome. Rheumatology 2000, 39, 307–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giampietro, P.F.; Peterson, M.; Schneider, R.; Davis, J.G.; Raggio, C.; Myers, E.; Burke, S.W.; Boachie-Adjei, O.; Mueller, C.M. Assessment of bone mineral density in adults and children with Marfan syndrome. Osteopors Int. 2003, 14, 559–563. [Google Scholar]
- Moura, B.; Tubach, F.; Sulpice, M.; Boileau, C.; Jondeau, G.; Muti, C.; Chevallier, B.; Ounnoughene, Y.; Le Parc, J.-M. Multidisciplinary Marfan Syndrome Clinic Group. Bone mineral density in Marfan syndrome. A large case-control study. Jt. Bone Spine. 2006, 73, 733–735. [Google Scholar] [CrossRef] [PubMed]
- Smaldone, S.; Clayton, N.; del Solar, M.; Pasqual-Gonzales, G.; Cheng, S.; Wentworth, B.; Ramirez, F. Fibrillin-1 regulates skeletal stem cell differentiation by modulating TGFβ activity within the marrow niche. J. Bone Miner. Res. 2016, 31, 86–97. [Google Scholar] [CrossRef] [Green Version]
- Nistala, H.; Lee-Arteaga, S.; Smaldone, S.; Siciliano, G.; Ramirez, F. Extracellular microfibrils modulate osteoblast-supported osteoclastogenesis by restricting TGFβ stimulation of RANKL production. J. Biol. Chem. 2010, 285, 34126–34133. [Google Scholar] [CrossRef] [Green Version]
- Tiedermann, K.; Boraschi-Diaz, I.; Rajakumar, I.; Kaur, J.; Roughley, P.; Reinhardt, D.P.; Komarova, S.V. Fibrillin-1 directly regulates osteoclast formation and function by a dual mechanism. J. Cell Sci. 2013, 126, 4187–4194. [Google Scholar] [CrossRef] [Green Version]
- Erkula, G.; Jones, K.V.; Sponseller, P.D.; Dietz, H.C.; Pyeritz, R.E. Growth and maturation in Marfan syndrome. Amer. J. Genet. 2002, 109, 100–115. [Google Scholar] [CrossRef]
- Long, F.; Linsenmayer, T.F. Regulation of growth region cartilage proliferation and differentiation by perichondrium. Development 1998, 125, 1067–1073. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, J.; Horton, J.; Sohn, P.; Serra, R. The perichondrium plays and important role in mediating the effects of TGF-β1 on endochrondral bone formation. Dev. Dyn. 2001, 221, 311–321. [Google Scholar] [CrossRef]
- McKusick, V.A. Heritable Disorders of the Connective Tissue, 4th ed.; Mosby, C.V., Ed.; Mosby: St. Louis, MO, USA, 1972; pp. 61–223. [Google Scholar]
- Loeys, B.L.; Mortier, G.; Dietz, H.C. Bone lessons from Marfan syndrome and related disorders: Fibrillin, TGF-β and BMP at the balance of too long and too short. Pediatr. Endocrinol. Rev. 2013, 10, 417–423. [Google Scholar]
- Keen, D.R.; Jordan, C.D.; Reinhardt, D.P.; Ridgway, C.C.; Ono, R.N.; Corson, G.M.; Fairhurst, M.L.; Sussman, M.D.; Memoli, V.A.; Sakai, L.Y. Fibrillin-1 in human cartilage: Developmental expression and formation of special banded fibers. J. Histochem. Cytochem. 1997, 45, 1069–1082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quarto, N.; Leonard, B.; Li, S.; Marchand, M.; Anderson, E.; Behr, B.; Francke, U.; Reijo-Pera, R.; Chiao, E.; Longaker, M.T. Skeletogenic phenotype of human Marfan embryonic stem cells faithfully peoncopied by patient-specific induced-pluripotent stem cells. Proc. Natl. Acad. Sci. USA 2012, 109, 215–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bassnett, S. Zinns zonule. Prog. Retin. Eye Res. 2021, 82, 100902. [Google Scholar] [CrossRef] [PubMed]
- Jones, W.; Rodriguez, J.; Bassnett, S. Targeted deletion of fibrillin-1 in the mouse eye results in ectopia lentis and other ocular phenotypes associated with Marfan syndrome. Dis. Model Mech. 2019, 12, dmm037283. [Google Scholar] [CrossRef] [Green Version]
- Mariko, B.; Pezet, M.; Escoubet, B.; Bouillot, S.; Andrieu, J.P.; Starcher, B.; Quaglino, E.D.; Jacob, M.P.; Huber, P.; Ramirez, F. Fibrillin-1 genetic deficiency leads to pathological aging of arteries in mice. J. Pathol. 2011, 224, 33–44. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Asano, K.; Cantalupo, A.; Sedes, L.; Ramirez, F. The Multiple Functions of Fibrillin-1 Microfibrils in Organismal Physiology. Int. J. Mol. Sci. 2022, 23, 1892. https://doi.org/10.3390/ijms23031892
Asano K, Cantalupo A, Sedes L, Ramirez F. The Multiple Functions of Fibrillin-1 Microfibrils in Organismal Physiology. International Journal of Molecular Sciences. 2022; 23(3):1892. https://doi.org/10.3390/ijms23031892
Chicago/Turabian StyleAsano, Keiichi, Anna Cantalupo, Lauriane Sedes, and Francesco Ramirez. 2022. "The Multiple Functions of Fibrillin-1 Microfibrils in Organismal Physiology" International Journal of Molecular Sciences 23, no. 3: 1892. https://doi.org/10.3390/ijms23031892
APA StyleAsano, K., Cantalupo, A., Sedes, L., & Ramirez, F. (2022). The Multiple Functions of Fibrillin-1 Microfibrils in Organismal Physiology. International Journal of Molecular Sciences, 23(3), 1892. https://doi.org/10.3390/ijms23031892