
Takotsubo Syndrome: Translational Implications and Pathomechanisms

 , and
, and
Abstract
:1. Introduction
2. Clinical Characteristics and Epidemiology
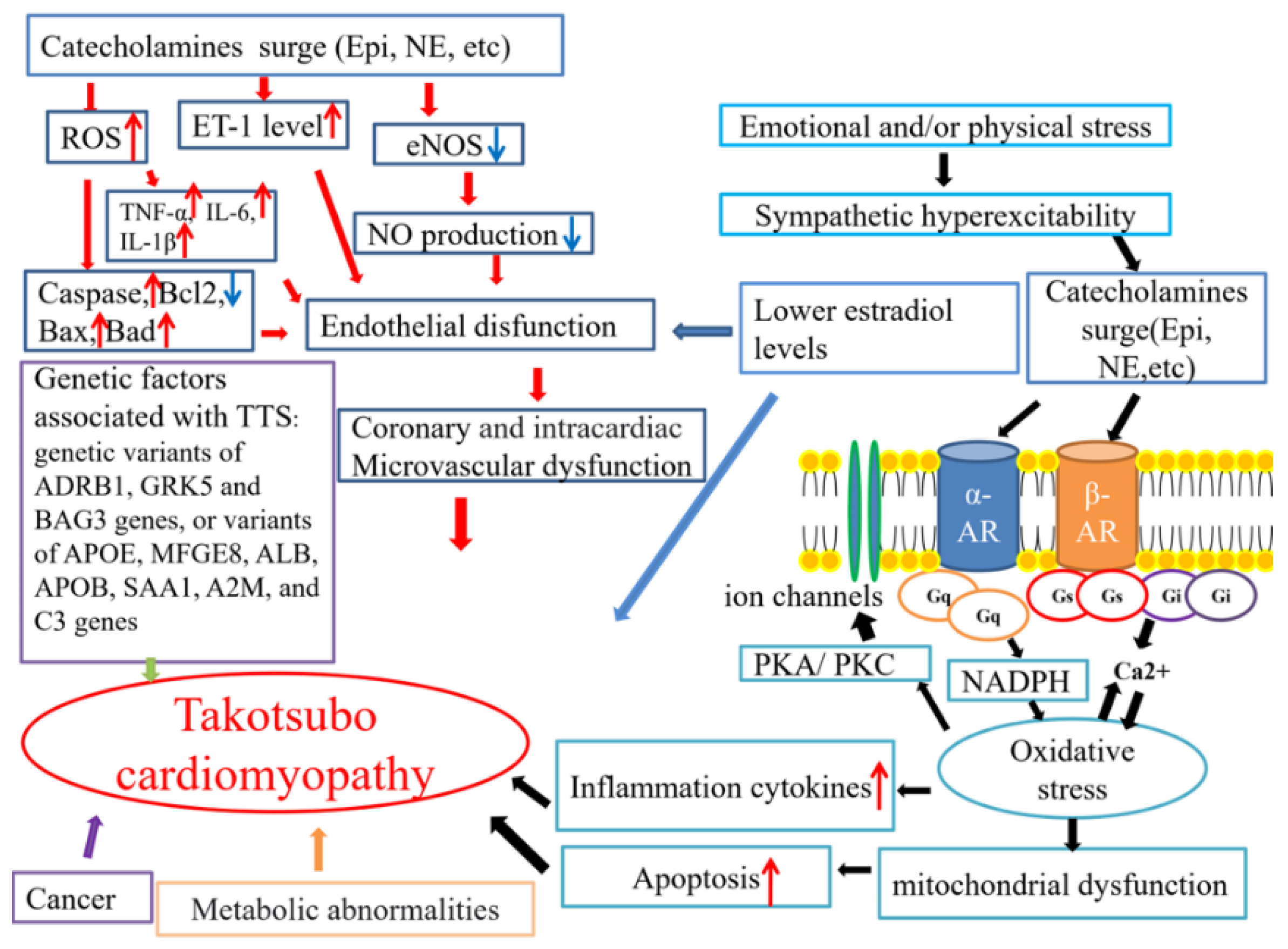
3. Pathophysiology
3.1. A Surge in Catecholamines Caused by Sympathetic Excess
3.1.1. Central Nervous System and Peripheral Nervous System
3.1.2. Emotional Stress and Physical Injury
3.1.3. The Role of Chromogranin-A
3.1.4. The Role of Alpha- or Beta-Adrenoceptors
3.1.5. The Role of Beta-Blockers
3.2. Coronary Artery Spasm
3.3. Coronary Microvascular Dysfunction
3.4. Endothelial Dysfunction
3.5. The Low Estradiol Level
3.6. Genetic Factors
3.7. Other Possible Contributors
3.7.1. Inflammation in TTS
3.7.2. Abnormal Myocardial Metabolism in TTS
3.7.3. Cancer
4. Experimental Models of TTS
4.1. Animal Models and Mechanistic Studies
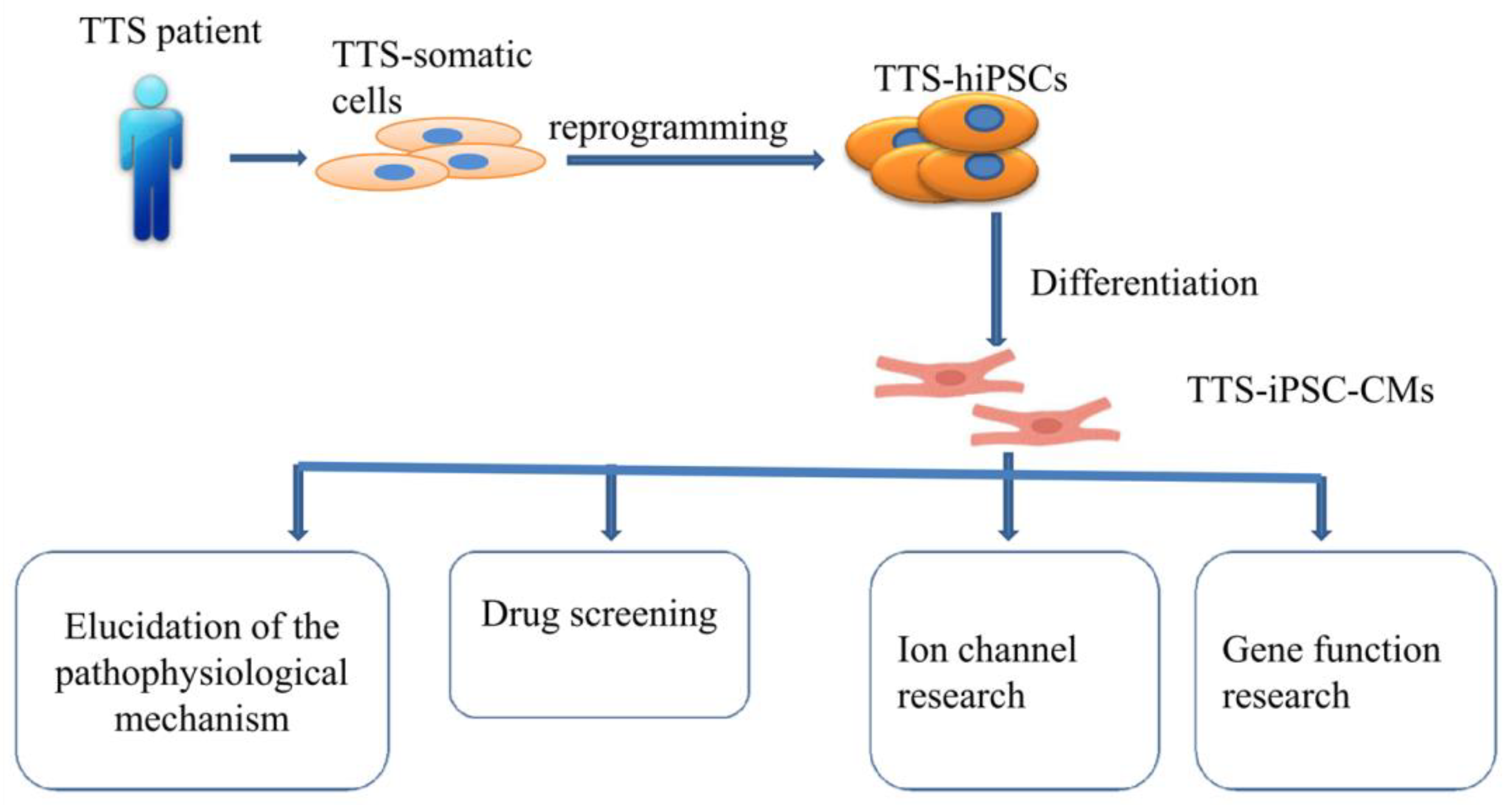
4.2. Human Cardiomyocytes Derived from Induced Pluripotent Stem Cells (hiPSC-CMs) Models
4.3. Other Models
4.4. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cramer, M.J.; De Boeck, B.; Melman, P.G.; Sieswerda, G.J. The ‘broken heart’ syndrome: What can be learned from the tears and distress? Neth. Heart J. 2007, 15, 283–285. [Google Scholar] [CrossRef] [PubMed]
- Sharkey, S.W.; Windenburg, D.C.; Lesser, J.R.; Maron, M.S.; Hauser, R.G.; Lesser, J.N.; Haas, T.S.; Hodges, J.S.; Maron, B.J. Natural history and expansive clinical profile of stress (tako-tsubo) cardiomyopathy. J. Am. Coll. Cardiol. 2010, 55, 333–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Topal, Y.; Topal, H.; Dogan, C.; Tiryaki, S.B.; Biteker, M. Takotsubo (stress) cardiomyopathy in childhood. Eur. J. Pediatr. 2020, 179, 619–625. [Google Scholar] [CrossRef] [PubMed]
- Dote, K.; Sato, H.; Tateishi, H.; Uchida, T.; Ishihara, M. Myocardial stunning due to simultaneous multivessel coronary spasms: A review of 5 cases. J. Cardiol. 1991, 21, 203–214. [Google Scholar]
- Lyon, A.R.; Bossone, E.; Schneider, B.; Sechtem, U.; Citro, R.; Underwood, S.R.; Sheppard, M.N.; Figtree, G.A.; Parodi, G.; Akashi, Y.J.; et al. Current state of knowledge on Takotsubo syndrome: A Position Statement from the Taskforce on Takotsubo Syndrome of the Heart Failure Association of the European Society of Cardiology. Eur. J. Heart Fail. 2016, 18, 8–27. [Google Scholar] [CrossRef] [Green Version]
- Prasad, A.; Lerman, A.; Rihal, C.S. Apical ballooning syndrome (Tako-Tsubo or stress cardiomyopathy): A mimic of acute myocardial infarction. Am. Heart J. 2008, 155, 408–417. [Google Scholar] [CrossRef]
- Ancona, F.; Bertoldi, L.F.; Ruggieri, F.; Cerri, M.; Magnoni, M.; Beretta, L.; Cianflone, D.; Camici, P.G. Takotsubo cardiomyopathy and neurogenic stunned myocardium: Similar albeit different. Eur. Heart J. 2016, 37, 2830–2832. [Google Scholar] [CrossRef] [Green Version]
- Madias, J.E. “Neurogenic stress cardiomyopathy in heart donors” is a form of Takotsubo syndrome. Int. J. Cardiol. 2015, 184, 612–613. [Google Scholar] [CrossRef]
- Mohamedali, B.; Bhat, G.; Zelinger, A. Frequency and pattern of left ventricular dysfunction in potential heart donors: Implications regarding use of dysfunctional hearts for successful transplantation. J. Am. Coll. Cardiol. 2012, 60, 235–236. [Google Scholar] [CrossRef] [Green Version]
- Moussouttas, M.; Mearns, E.; Walters, A.; DeCaro, M. Plasma Catecholamine Profile of Subarachnoid Hemorrhage Patients with Neurogenic Cardiomyopathy. Cerebrovasc. Dis. Extra 2015, 5, 57–67. [Google Scholar] [CrossRef]
- Tavazzi, G.; Zanierato, M.; Via, G.; Iotti, G.A.; Procaccio, F. Are Neurogenic Stress Cardiomyopathy and Takotsubo Different Syndromes With Common Pathways?: Etiopathological Insights on Dysfunctional Hearts. JACC Heart Fail. 2017, 5, 940–942. [Google Scholar] [CrossRef] [PubMed]
- Aweimer, A.; El-Battrawy, I.; Akin, I.; Borggrefe, M.; Mugge, A.; Patsalis, P.C.; Urban, A.; Kummer, M.; Vasileva, S.; Stachon, A.; et al. Abnormal thyroid function is common in takotsubo syndrome and depends on two distinct mechanisms: Results of a multicentre observational study. J. Intern. Med. 2021, 289, 675–687. [Google Scholar] [CrossRef] [PubMed]
- Naegele, M.; Flammer, A.J.; Enseleit, F.; Roas, S.; Frank, M.; Hirt, A.; Kaiser, P.; Cantatore, S.; Templin, C.; Frohlich, G.; et al. Endothelial function and sympathetic nervous system activity in patients with Takotsubo syndrome. Int. J. Cardiol. 2016, 224, 226–230. [Google Scholar] [CrossRef]
- El-Battrawy, I.; Zhao, Z.; Lan, H.; Schunemann, J.D.; Sattler, K.; Buljubasic, F.; Patocskai, B.; Li, X.; Yucel, G.; Lang, S.; et al. Estradiol protection against toxic effects of catecholamine on electrical properties in human-induced pluripotent stem cell derived cardiomyocytes. Int. J. Cardiol. 2018, 254, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Scally, C.; Abbas, H.; Ahearn, T.; Srinivasan, J.; Mezincescu, A.; Rudd, A.; Spath, N.; Yucel-Finn, A.; Yuecel, R.; Oldroyd, K.; et al. Myocardial and Systemic Inflammation in Acute Stress-Induced (Takotsubo) Cardiomyopathy. Circulation 2019, 139, 1581–1592. [Google Scholar] [CrossRef]
- Paur, H.; Wright, P.T.; Sikkel, M.B.; Tranter, M.H.; Mansfield, C.; O’Gara, P.; Stuckey, D.J.; Nikolaev, V.O.; Diakonov, I.; Pannell, L.; et al. High levels of circulating epinephrine trigger apical cardiodepression in a beta2-adrenergic receptor/Gi-dependent manner: A new model of Takotsubo cardiomyopathy. Circulation 2012, 126, 697–706. [Google Scholar] [CrossRef] [Green Version]
- Orphanou, N.; Eftychiou, C.; Papasavvas, E.; Ioannides, M.; Avraamides, P. Syncope in a hypertrophic heart at a wedding party: Can happiness break a thick heart? Takotsubo cardiomyopathy complicated with left ventricular outflow tract obstruction in a hypertrophic heart. Oxf Med. Case Rep. 2020, 2020, omaa036. [Google Scholar] [CrossRef]
- Bonacchi, M.; Vannini, A.; Harmelin, G.; Batacchi, S.; Bugetti, M.; Sani, G.; Peris, A. Inverted-Takotsubo cardiomyopathy: Severe refractory heart failure in poly-trauma patients saved by emergency extracorporeal life support. Interact. Cardiovasc. Thorac. Surg. 2015, 20, 365–371. [Google Scholar] [CrossRef] [Green Version]
- Kow, K.; Watson, T.J.; Foo, D.; Ho, H.H. Clinical characteristics and outcomes of South-East Asian patients with Takotsubo (stress-induced) cardiomyopathy. Int. J. Cardiol. Heart Vasc. 2018, 21, 29–31. [Google Scholar] [CrossRef]
- Templin, C.; Ghadri, J.R.; Diekmann, J.; Napp, L.C.; Bataiosu, D.R.; Jaguszewski, M.; Cammann, V.L.; Sarcon, A.; Geyer, V.; Neumann, C.A.; et al. Clinical Features and Outcomes of Takotsubo (Stress) Cardiomyopathy. N. Engl. J. Med. 2015, 373, 929–938. [Google Scholar] [CrossRef] [Green Version]
- Doyen, D.; Moschietto, S.; Squara, F.; Moceri, P.; Hyvernat, H.; Ferrari, E.; Dellamonica, J.; Bernardin, G. Incidence, clinical features and outcome of Takotsubo syndrome in the intensive care unit. Arch. Cardiovasc. Dis. 2020, 113, 176–188. [Google Scholar] [CrossRef] [PubMed]
- Minhas, A.S.; Hughey, A.B.; Kolias, T.J. Nationwide Trends in Reported Incidence of Takotsubo Cardiomyopathy from 2006 to 2012. Am. J. Cardiol. 2015, 116, 1128–1131. [Google Scholar] [CrossRef] [PubMed]
- Ghadri, J.R.; Wittstein, I.S.; Prasad, A.; Sharkey, S.; Dote, K.; Akashi, Y.J.; Cammann, V.L.; Crea, F.; Galiuto, L.; Desmet, W.; et al. International Expert Consensus Document on Takotsubo Syndrome (Part I): Clinical Characteristics, Diagnostic Criteria, and Pathophysiology. Eur. Heart J. 2018, 39, 2032–2046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Budnik, M.; Nowak, R.; Fijalkowski, M.; Kochanowski, J.; Nargiello, E.; Piatkowski, R.; Peller, M.; Kucharz, J.; Jaguszewski, M.; Gruchala, M.; et al. Sex-dependent differences in clinical characteristics and in-hospital outcomes in patients with takotsubo syndrome. Pol. Arch. Intern. Med. 2020, 130, 25–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lach, R.; Schön, J.; Krolopp, T.; Arndt, S.; Langer, B.; Grellmann, W. Depth-Sensing Macroindentation Test and Stepped Isothermal Method–Accelerated Assessment of the Local Retardation Behaviour of Thermoplastic Polymers. Macromol. Symp. 2016, 366, 60–65. [Google Scholar] [CrossRef]
- Park, J.H.; Kang, S.J.; Song, J.K.; Kim, H.K.; Lim, C.M.; Kang, D.H.; Koh, Y. Left ventricular apical ballooning due to severe physical stress in patients admitted to the medical ICU. Chest 2005, 128, 296–302. [Google Scholar] [CrossRef]
- Rozema, T.; Klein, L.R. Takotsubo cardiomyopathy: A case report and literature review. Cardiol. Young 2016, 26, 406–409. [Google Scholar] [CrossRef]
- Chinali, M.; Formigari, R.; Grutter, G. Takotsubo cardiomyopathy in a young adult with transplanted heart: What happened to denervation? ESC Heart Fail. 2018, 5, 197–200. [Google Scholar] [CrossRef]
- Cammann, V.L.; Szawan, K.A.; Stahli, B.E.; Kato, K.; Budnik, M.; Wischnewsky, M.; Dreiding, S.; Levinson, R.A.; Di Vece, D.; Gili, S.; et al. Age-Related Variations in Takotsubo Syndrome. J. Am. Coll. Cardiol. 2020, 75, 1869–1877. [Google Scholar] [CrossRef]
- Deshmukh, A.; Kumar, G.; Pant, S.; Rihal, C.; Murugiah, K.; Mehta, J.L. Prevalence of Takotsubo cardiomyopathy in the United States. Am. Heart J. 2012, 164, 66–71.e61. [Google Scholar] [CrossRef]
- Desai, A.; Noor, A.; Joshi, S.; Kim, A.S. Takotsubo cardiomyopathy in cancer patients. Cardiooncology 2019, 5, 7. [Google Scholar] [CrossRef] [PubMed]
- Giza, D.E.; Lopez-Mattei, J.; Vejpongsa, P.; Munoz, E.; Iliescu, G.; Kitkungvan, D.; Hassan, S.A.; Kim, P.; Ewer, M.S.; Iliescu, C. Stress-Induced Cardiomyopathy in Cancer Patients. Am. J. Cardiol. 2017, 120, 2284–2288. [Google Scholar] [CrossRef] [PubMed]
- Cammann, V.L.; Sarcon, A.; Ding, K.J.; Seifert, B.; Kato, K.; Di Vece, D.; Szawan, K.A.; Gili, S.; Jurisic, S.; Bacchi, B.; et al. Clinical Features and Outcomes of Patients With Malignancy and Takotsubo Syndrome: Observations From the International Takotsubo Registry. J. Am. Heart Assoc. 2019, 8, e010881. [Google Scholar] [CrossRef] [Green Version]
- Joy, P.S.; Guddati, A.K.; Shapira, I. Outcomes of Takotsubo cardiomyopathy in hospitalized cancer patients. J. Cancer Res. Clin. Oncol. 2018, 144, 1539–1545. [Google Scholar] [CrossRef] [PubMed]
- Sattler, K.; El-Battrawy, I.; Lang, S.; Zhou, X.; Schramm, K.; Tulumen, E.; Kronbach, F.; Roger, S.; Behnes, M.; Kuschyk, J.; et al. Prevalence of cancer in Takotsubo cardiomyopathy: Short and long-term outcome. Int. J. Cardiol. 2017, 238, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Moller, C.; Stiermaier, T.; Graf, T.; Eitel, C.; Thiele, H.; Burgdorf, C.; Eitel, I. Prevalence and long-term prognostic impact of malignancy in patients with Takotsubo syndrome. Eur. J. Heart Fail. 2018, 20, 816–818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Battrawy, I.; Santoro, F.; Stiermaier, T.; Moller, C.; Guastafierro, F.; Novo, G.; Novo, S.; Santangelo, A.; Mariano, E.; Romeo, F.; et al. Prevalence, management, and outcome of adverse rhythm disorders in takotsubo syndrome: Insights from the international multicenter GEIST registry. Heart Fail. Rev. 2020, 25, 505–511. [Google Scholar] [CrossRef]
- Di Vece, D.; Silverio, A.; Bellino, M.; Galasso, G.; Vecchione, C.; La Canna, G.; Citro, R. Dynamic Left Intraventricular Obstruction Phenotype in Takotsubo Syndrome. J. Clin. Med. 2021, 10, 3235. [Google Scholar] [CrossRef]
- Conradi, P.M.; van Loon, R.B.; Handoko, M.L. Dynamic left ventricular outflow tract obstruction in Takotsubo cardiomyopathy resulting in cardiogenic shock. BMJ Case Rep. 2021, 14, e240010. [Google Scholar] [CrossRef]
- Ortuno, S.; Jozwiak, M.; Mira, J.P.; Nguyen, L.S. Case Report: Takotsubo Syndrome Associated With Novel Coronavirus Disease 2019. Front. Cardiovasc. Med. 2021, 8, 614562. [Google Scholar] [CrossRef]
- Cau, R.; Bassareo, P.; Deidda, M.; Caredda, G.; Suri, J.S.; Pontone, G.; Saba, L. Could CMR Tissue-Tracking and Parametric Mapping Distinguish Between Takotsubo Syndrome and Acute Myocarditis? A Pilot Study. Acad. Radiol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Eitel, I.; von Knobelsdorff-Brenkenhoff, F.; Bernhardt, P.; Carbone, I.; Muellerleile, K.; Aldrovandi, A.; Francone, M.; Desch, S.; Gutberlet, M.; Strohm, O.; et al. Clinical characteristics and cardiovascular magnetic resonance findings in stress (takotsubo) cardiomyopathy. JAMA 2011, 306, 277–286. [Google Scholar] [CrossRef] [PubMed]
- Taghavi, S.; Chenaghlou, M.; Mirtajaddini, M.; Naderi, N.; Amin, A. Takotsubo syndrome without major stress mimicking myocarditis. Anatol. J. Cardiol. 2020, 23, 349–350. [Google Scholar] [CrossRef] [PubMed]
- Scantlebury, D.C.; Prasad, A. Diagnosis of Takotsubo cardiomyopathy. Circ. J. 2014, 78, 2129–2139. [Google Scholar] [CrossRef] [Green Version]
- Redfors, B.; Jha, S.; Thorleifsson, S.; Jernberg, T.; Angeras, O.; Frobert, O.; Petursson, P.; Tornvall, P.; Sarno, G.; Ekenback, C.; et al. Short- and Long-Term Clinical Outcomes for Patients With Takotsubo Syndrome and Patients With Myocardial Infarction: A Report From the Swedish Coronary Angiography and Angioplasty Registry. J. Am. Heart Assoc. 2021, 10, e017290. [Google Scholar] [CrossRef]
- Van Vliet, P.D.; Burchell, H.B.; Titus, J.L. Focal myocarditis associated with pheochromocytoma. N. Engl. J. Med. 1966, 274, 1102–1108. [Google Scholar] [CrossRef]
- Szakacs, J.E.; Cannon, A. L-Norepinephrine myocarditis. Am. J. Clin. Pathol. 1958, 30, 425–434. [Google Scholar] [CrossRef]
- Samuels, M.A. The brain-heart connection. Circulation 2007, 116, 77–84. [Google Scholar] [CrossRef]
- Ghadri, J.R.; Sarcon, A.; Diekmann, J.; Bataiosu, D.R.; Cammann, V.L.; Jurisic, S.; Napp, L.C.; Jaguszewski, M.; Scherff, F.; Brugger, P.; et al. Happy heart syndrome: Role of positive emotional stress in takotsubo syndrome. Eur. Heart J. 2016, 37, 2823–2829. [Google Scholar] [CrossRef] [Green Version]
- Hiestand, T.; Hanggi, J.; Klein, C.; Topka, M.S.; Jaguszewski, M.; Ghadri, J.R.; Luscher, T.F.; Jancke, L.; Templin, C. Takotsubo Syndrome Associated With Structural Brain Alterations of the Limbic System. J. Am. Coll. Cardiol. 2018, 71, 809–811. [Google Scholar] [CrossRef]
- Scheitz, J.F.; Ghadri, J.R.; Templin, C. Brain-heart interaction revisited: Takotsubo syndrome secondary to seizures. Int. J. Cardiol. 2020, 299, 71–72. [Google Scholar] [CrossRef] [PubMed]
- Templin, C.; Hanggi, J.; Klein, C.; Topka, M.S.; Hiestand, T.; Levinson, R.A.; Jurisic, S.; Luscher, T.F.; Ghadri, J.R.; Jancke, L. Altered limbic and autonomic processing supports brain-heart axis in Takotsubo syndrome. Eur. Heart J. 2019, 40, 1183–1187. [Google Scholar] [CrossRef] [PubMed]
- Tawakol, A.; Ishai, A.; Takx, R.A.; Figueroa, A.L.; Ali, A.; Kaiser, Y.; Truong, Q.A.; Solomon, C.J.; Calcagno, C.; Mani, V.; et al. Relation between resting amygdalar activity and cardiovascular events: A longitudinal and cohort study. Lancet 2017, 389, 834–845. [Google Scholar] [CrossRef] [Green Version]
- De Bosscher, K.; Van Craenenbroeck, K.; Meijer, O.C.; Haegeman, G. Selective transrepression versus transactivation mechanisms by glucocorticoid receptor modulators in stress and immune systems. Eur. J. Pharmacol. 2008, 583, 290–302. [Google Scholar] [CrossRef] [PubMed]
- Nikkheslat, N.; Zunszain, P.A.; Horowitz, M.A.; Barbosa, I.G.; Parker, J.A.; Myint, A.M.; Schwarz, M.J.; Tylee, A.T.; Carvalho, L.A.; Pariante, C.M. Insufficient glucocorticoid signaling and elevated inflammation in coronary heart disease patients with comorbid depression. Brain Behav. Immun. 2015, 48, 8–18. [Google Scholar] [CrossRef] [Green Version]
- Harrison, N.A.; Cooper, E.; Voon, V.; Miles, K.; Critchley, H.D. Central autonomic network mediates cardiovascular responses to acute inflammation: Relevance to increased cardiovascular risk in depression? Brain Behav Immun 2013, 31, 189–196. [Google Scholar] [CrossRef] [Green Version]
- Miller, A.H.; Pariante, C.M.; Pearce, B.D. Effects of cytokines on glucocorticoid receptor expression and function. Glucocorticoid resistance and relevance to depression. Adv. Exp. Med. Biol. 1999, 461, 107–116. [Google Scholar] [CrossRef]
- D’Aloia, A.; Caretta, G.; Vizzardi, E.; Zanini, G.; Bugatti, S.; Bonadei, I.; Dei Cas, L. Heart failure syndrome due to dobutamine stress echocardiography: Tako-Tsubo induced-cardiomiopathy. Panminerva Med. 2012, 54, 53–55. [Google Scholar]
- Abraham, J.; Mudd, J.O.; Kapur, N.K.; Klein, K.; Champion, H.C.; Wittstein, I.S. Stress cardiomyopathy after intravenous administration of catecholamines and beta-receptor agonists. J. Am. Coll. Cardiol. 2009, 53, 1320–1325. [Google Scholar] [CrossRef] [Green Version]
- Kido, K.; Guglin, M. Drug-Induced Takotsubo Cardiomyopathy. J. Cardiovasc. Pharmacol. Ther. 2017, 22, 552–563. [Google Scholar] [CrossRef]
- Wittstein, I.S.; Thiemann, D.R.; Lima, J.A.; Baughman, K.L.; Schulman, S.P.; Gerstenblith, G.; Wu, K.C.; Rade, J.J.; Bivalacqua, T.J.; Champion, H.C. Neurohumoral features of myocardial stunning due to sudden emotional stress. N. Engl. J. Med. 2005, 352, 539–548. [Google Scholar] [CrossRef] [PubMed]
- Mori, H.; Ishikawa, S.; Kojima, S.; Hayashi, J.; Watanabe, Y.; Hoffman, J.I.; Okino, H. Increased responsiveness of left ventricular apical myocardium to adrenergic stimuli. Cardiovasc. Res. 1993, 27, 192–198. [Google Scholar] [CrossRef] [PubMed]
- Redfors, B.; Ali, A.; Shao, Y.; Lundgren, J.; Gan, L.M.; Omerovic, E. Different catecholamines induce different patterns of takotsubo-like cardiac dysfunction in an apparently afterload dependent manner. Int. J. Cardiol. 2014, 174, 330–336. [Google Scholar] [CrossRef] [PubMed]
- Moftaquir-Handaj, A.; Barbe, F.; Barbarino-Monnier, P.; Aunis, D.; Boutroy, M.J. Circulating chromogranin A and catecholamines in human fetuses at uneventful birth. Pediatr. Res. 1995, 37, 101–105. [Google Scholar] [CrossRef] [Green Version]
- Tarantino, N.; Santoro, F.; Di Biase, L.; Di Terlizzi, V.; Vitale, E.; Barone, R.; Della Rocca, D.G.; De Leon De La Cruz, N.S.; Di Biase, M.; Brunetti, N.D. Chromogranin-A serum levels in patients with takotsubo syndrome and ST elevation acute myocardial infarction. Int. J. Cardiol. 2020, 320, 12–17. [Google Scholar] [CrossRef]
- D’Amico M, A.; Ghinassi, B.; Izzicupo, P.; Manzoli, L.; Di Baldassarre, A. Biological function and clinical relevance of chromogranin A and derived peptides. Endocr. Connect. 2014, 3, R45–R54. [Google Scholar] [CrossRef] [Green Version]
- Pieroni, M.; Corti, A.; Tota, B.; Curnis, F.; Angelone, T.; Colombo, B.; Cerra, M.C.; Bellocci, F.; Crea, F.; Maseri, A. Myocardial production of chromogranin A in human heart: A new regulatory peptide of cardiac function. Eur. Heart J. 2007, 28, 1117–1127. [Google Scholar] [CrossRef] [Green Version]
- Angelone, T.; Mazza, R.; Cerra, M.C. Chromogranin-A: A multifaceted cardiovascular role in health and disease. Curr. Med. Chem. 2012, 19, 4042–4050. [Google Scholar] [CrossRef]
- Kume, T.; Kawamoto, T.; Okura, H.; Toyota, E.; Neishi, Y.; Watanabe, N.; Hayashida, A.; Okahashi, N.; Yoshimura, Y.; Saito, K.; et al. Local release of catecholamines from the hearts of patients with tako-tsubo-like left ventricular dysfunction. Circ. J. 2008, 72, 106–108. [Google Scholar] [CrossRef] [Green Version]
- Boland, T.A.; Lee, V.H.; Bleck, T.P. Stress-induced cardiomyopathy. Crit. Care Med. 2015, 43, 686–693. [Google Scholar] [CrossRef]
- Enache, I.; Radu, R.A.; Terecoasa, E.O.; Dorobat, B.; Tiu, C. Stress cardiomyopathy misinterpreted as ST-segment elevation myocardial infarction in a patient with aneurysmal subarachnoid hemorrhage: A case report. Rom. J. Intern. Med. 2020, 58, 173–177. [Google Scholar] [CrossRef] [PubMed]
- Heubach, J.F.; Ravens, U.; Kaumann, A.J. Epinephrine activates both Gs and Gi pathways, but norepinephrine activates only the Gs pathway through human beta2-adrenoceptors overexpressed in mouse heart. Mol. Pharmacol. 2004, 65, 1313–1322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Lisa, F.; Kaludercic, N.; Paolocci, N. beta(2)-Adrenoceptors, NADPH oxidase, ROS and p38 MAPK: Another ‘radical’ road to heart failure? Br. J. Pharmacol. 2011, 162, 1009–1011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harding, S.E.; Gong, H. Beta-adrenoceptor blockers as agonists: Coupling of beta2-adrenoceptors to multiple G-proteins in the failing human heart. Congest. Heart Fail. 2004, 10, 181–185; quiz 186–187. [Google Scholar] [CrossRef] [PubMed]
- Evans, B.A.; Sato, M.; Sarwar, M.; Hutchinson, D.S.; Summers, R.J. Ligand-directed signalling at beta-adrenoceptors. Br. J. Pharmacol. 2010, 159, 1022–1038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chilmonczyk, Z.; Bojarski, A.J.; Sylte, I. Ligand-directed trafficking of receptor stimulus. Pharmacol. Rep. 2014, 66, 1011–1021. [Google Scholar] [CrossRef]
- Kenakin, T.; Christopoulos, A. Signalling bias in new drug discovery: Detection, quantification and therapeutic impact. Nat. Rev. Drug Discov. 2013, 12, 205–216. [Google Scholar] [CrossRef] [Green Version]
- Sattler, S.; Couch, L.S.; Harding, S.E. Takotsubo Syndrome: Latest Addition to the Expanding Family of Immune-Mediated Diseases? JACC Basic Transl. Sci. 2018, 3, 779–781. [Google Scholar] [CrossRef]
- Chesley, A.; Lundberg, M.S.; Asai, T.; Xiao, R.P.; Ohtani, S.; Lakatta, E.G.; Crow, M.T. The beta(2)-adrenergic receptor delivers an antiapoptotic signal to cardiac myocytes through G(i)-dependent coupling to phosphatidylinositol 3′-kinase. Circ. Res. 2000, 87, 1172–1179. [Google Scholar] [CrossRef] [Green Version]
- Gong, H.; Adamson, D.L.; Ranu, H.K.; Koch, W.J.; Heubach, J.F.; Ravens, U.; Zolk, O.; Harding, S.E. The effect of Gi-protein inactivation on basal, and beta(1)- and beta(2)AR-stimulated contraction of myocytes from transgenic mice overexpressing the beta(2)-adrenoceptor. Br. J. Pharmacol. 2000, 131, 594–600. [Google Scholar] [CrossRef] [Green Version]
- Heubach, J.F.; Blaschke, M.; Harding, S.E.; Ravens, U.; Kaumann, A.J. Cardiostimulant and cardiodepressant effects through overexpressed human beta2-adrenoceptors in murine heart: Regional differences and functional role of beta1-adrenoceptors. Naunyn Schmiedebergs Arch. Pharmacol. 2003, 367, 380–390. [Google Scholar] [CrossRef] [PubMed]
- Foerster, K.; Groner, F.; Matthes, J.; Koch, W.J.; Birnbaumer, L.; Herzig, S. Cardioprotection specific for the G protein Gi2 in chronic adrenergic signaling through beta 2-adrenoceptors. Proc. Natl. Acad. Sci. USA 2003, 100, 14475–14480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, W.Z.; Zheng, M.; Koch, W.J.; Lefkowitz, R.J.; Kobilka, B.K.; Xiao, R.P. Dual modulation of cell survival and cell death by beta(2)-adrenergic signaling in adult mouse cardiac myocytes. Proc. Natl. Acad. Sci. USA 2001, 98, 1607–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Communal, C.; Colucci, W.S.; Singh, K. p38 mitogen-activated protein kinase pathway protects adult rat ventricular myocytes against beta -adrenergic receptor-stimulated apoptosis. Evidence for Gi-dependent activation. J. Biol. Chem. 2000, 275, 19395–19400. [Google Scholar] [CrossRef] [Green Version]
- Nef, H.M.; Mollmann, H.; Hilpert, P.; Troidl, C.; Voss, S.; Rolf, A.; Behrens, C.B.; Weber, M.; Hamm, C.W.; Elsasser, A. Activated cell survival cascade protects cardiomyocytes from cell death in Tako-Tsubo cardiomyopathy. Eur. J. Heart Fail. 2009, 11, 758–764. [Google Scholar] [CrossRef]
- Ali, A.; Redfors, B.; Lundgren, J.; Alkhoury, J.; Oras, J.; Gan, L.M.; Omerovic, E. Effects of pretreatment with cardiostimulants and beta-blockers on isoprenaline-induced takotsubo-like cardiac dysfunction in rats. Int. J. Cardiol. 2019, 281, 99–104. [Google Scholar] [CrossRef]
- Akashi, Y.J.; Goldstein, D.S.; Barbaro, G.; Ueyama, T. Takotsubo cardiomyopathy: A new form of acute, reversible heart failure. Circulation 2008, 118, 2754–2762. [Google Scholar] [CrossRef] [Green Version]
- Yoshikawa, T. Takotsubo cardiomyopathy, a new concept of cardiomyopathy: Clinical features and pathophysiology. Int. J. Cardiol. 2015, 182, 297–303. [Google Scholar] [CrossRef]
- Isogai, T.; Matsui, H.; Tanaka, H.; Fushimi, K.; Yasunaga, H. Early beta-blocker use and in-hospital mortality in patients with Takotsubo cardiomyopathy. Heart 2016, 102, 1029–1035. [Google Scholar] [CrossRef]
- Evison, I.; Watson, G.; Chan, C.; Bridgman, P. The effects of beta-blockers in patients with stress cardiomyopathy. Intern. Med. J. 2021, 51, 411–413. [Google Scholar] [CrossRef]
- Kurisu, S.; Kihara, Y. Clinical management of takotsubo cardiomyopathy. Circ. J. 2014, 78, 1559–1566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, K.; Carson, K.; Usmani, Z.; Sawhney, G.; Shah, R.; Horowitz, J. Systematic review and meta-analysis of incidence and correlates of recurrence of takotsubo cardiomyopathy. Int. J. Cardiol. 2014, 174, 696–701. [Google Scholar] [CrossRef] [PubMed]
- Brunetti, N.D.; Santoro, F.; De Gennaro, L.; Correale, M.; Gaglione, A.; Di Biase, M. Drug treatment rates with beta-blockers and ACE-inhibitors/angiotensin receptor blockers and recurrences in takotsubo cardiomyopathy: A meta-regression analysis. Int. J. Cardiol. 2016, 214, 340–342. [Google Scholar] [CrossRef] [PubMed]
- Brunetti, N.D.; Santoro, F.; De Gennaro, L.; Correale, M.; Gaglione, A.; Di Biase, M.; Madias, J.E. Combined therapy with beta-blockers and ACE-inhibitors/angiotensin receptor blockers and recurrence of Takotsubo (stress) cardiomyopathy: A meta-regression study. Int. J. Cardiol. 2017, 230, 281–283. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Fan, X.; Yang, Z.; Cyganek, L.; Li, X.; Yuecel, G.; Lan, H.; Li, Y.; Wendel, A.; Lang, S.; et al. Alpha 1-adrenoceptor signalling contributes to toxic effects of catecholamine on electrical properties in cardiomyocytes. Europace 2021, 23, 1137–1148. [Google Scholar] [CrossRef]
- Singh, S.; Desai, R.; Gandhi, Z.; Fong, H.K.; Doreswamy, S.; Desai, V.; Chockalingam, A.; Mehta, P.K.; Sachdeva, R.; Kumar, G. Takotsubo Syndrome in Patients with COVID-19: A Systematic Review of Published Cases. SN Compr. Clin. Med. 2020, 1–7. [Google Scholar] [CrossRef]
- Fu, Y.C.; Chi, C.S.; Yin, S.C.; Hwang, B.; Chiu, Y.T.; Hsu, S.L. Norepinephrine induces apoptosis in neonatal rat endothelial cells via down-regulation of Bcl-2 and activation of beta-adrenergic and caspase-2 pathways. Cardiovasc. Res. 2004, 61, 143–151. [Google Scholar] [CrossRef]
- Romeo, F.; Li, D.; Shi, M.; Mehta, J.L. Carvedilol prevents epinephrine-induced apoptosis in human coronary artery endothelial cells: Modulation of Fas/Fas ligand and caspase-3 pathway. Cardiovasc. Res. 2000, 45, 788–794. [Google Scholar] [CrossRef] [Green Version]
- Sato, K.; Takahashi, J.; Amano, K.; Shimokawa, H. A case of recurrent takotsubo-like cardiomyopathy associated with pheochromocytoma exhibiting different patterns of left ventricular wall motion abnormality and coronary vasospasm: A case report. Eur. Heart J. Case Rep. 2020, 4, 1–5. [Google Scholar] [CrossRef]
- Uchida, Y.; Egami, H.; Uchida, Y.; Sakurai, T.; Kanai, M.; Shirai, S.; Nakagawa, O.; Oshima, T. Possible participation of endothelial cell apoptosis of coronary microvessels in the genesis of Takotsubo cardiomyopathy. Clin. Cardiol. 2010, 33, 371–377. [Google Scholar] [CrossRef]
- Grani, C.; Grunwald, C.; Windecker, S.; Siontis, G.C.M. Coronary Artery Anomaly in Takotsubo Cardiomyopathy: Cause or Innocent Bystander? Tex. Heart Inst. J. 2020, 47, 44–46. [Google Scholar] [CrossRef] [PubMed]
- Tsuchihashi, K.; Ueshima, K.; Uchida, T.; Oh-mura, N.; Kimura, K.; Owa, M.; Yoshiyama, M.; Miyazaki, S.; Haze, K.; Ogawa, H.; et al. Transient left ventricular apical ballooning without coronary artery stenosis: A novel heart syndrome mimicking acute myocardial infarction. Angina Pectoris-Myocardial Infarction Investigations in Japan. J. Am. Coll. Cardiol. 2001, 38, 11–18. [Google Scholar] [CrossRef] [Green Version]
- Jin, Y.; Li, Q.; Guo, X. Alternate recurrent coronary artery spasm and stress cardiomyopathy: A case report. BMC Cardiovasc. Disord. 2020, 20, 476. [Google Scholar] [CrossRef] [PubMed]
- Lombardi, M.; Vergallo, R.; Liuzzo, G.; Crea, F. A case report of coronary artery spasm and takotsubo syndrome: Exploring the hidden side of the moon. Eur. Heart J. Case Rep. 2021, 5, ytaa477. [Google Scholar] [CrossRef] [PubMed]
- Montone, R.A.; Niccoli, G.; Fracassi, F.; Russo, M.; Gurgoglione, F.; Camma, G.; Lanza, G.A.; Crea, F. Patients with acute myocardial infarction and non-obstructive coronary arteries: Safety and prognostic relevance of invasive coronary provocative tests. Eur. Heart J. 2018, 39, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.M.; Lerman, A.; Lennon, R.J.; Prasad, A. Impaired coronary microvascular reactivity in women with apical ballooning syndrome (Takotsubo/stress cardiomyopathy). Eur. Heart J. Acute Cardiovasc. Care 2013, 2, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Verna, E.; Provasoli, S.; Ghiringhelli, S.; Morandi, F.; Salerno-Uriarte, J. Abnormal coronary vasoreactivity in transient left ventricular apical ballooning (tako-tsubo) syndrome. Int. J. Cardiol. 2018, 250, 4–10. [Google Scholar] [CrossRef]
- Khalid, N.; Iqbal, I.; Coram, R.; Raza, T.; Fahsah, I.; Ikram, S. Thrombolysis In Myocardial Infarction Frame Count in Takotsubo Cardiomyopathy. Int. J. Cardiol. 2015, 191, 107–108. [Google Scholar] [CrossRef]
- Khalid, N.; Chhabra, L. Takotsubo cardiomyopathy and microcirculatory dysfunction. Nat. Rev. Cardiol 2015, 12, 497. [Google Scholar] [CrossRef]
- Flammer, A.J.; Luscher, T.F. Human endothelial dysfunction: EDRFs. Pflugers Arch. 2010, 459, 1005–1013. [Google Scholar] [CrossRef] [Green Version]
- Munzel, T.; Gori, T.; Bruno, R.M.; Taddei, S. Is oxidative stress a therapeutic target in cardiovascular disease? Eur. Heart J. 2010, 31, 2741–2748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gori, T.; Munzel, T. Oxidative stress and endothelial dysfunction: Therapeutic implications. Ann. Med. 2011, 43, 259–272. [Google Scholar] [CrossRef] [PubMed]
- Martin, E.A.; Prasad, A.; Rihal, C.S.; Lerman, L.O.; Lerman, A. Endothelial function and vascular response to mental stress are impaired in patients with apical ballooning syndrome. J. Am. Coll. Cardiol. 2010, 56, 1840–1846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spieker, L.E.; Hurlimann, D.; Ruschitzka, F.; Corti, R.; Enseleit, F.; Shaw, S.; Hayoz, D.; Deanfield, J.E.; Luscher, T.F.; Noll, G. Mental stress induces prolonged endothelial dysfunction via endothelin-A receptors. Circulation 2002, 105, 2817–2820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yaribeygi, H.; Atkin, S.L.; Sahebkar, A. A review of the molecular mechanisms of hyperglycemia-induced free radical generation leading to oxidative stress. J. Cell Physiol. 2019, 234, 1300–1312. [Google Scholar] [CrossRef] [PubMed]
- Munzel, T.; Templin, C.; Cammann, V.L.; Hahad, O. Takotsubo Syndrome: Impact of endothelial dysfunction and oxidative stress. Free Radic. Biol. Med. 2021, 169, 216–223. [Google Scholar] [CrossRef]
- Jaguszewski, M.; Osipova, J.; Ghadri, J.R.; Napp, L.C.; Widera, C.; Franke, J.; Fijalkowski, M.; Nowak, R.; Fijalkowska, M.; Volkmann, I.; et al. A signature of circulating microRNAs differentiates takotsubo cardiomyopathy from acute myocardial infarction. Eur Heart J. 2014, 35, 999–1006. [Google Scholar] [CrossRef] [Green Version]
- Amadio, P.; Porro, B.; Cavalca, V.; Barbieri, S.S.; Eligini, S.; Fiorelli, S.; Di Minno, A.; Gorini, A.; Giuliani, M.; Werba, J.P.; et al. Persistent long-term platelet activation and endothelial perturbation in women with Takotsubo syndrome. Biomed. Pharmacother. 2021, 136, 111259. [Google Scholar] [CrossRef]
- Ueyama, T.; Hano, T.; Kasamatsu, K.; Yamamoto, K.; Tsuruo, Y.; Nishio, I. Estrogen attenuates the emotional stress-induced cardiac responses in the animal model of Tako-tsubo (Ampulla) cardiomyopathy. J. Cardiovasc. Pharmacol. 2003, 42 (Suppl. S1), S117–S119. [Google Scholar] [CrossRef]
- Moller, C.; Stiermaier, T.; Brabant, G.; Graf, T.; Thiele, H.; Eitel, I. Comprehensive assessment of sex hormones in Takotsubo syndrome. Int. J. Cardiol. 2018, 250, 11–15. [Google Scholar] [CrossRef]
- Celano, C.M.; Torri, A.; Seiner, S. Takotsubo cardiomyopathy after electroconvulsive therapy: A case report and review. J. ECT 2011, 27, 221–223. [Google Scholar] [CrossRef] [PubMed]
- Satterthwaite, T.D.; Cristancho, M.A.; Alici, Y.; Weiss, D.; O’Reardon, J.P. Electroconvulsive therapy in a 72-year-old woman with a history of Takotsubo cardiomyopathy: A case report and review of the literature. Brain Stimul. 2009, 2, 238–240. [Google Scholar] [CrossRef] [PubMed]
- Binhas, M.; Liger, C.; Sedaghati, A.; Gilton, A.; Dhonneur, G. When retrial of ECT is possible after ECT-induced Takotsubo cardiomyopathy? Ann. Fr. Anesth. Reanim. 2013, 32, 723–724. [Google Scholar] [CrossRef] [PubMed]
- Kumar, G.; Holmes, D.R., Jr.; Prasad, A. “Familial” apical ballooning syndrome (Takotsubo cardiomyopathy). Int. J. Cardiol. 2010, 144, 444–445. [Google Scholar] [CrossRef]
- Pison, L.; De Vusser, P.; Mullens, W. Apical ballooning in relatives. Heart 2004, 90, e67. [Google Scholar] [CrossRef]
- Ferradini, V.; Vacca, D.; Belmonte, B.; Mango, R.; Scola, L.; Novelli, G.; Balistreri, C.R.; Sangiuolo, F. Genetic and Epigenetic Factors of Takotsubo Syndrome: A Systematic Review. Int. J. Mol. Sci. 2021, 22, 9875. [Google Scholar] [CrossRef]
- Elesber, A.A.; Prasad, A.; Lennon, R.J.; Wright, R.S.; Lerman, A.; Rihal, C.S. Four-year recurrence rate and prognosis of the apical ballooning syndrome. J. Am. Coll. Cardiol. 2007, 50, 448–452. [Google Scholar] [CrossRef] [Green Version]
- Spinelli, L.; Trimarco, V.; Di Marino, S.; Marino, M.; Iaccarino, G.; Trimarco, B. L41Q polymorphism of the G protein coupled receptor kinase 5 is associated with left ventricular apical ballooning syndrome. Eur. J. Heart Fail. 2010, 12, 13–16. [Google Scholar] [CrossRef] [Green Version]
- Cherian, J.; Angelis, D.; Filiberti, A.; Saperia, G. Can takotsubo cardiomyopathy be familial? Int. J. Cardiol. 2007, 121, 74–75. [Google Scholar] [CrossRef]
- Ueyama, T.; Yoshida, K.; Senba, E. Emotional stress induces immediate-early gene expression in rat heart via activation of alpha- and beta-adrenoceptors. Am. J. Physiol. 1999, 277, H1553–H1561. [Google Scholar] [CrossRef]
- Eitel, I.; Moeller, C.; Munz, M.; Stiermaier, T.; Meitinger, T.; Thiele, H.; Erdmann, J. Genome-wide association study in takotsubo syndrome—Preliminary results and future directions. Int. J. Cardiol. 2017, 236, 335–339. [Google Scholar] [CrossRef] [PubMed]
- Borchert, T.; Hubscher, D.; Guessoum, C.I.; Lam, T.D.; Ghadri, J.R.; Schellinger, I.N.; Tiburcy, M.; Liaw, N.Y.; Li, Y.; Haas, J.; et al. Catecholamine-Dependent beta-Adrenergic Signaling in a Pluripotent Stem Cell Model of Takotsubo Cardiomyopathy. J. Am. Coll. Cardiol. 2017, 70, 975–991. [Google Scholar] [CrossRef] [PubMed]
- Eckey, K.; Strutz-Seebohm, N.; Katz, G.; Fuhrmann, G.; Henrion, U.; Pott, L.; Linke, W.A.; Arad, M.; Lang, F.; Seebohm, G. Modulation of human ether a gogo related channels by CASQ2 contributes to etiology of catecholaminergic polymorphic ventricular tachycardia (CPVT). Cell Physiol. Biochem. 2010, 26, 503–512. [Google Scholar] [CrossRef] [PubMed]
- Eitel, I.; Lucke, C.; Grothoff, M.; Sareban, M.; Schuler, G.; Thiele, H.; Gutberlet, M. Inflammation in takotsubo cardiomyopathy: Insights from cardiovascular magnetic resonance imaging. Eur. Radiol. 2010, 20, 422–431. [Google Scholar] [CrossRef] [PubMed]
- Oras, J.; Redfors, B.; Ali, A.; Lundgren, J.; Sihlbom, C.; Thorsell, A.; Seeman-Lodding, H.; Omerovic, E.; Ricksten, S.E. Anaesthetic-induced cardioprotection in an experimental model of the Takotsubo syndrome - isoflurane vs. propofol. Acta Anaesthesiol. Scand. 2017, 61, 309–321. [Google Scholar] [CrossRef]
- Santoro, F.; Tarantino, N.; Ferraretti, A.; Ieva, R.; Musaico, F.; Guastafierro, F.; Di Martino, L.; Di Biase, M.; Brunetti, N.D. Serum interleukin 6 and 10 levels in Takotsubo cardiomyopathy: Increased admission levels may predict adverse events at follow-up. Atherosclerosis 2016, 254, 28–34. [Google Scholar] [CrossRef]
- Pirzer, R.; Elmas, E.; Haghi, D.; Lippert, C.; Kralev, S.; Lang, S.; Borggrefe, M.; Kalsch, T. Platelet and monocyte activity markers and mediators of inflammation in Takotsubo cardiomyopathy. Heart Vessel. 2012, 27, 186–192. [Google Scholar] [CrossRef]
- Raymond, R.J.; Dehmer, G.J.; Theoharides, T.C.; Deliargyris, E.N. Elevated interleukin-6 levels in patients with asymptomatic left ventricular systolic dysfunction. Am. Heart J. 2001, 141, 435–438. [Google Scholar] [CrossRef]
- Tsutamoto, T.; Hisanaga, T.; Wada, A.; Maeda, K.; Ohnishi, M.; Fukai, D.; Mabuchi, N.; Sawaki, M.; Kinoshita, M. Interleukin-6 spillover in the peripheral circulation increases with the severity of heart failure, and the high plasma level of interleukin-6 is an important prognostic predictor in patients with congestive heart failure. J. Am. Coll. Cardiol. 1998, 31, 391–398. [Google Scholar] [CrossRef] [Green Version]
- Santoro, F.; Costantino, M.D.; Guastafierro, F.; Triggiani, G.; Ferraretti, A.; Tarantino, N.; Saguner, A.; Di Biase, M.; Brunetti, N.D. Inflammatory patterns in Takotsubo cardiomyopathy and acute coronary syndrome: A propensity score matched analysis. Atherosclerosis 2018, 274, 157–161. [Google Scholar] [CrossRef]
- Wilson, H.M.; Cheyne, L.; Brown, P.A.J.; Kerr, K.; Hannah, A.; Srinivasan, J.; Duniak, N.; Horgan, G.; Dawson, D.K. Characterization of the Myocardial Inflammatory Response in Acute Stress-Induced (Takotsubo) Cardiomyopathy. JACC Basic. Transl. Sci. 2018, 3, 766–778. [Google Scholar] [CrossRef] [PubMed]
- Radfar, A.; Abohashem, S.; Osborne, M.T.; Wang, Y.; Dar, T.; Hassan, M.Z.O.; Ghoneem, A.; Naddaf, N.; Patrich, T.; Abbasi, T.; et al. Stress-associated neurobiological activity associates with the risk for and timing of subsequent Takotsubo syndrome. Eur. Heart J. 2021, 42, 1898–1908. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.H.; Neil, C.J.; Sverdlov, A.L.; Ngo, D.T.; Chan, W.P.; Heresztyn, T.; Chirkov, Y.Y.; Tsikas, D.; Frenneaux, M.P.; Horowitz, J.D. Enhanced NO signaling in patients with Takotsubo cardiomyopathy: Short-term pain, long-term gain? Cardiovasc. Drugs Ther. 2013, 27, 541–547. [Google Scholar] [CrossRef] [PubMed]
- Ferro, A.; Coash, M.; Yamamoto, T.; Rob, J.; Ji, Y.; Queen, L. Nitric oxide-dependent beta2-adrenergic dilatation of rat aorta is mediated through activation of both protein kinase A and Akt. Br. J. Pharmacol. 2004, 143, 397–403. [Google Scholar] [CrossRef] [Green Version]
- Liang, Y.; Liu, D.; Ochs, T.; Tang, C.; Chen, S.; Zhang, S.; Geng, B.; Jin, H.; Du, J. Endogenous sulfur dioxide protects against isoproterenol-induced myocardial injury and increases myocardial antioxidant capacity in rats. Lab. Invest. 2011, 91, 12–23. [Google Scholar] [CrossRef] [Green Version]
- Mukhopadhyay, P.; Rajesh, M.; Batkai, S.; Kashiwaya, Y.; Hasko, G.; Liaudet, L.; Szabo, C.; Pacher, P. Role of superoxide, nitric oxide, and peroxynitrite in doxorubicin-induced cell death in vivo and in vitro. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H1466–H1483. [Google Scholar] [CrossRef] [Green Version]
- Luo, X.; Kraus, W.L. On PAR with PARP: Cellular stress signaling through poly(ADP-ribose) and PARP-1. Genes Dev. 2012, 26, 417–432. [Google Scholar] [CrossRef] [Green Version]
- Virag, L.; Szabo, C. The therapeutic potential of poly(ADP-ribose) polymerase inhibitors. Pharmacol. Rev. 2002, 54, 375–429. [Google Scholar] [CrossRef]
- Ying, W.; Chen, Y.; Alano, C.C.; Swanson, R.A. Tricarboxylic acid cycle substrates prevent PARP-mediated death of neurons and astrocytes. J. Cereb. Blood Flow Metab. 2002, 22, 774–779. [Google Scholar] [CrossRef] [Green Version]
- Surikow, S.Y.; Nguyen, T.H.; Stafford, I.; Chapman, M.; Chacko, S.; Singh, K.; Licari, G.; Raman, B.; Kelly, D.J.; Zhang, Y.; et al. Nitrosative Stress as a Modulator of Inflammatory Change in a Model of Takotsubo Syndrome. JACC Basic Transl. Sci. 2018, 3, 213–226. [Google Scholar] [CrossRef]
- Andrabi, S.A.; Umanah, G.K.; Chang, C.; Stevens, D.A.; Karuppagounder, S.S.; Gagne, J.P.; Poirier, G.G.; Dawson, V.L.; Dawson, T.M. Poly(ADP-ribose) polymerase-dependent energy depletion occurs through inhibition of glycolysis. Proc. Natl. Acad. Sci. USA 2014, 111, 10209–10214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ueyama, T.; Kawabe, T.; Hano, T.; Tsuruo, Y.; Ueda, K.; Ichinose, M.; Kimura, H.; Yoshida, K. Upregulation of heme oxygenase-1 in an animal model of Takotsubo cardiomyopathy. Circ. J. 2009, 73, 1141–1146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishikura, F.; Takano, Y.; Ueyama, T. Amlodipine has a preventive effect on temporal left ventricular hypokinesia after emotional stress compared with an angiotensin II receptor blocker. J. Med. Ultrason. (2001) 2013, 40, 3–7. [Google Scholar] [CrossRef] [PubMed]
- Ueyama, T.; Ishikura, F.; Matsuda, A.; Asanuma, T.; Ueda, K.; Ichinose, M.; Kasamatsu, K.; Hano, T.; Akasaka, T.; Tsuruo, Y.; et al. Chronic estrogen supplementation following ovariectomy improves the emotional stress-induced cardiovascular responses by indirect action on the nervous system and by direct action on the heart. Circ. J. 2007, 71, 565–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nef, H.M.; Mollmann, H.; Troidl, C.; Kostin, S.; Voss, S.; Hilpert, P.; Behrens, C.B.; Rolf, A.; Rixe, J.; Weber, M.; et al. Abnormalities in intracellular Ca2+ regulation contribute to the pathomechanism of Tako-Tsubo cardiomyopathy. Eur. Heart J. 2009, 30, 2155–2164. [Google Scholar] [CrossRef] [Green Version]
- Nef, H.M.; Mollmann, H.; Kostin, S.; Troidl, C.; Voss, S.; Weber, M.; Dill, T.; Rolf, A.; Brandt, R.; Hamm, C.W.; et al. Tako-Tsubo cardiomyopathy: Intraindividual structural analysis in the acute phase and after functional recovery. Eur. Heart J. 2007, 28, 2456–2464. [Google Scholar] [CrossRef] [Green Version]
- Nunez-Gil, I.J.; Andres, M.; Benito, B.; Bernardo, E.; Vedia, O.; Ferreira-Gonzalez, I.; Barba, I. Serum Metabolomic Analysis Suggests Impairment of Myocardial Energy Production in Takotsubo Syndrome. Metabolites 2021, 11, 439. [Google Scholar] [CrossRef]
- Matsuo, S.; Nakajima, K.; Kinuya, S.; Yamagishi, M. Diagnostic utility of 123I-BMIPP imaging in patients with Takotsubo cardiomyopathy. J. Cardiol. 2014, 64, 49–56. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, T.H.; Neil, C.J.; Sverdlov, A.L.; Mahadavan, G.; Chirkov, Y.Y.; Kucia, A.M.; Stansborough, J.; Beltrame, J.F.; Selvanayagam, J.B.; Zeitz, C.J.; et al. N-terminal pro-brain natriuretic protein levels in takotsubo cardiomyopathy. Am. J. Cardiol. 2011, 108, 1316–1321. [Google Scholar] [CrossRef]
- El-Sayed, A.M.; Brinjikji, W.; Salka, S. Demographic and co-morbid predictors of stress (takotsubo) cardiomyopathy. Am. J. Cardiol. 2012, 110, 1368–1372. [Google Scholar] [CrossRef]
- Brunetti, N.D.; Tarantino, N.; Guastafierro, F.; De Gennaro, L.; Correale, M.; Stiermaier, T.; Moller, C.; Di Biase, M.; Eitel, I.; Santoro, F. Malignancies and outcome in Takotsubo syndrome: A meta-analysis study on cancer and stress cardiomyopathy. Heart Fail. Rev. 2019, 24, 481–488. [Google Scholar] [CrossRef] [PubMed]
- Desai, R.; Abbas, S.A.; Goyal, H.; Durairaj, A.; Fong, H.K.; Hung, O.; Sachdeva, R.; Barac, A.; Yusuf, S.W.; Kumar, G. Frequency of Takotsubo Cardiomyopathy in Adult Patients Receiving Chemotherapy (from a 5-Year Nationwide Inpatient Study). Am. J. Cardiol. 2019, 123, 667–673. [Google Scholar] [CrossRef] [PubMed]
- Tornvall, P.; Collste, O.; Ehrenborg, E.; Jarnbert-Petterson, H. A Case-Control Study of Risk Markers and Mortality in Takotsubo Stress Cardiomyopathy. J. Am. Coll. Cardiol. 2016, 67, 1931–1936. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Xie, B.; Tse, G.; Roever, L.; Xia, Y.; Li, G.; Wang, Y.; Liu, T. Malignancy predicts outcome of Takotsubo syndrome: A systematic review and meta-analysis. Heart Fail. Rev. 2020, 25, 513–522. [Google Scholar] [CrossRef]
- Nunez-Gil, I.J.; Vedia, O.; Almendro-Delia, M.; Raposeiras-Roubin, S.; Sionis, A.; Martin-Garcia, A.C.; Martin-Garcia, A.; Andres, M.; Blanco, E.; Martin-de-Miguel, I.; et al. Takotsubo syndrome and cancer, clinical and prognostic implications, insights of RETAKO. Med. Clin. 2020, 155, 521–528. [Google Scholar] [CrossRef]
- Burgdorf, C.; Kurowski, V.; Bonnemeier, H.; Schunkert, H.; Radke, P.W. Long-term prognosis of the transient left ventricular dysfunction syndrome (Tako-Tsubo cardiomyopathy): Focus on malignancies. Eur. J. Heart Fail. 2008, 10, 1015–1019. [Google Scholar] [CrossRef]
- Budnik, M.; Kucharz, J.; Wiechno, P.; Demkow, T.; Kochanowski, J.; Gorska, E.; Opolski, G. Chemotherapy-Induced Takotsubo Syndrome. Adv. Exp. Med. Biol. 2018, 1114, 19–29. [Google Scholar] [CrossRef]
- Finsterer, J.; Stollberger, C.; Pulgram, T. Paraneoplastic takotsubo syndrome with ventricular thrombus and stroke. Herz 2015, 40, 632–634. [Google Scholar] [CrossRef]
- Singh, S.B.; Harle, I.A. Takotsubo cardiomyopathy secondary in part to cancer-related pain crisis: A case report. J. Pain Symptom Manag. 2014, 48, 137–142. [Google Scholar] [CrossRef]
- Burgy, M.; Brossat, H.; Barthelemy, P.; Imperiale, A.; Trinh, A.; Hazam, C.A.; Bergerat, J.P.; Mathelin, C. First report of trastuzumab treatment after postoperative Takotsubo cardiomyopathy. Anticancer Res. 2014, 34, 3579–3582. [Google Scholar]
- Malley, T.; Watson, E. A case of Takotsubo cardiomyopathy after chemotherapy. Oxf. Med. Case Rep. 2016, 2016, 55–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santoro, F.; Ferraretti, A.; Musaico, F.; Di Martino, L.; Tarantino, N.; Ieva, R.; Di Biase, M.; Brunetti, N.D. Carbohydrate-antigen-125 levels predict hospital stay duration and adverse events at long-term follow-up in Takotsubo cardiomyopathy. Intern. Emerg. Med. 2016, 11, 687–694. [Google Scholar] [CrossRef] [PubMed]
- Santoro, F.; Zimotti, T.; Mallardi, A.; Leopizzi, A.; Vitale, E.; Tarantino, N.; Ferraretti, A.; Solimando, A.G.; Racanelli, V.; Iacoviello, M.; et al. Prognostic role of neoplastic markers in Takotsubo syndrome. Sci. Rep. 2021, 11, 16548. [Google Scholar] [CrossRef] [PubMed]
- Coen, M.; Rigamonti, F.; Roth, A.; Koessler, T. Chemotherapy-induced Takotsubo cardiomyopathy, a case report and review of the literature. BMC Cancer 2017, 17, 394. [Google Scholar] [CrossRef]
- Desai, R.; Desai, A.; Abbas, S.A.; Patel, U.; Bansod, S.; Damarlapally, N.; Doshi, R.; Savani, S.; Gangani, K.; Sachdeva, R.; et al. National prevalence, trends and outcomes of takotsubo syndrome in hospitalizations with prior history of mediastinal/intrathoracic cancer and radiation therapy. Int. J. Cardiol. 2020, 309, 14–18. [Google Scholar] [CrossRef]
- Y-Hassan, S.; Tornvall, P.; Tornerud, M.; Henareh, L. Capecitabine caused cardiogenic shock through induction of global Takotsubo syndrome. Cardiovasc. Revasc. Med. 2013, 14, 57–61. [Google Scholar] [CrossRef]
- Goel, S.; Sharma, A.; Garg, A.; Chandra, A.; Shetty, V. Chemotherapy induced Takotsubo cardiomyopathy. World J. Clin. Cases 2014, 2, 565–568. [Google Scholar] [CrossRef]
- Pai, V.B.; Nahata, M.C. Cardiotoxicity of chemotherapeutic agents: Incidence, treatment and prevention. Drug Saf. 2000, 22, 263–302. [Google Scholar] [CrossRef]
- Smith, S.A.; Auseon, A.J. Chemotherapy-induced takotsubo cardiomyopathy. Heart Fail. Clin. 2013, 9, 233–242. [Google Scholar] [CrossRef]
- Grunwald, M.R.; Howie, L.; Diaz, L.A., Jr. Takotsubo cardiomyopathy and Fluorouracil: Case report and review of the literature. J. Clin. Oncol. 2012, 30, e11-14. [Google Scholar] [CrossRef]
- Stewart, T.; Pavlakis, N.; Ward, M. Cardiotoxicity with 5-fluorouracil and capecitabine: More than just vasospastic angina. Intern. Med. J. 2010, 40, 303–307. [Google Scholar] [CrossRef] [PubMed]
- Kuroda, R.; Shintani-Ishida, K.; Unuma, K.; Yoshida, K. Immobilization Stress With alpha2-Adrenergic Stimulation Induces Regional and Transient Reduction of Cardiac Contraction Through Gi Coupling in Rats. Int. Heart J. 2015, 56, 537–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ueyama, T.; Yamamoto, Y.; Ueda, K.; Kawabe, T.; Hano, T.; Ito, T.; Tsuruo, Y.; Ichinose, M.; Yoshida, K. Cardiac and vascular gene profiles in an animal model of takotsubo cardiomyopathy. Heart Vessel. 2011, 26, 321–337. [Google Scholar] [CrossRef] [PubMed]
- Ueyama, T. Emotional stress-induced Tako-tsubo cardiomyopathy: Animal model and molecular mechanism. Ann. N. Y. Acad. Sci. 2004, 1018, 437–444. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Sun, Y.; Liu, X.; Yang, Y.; Sun, T.; Krittanawong, C.; El-Am, E.A.; Liu, G.; Yang, J.; Ma, N. Speckle tracking echocardiography in early detection of myocardial injury in a rat model with stress cardiomyopathy. Med. Ultrason. 2019, 21, 441–448. [Google Scholar] [CrossRef] [Green Version]
- Takano, Y.; Ueyama, T.; Ishikura, F. Azelnidipine, unique calcium channel blocker could prevent stress-induced cardiac dysfunction like alpha.beta blocker. J. Cardiol. 2012, 60, 18–22. [Google Scholar] [CrossRef] [Green Version]
- Ueyama, T.; Kasamatsu, K.; Hano, T.; Tsuruo, Y.; Ishikura, F. Catecholamines and estrogen are involved in the pathogenesis of emotional stress-induced acute heart attack. Ann. N. Y. Acad. Sci. 2008, 1148, 479–485. [Google Scholar] [CrossRef]
- Ueyama, T.; Kasamatsu, K.; Hano, T.; Yamamoto, K.; Tsuruo, Y.; Nishio, I. Emotional stress induces transient left ventricular hypocontraction in the rat via activation of cardiac adrenoceptors: A possible animal model of ‘tako-tsubo’ cardiomyopathy. Circ. J. 2002, 66, 712–713. [Google Scholar] [CrossRef] [Green Version]
- Kvetnansky, R.; Pacak, K.; Fukuhara, K.; Viskupic, E.; Hiremagalur, B.; Nankova, B.; Goldstein, D.S.; Sabban, E.L.; Kopin, I.J. Sympathoadrenal system in stress. Interaction with the hypothalamic-pituitary-adrenocortical system. Ann. N. Y. Acad. Sci. 1995, 771, 131–158. [Google Scholar] [CrossRef]
- Kolodzinska, A.; Czarzasta, K.; Szczepankiewicz, B.; Budnik, M.; Glowczynska, R.; Fojt, A.; Ilczuk, T.; Krasuski, K.; Borodzicz, S.; Cudnoch-Jedrzejewska, A.; et al. Isoprenaline induced Takotsubo syndrome: Histopathological analyses of female rat hearts. Cardiol. J. 2020. [Google Scholar] [CrossRef] [Green Version]
- Wright, P.T.; Bhogal, N.K.; Diakonov, I.; Pannell, L.M.K.; Perera, R.K.; Bork, N.I.; Schobesberger, S.; Lucarelli, C.; Faggian, G.; Alvarez-Laviada, A.; et al. Cardiomyocyte Membrane Structure and cAMP Compartmentation Produce Anatomical Variation in beta2AR-cAMP Responsiveness in Murine Hearts. Cell Rep. 2018, 23, 459–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Izumi, Y.; Okatani, H.; Shiota, M.; Nakao, T.; Ise, R.; Kito, G.; Miura, K.; Iwao, H. Effects of metoprolol on epinephrine-induced takotsubo-like left ventricular dysfunction in non-human primates. Hypertens. Res. 2009, 32, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Takato, T.; Ashida, T.; Seko, Y.; Fujii, J.; Kawai, S. Ventricular tachyarrhythmia-related basal cardiomyopathy in rabbits with vagal stimulation--a novel experimental model for inverted Takotsubo-like cardiomyopathy. J. Cardiol. 2010, 56, 85–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sachdeva, J.; Dai, W.; Kloner, R.A. Functional and histological assessment of an experimental model of Takotsubo’s cardiomyopathy. J. Am. Heart Assoc. 2014, 3, e000921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shao, Y.; Redfors, B.; Stahlman, M.; Tang, M.S.; Miljanovic, A.; Mollmann, H.; Troidl, C.; Szardien, S.; Hamm, C.; Nef, H.; et al. A mouse model reveals an important role for catecholamine-induced lipotoxicity in the pathogenesis of stress-induced cardiomyopathy. Eur. J. Heart Fail. 2013, 15, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Kolodzinska, A.; Czarzasta, K.; Szczepankiewicz, B.; Glowczynska, R.; Fojt, A.; Ilczuk, T.; Budnik, M.; Krasuski, K.; Folta, M.; Cudnoch-Jedrzejewska, A.; et al. Toll-like receptor expression and apoptosis morphological patterns in female rat hearts with takotsubo syndrome induced by isoprenaline. Life Sci. 2018, 199, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Godsman, N.; Kohlhaas, M.; Nickel, A.; Cheyne, L.; Mingarelli, M.; Schweiger, L.; Hepburn, C.; Munts, C.; Welch, A.; Delibegovic, M.; et al. Metabolic alterations in a rat model of Takotsubo syndrome. Cardiovasc. Res. 2021. [Google Scholar] [CrossRef]
- Qi, C.; Shao, Y.; Liu, X.; Wang, D.; Li, X. The cardioprotective effects of icariin on the isoprenaline-induced takotsubo-like rat model: Involvement of reactive oxygen species and the TLR4/NF-kappaB signaling pathway. Int. Immunopharmacol. 2019, 74, 105733. [Google Scholar] [CrossRef]
- Mao, S.; Luo, X.; Li, Y.; He, C.; Huang, F.; Su, C. Role of PI3K/AKT/mTOR Pathway Associated Oxidative Stress and Cardiac Dysfunction in Takotsubo Syndrome. Curr. Neurovasc. Res. 2020, 17, 35–43. [Google Scholar] [CrossRef]
- Perez-Trevino, P.; Sepulveda-Leal, J.; Altamirano, J. Simultaneous assessment of calcium handling and contractility dynamics in isolated ventricular myocytes of a rat model of post-acute isoproterenol-induced cardiomyopathy. Cell Calcium. 2020, 86, 102138. [Google Scholar] [CrossRef]
- Willis, B.C.; Salazar-Cantu, A.; Silva-Platas, C.; Fernandez-Sada, E.; Villegas, C.A.; Rios-Argaiz, E.; Gonzalez-Serrano, P.; Sanchez, L.A.; Guerrero-Beltran, C.E.; Garcia, N.; et al. Impaired oxidative metabolism and calcium mishandling underlie cardiac dysfunction in a rat model of post-acute isoproterenol-induced cardiomyopathy. Am. J. Physiol. Heart Circ. Physiol. 2015, 308, H467–H477. [Google Scholar] [CrossRef] [Green Version]
- Oras, J.; Redfors, B.; Ali, A.; Alkhoury, J.; Seeman-Lodding, H.; Omerovic, E.; Ricksten, S.E. Early treatment with isoflurane attenuates left ventricular dysfunction and improves survival in experimental Takotsubo. Acta Anaesthesiol. Scand. 2017, 61, 399–407. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Jin, S.; Teng, X.; Duan, X.; Chen, Y.; Wu, Y. Hydrogen sulfide attenuates cardiac injury in takotsubo cardiomyopathy by alleviating oxidative stress. Nitric Oxide 2017, 67, 10–25. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Zhang, H.; Ong’achwa Machuki, J.; Zhang, T.; Han, L.; Sang, L.; Wu, L.; Zhao, Z.; James Turley, M.; Hu, X.; et al. GPER mediates estrogen cardioprotection against epinephrine-induced stress. J. Endocrinol. 2021, 249, 209–222. [Google Scholar] [CrossRef] [PubMed]
- Qi, C.; Liu, X.; Xiong, T.; Wang, D. Tempol prevents isoprenaline-induced takotsubo syndrome via the reactive oxygen species/mitochondrial/anti-apoptosis /p38 MAPK pathway. Eur. J. Pharmacol. 2020, 886, 173439. [Google Scholar] [CrossRef]
- Walsh-Wilkinson, E.; Arsenault, M.; Couet, J. Segmental analysis by speckle-tracking echocardiography of the left ventricle response to isoproterenol in male and female mice. PeerJ 2021, 9, e11085. [Google Scholar] [CrossRef]
- Herraez, P.; Espinosa de los Monteros, A.; Fernandez, A.; Edwards, J.F.; Sacchini, S.; Sierra, E. Capture myopathy in live-stranded cetaceans. Vet. J. 2013, 196, 181–188. [Google Scholar] [CrossRef]
- Harthoorn, A.M.; van der Walt, K.; Young, E. Possible therapy for capture myopathy in captured wild animals. Nature 1974, 247, 577. [Google Scholar] [CrossRef]
- El-Battrawy, I.; Zhao, Z.; Lan, H.; Cyganek, L.; Tombers, C.; Li, X.; Buljubasic, F.; Lang, S.; Tiburcy, M.; Zimmermann, W.H.; et al. Electrical dysfunctions in human-induced pluripotent stem cell-derived cardiomyocytes from a patient with an arrhythmogenic right ventricular cardiomyopathy. Europace 2018, 20, f46–f56. [Google Scholar] [CrossRef]
- El-Battrawy, I.; Besler, J.; Ansari, U.; Liebe, V.; Schimpf, R.; Tulumen, E.; Rudic, B.; Lang, S.; Odening, K.; Cyganek, L.; et al. Long-term follow-up of implantable cardioverter-defibrillators in Short QT syndrome. Clin. Res. Cardiol. 2019, 108, 1140–1146. [Google Scholar] [CrossRef]
- El-Battrawy, I.; Albers, S.; Cyganek, L.; Zhao, Z.; Lan, H.; Li, X.; Xu, Q.; Kleinsorge, M.; Huang, M.; Liao, Z.; et al. A cellular model of Brugada syndrome with SCN10A variants using human-induced pluripotent stem cell-derived cardiomyocytes. Europace 2019, 21, 1410–1421. [Google Scholar] [CrossRef] [PubMed]
- El-Battrawy, I.; Muller, J.; Zhao, Z.; Cyganek, L.; Zhong, R.; Zhang, F.; Kleinsorge, M.; Lan, H.; Li, X.; Xu, Q.; et al. Studying Brugada Syndrome With an SCN1B Variants in Human-Induced Pluripotent Stem Cell-Derived Cardiomyocytes. Front. Cell Dev. Biol. 2019, 7, 261. [Google Scholar] [CrossRef] [PubMed]
- Lan, H.; Xu, Q.; El-Battrawy, I.; Zhong, R.; Li, X.; Lang, S.; Cyganek, L.; Borggrefe, M.; Zhou, X.; Akin, I. Ionic Mechanisms of Disopyramide Prolonging Action Potential Duration in Human-Induced Pluripotent Stem Cell-Derived Cardiomyocytes From a Patient With Short QT Syndrome Type 1. Front. Pharmacol. 2020, 11, 554422. [Google Scholar] [CrossRef] [PubMed]
- El-Battrawy, I.; Lan, H.; Cyganek, L.; Zhao, Z.; Li, X.; Buljubasic, F.; Lang, S.; Yucel, G.; Sattler, K.; Zimmermann, W.H.; et al. Modeling Short QT Syndrome Using Human-Induced Pluripotent Stem Cell-Derived Cardiomyocytes. J. Am. Heart Assoc. 2018, 7. [Google Scholar] [CrossRef] [Green Version]
- Buljubasic, F.; El-Battrawy, I.; Lan, H.; Lomada, S.K.; Chatterjee, A.; Zhao, Z.; Li, X.; Zhong, R.; Xu, Q.; Huang, M.; et al. Nucleoside Diphosphate Kinase B Contributes to Arrhythmogenesis in Human-Induced Pluripotent Stem Cell-Derived Cardiomyocytes from a Patient with Arrhythmogenic Right Ventricular Cardiomyopathy. J. Clin. Med. 2020, 9, 486. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.; Li, X.; El-Battrawy, I.; Lan, H.; Zhong, R.; Xu, Q.; Huang, M.; Liao, Z.; Lang, S.; Zimmermann, W.H.; et al. Drug Testing in Human-Induced Pluripotent Stem Cell-Derived Cardiomyocytes From a Patient With Short QT Syndrome Type 1. Clin. Pharmacol. Ther. 2019, 106, 642–651. [Google Scholar] [CrossRef]
- Yucel, G.; Zhao, Z.; El-Battrawy, I.; Lan, H.; Lang, S.; Li, X.; Buljubasic, F.; Zimmermann, W.H.; Cyganek, L.; Utikal, J.; et al. Lipopolysaccharides induced inflammatory responses and electrophysiological dysfunctions in human-induced pluripotent stem cell derived cardiomyocytes. Sci. Rep. 2017, 7, 2935. [Google Scholar] [CrossRef]
- Fan, X.; Yang, G.; Kowitz, J.; Duru, F.; Saguner, A.M.; Akin, I.; Zhou, X.; El-Battrawy, I. Preclinical short QT syndrome models: Studying the phenotype and drug-screening. Europace 2021. [Google Scholar] [CrossRef]
- El-Battrawy, I.; Lan, H.; Cyganek, L.; Maywald, L.; Zhong, R.; Zhang, F.; Xu, Q.; Lee, J.; Duperrex, E.; Hierlemann, A.; et al. Deciphering the pathogenic role of a variant with uncertain significance for short QT and Brugada syndromes using gene-edited human-induced pluripotent stem cell-derived cardiomyocytes and preclinical drug screening. Clin. Transl. Med. 2021, 11, e646. [Google Scholar] [CrossRef]
- El-Battrawy, I.; Lang, S.; Ansari, U.; Tulumen, E.; Schramm, K.; Fastner, C.; Zhou, X.; Hoffmann, U.; Borggrefe, M.; Akin, I. Prevalence of malignant arrhythmia and sudden cardiac death in takotsubo syndrome and its management. Europace 2018, 20, 843–850. [Google Scholar] [CrossRef]
- Ebert, A.; Joshi, A.U.; Andorf, S.; Dai, Y.; Sampathkumar, S.; Chen, H.; Li, Y.; Garg, P.; Toischer, K.; Hasenfuss, G.; et al. Proteasome-Dependent Regulation of Distinct Metabolic States During Long-Term Culture of Human iPSC-Derived Cardiomyocytes. Circ. Res. 2019, 125, 90–103. [Google Scholar] [CrossRef] [PubMed]
- Correia, C.; Koshkin, A.; Duarte, P.; Hu, D.; Teixeira, A.; Domian, I.; Serra, M.; Alves, P.M. Distinct carbon sources affect structural and functional maturation of cardiomyocytes derived from human pluripotent stem cells. Sci. Rep. 2017, 7, 8590. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Nunes, S.S. Maturation of Human Stem Cell-derived Cardiomyocytes in Biowires Using Electrical Stimulation. J. Vis. Exp. 2017. [Google Scholar] [CrossRef] [PubMed]
- Ruan, J.L.; Tulloch, N.L.; Razumova, M.V.; Saiget, M.; Muskheli, V.; Pabon, L.; Reinecke, H.; Regnier, M.; Murry, C.E. Mechanical Stress Conditioning and Electrical Stimulation Promote Contractility and Force Maturation of Induced Pluripotent Stem Cell-Derived Human Cardiac Tissue. Circulation 2016, 134, 1557–1567. [Google Scholar] [CrossRef] [PubMed]
- Herron, T.J.; Rocha, A.M.; Campbell, K.F.; Ponce-Balbuena, D.; Willis, B.C.; Guerrero-Serna, G.; Liu, Q.; Klos, M.; Musa, H.; Zarzoso, M.; et al. Extracellular Matrix-Mediated Maturation of Human Pluripotent Stem Cell-Derived Cardiac Monolayer Structure and Electrophysiological Function. Circ. Arrhythm. Electrophysiol. 2016, 9, e003638. [Google Scholar] [CrossRef] [Green Version]
- Nunes, S.S.; Miklas, J.W.; Liu, J.; Aschar-Sobbi, R.; Xiao, Y.; Zhang, B.; Jiang, J.; Masse, S.; Gagliardi, M.; Hsieh, A.; et al. Biowire: A platform for maturation of human pluripotent stem cell-derived cardiomyocytes. Nat. Methods 2013, 10, 781–787. [Google Scholar] [CrossRef] [Green Version]
- Land, S.; Niederer, S.A.; Louch, W.E.; Roe, A.T.; Aronsen, J.M.; Stuckey, D.J.; Sikkel, M.B.; Tranter, M.H.; Lyon, A.R.; Harding, S.E.; et al. Computational modeling of Takotsubo cardiomyopathy: Effect of spatially varying beta-adrenergic stimulation in the rat left ventricle. Am. J. Physiol. Heart Circ. Physiol. 2014, 307, H1487–H1496. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Model | Method | Main Finding | |
|---|---|---|---|
| Animal model | Rats [153,182,183,186,187,188,189] | Immobilization (IMO) | (1) The activation of β1 adrenergic receptors in the heart and the activation of α1 adrenergic receptors in the aorta were the primary cause of TTS (2) The reduction of estrogen may interfere with coronary microcirculation and may be involved in the primary cause of TTS by indirect action on the nervous system and by direct action on the heart (3) α2AR/Gi-dependent signaling attenuated myosin-binding protein-C(MyBP-C) phosphorylation and contractility in the anterior wall (AW) through an epinephrine surge in TTS rats |
| Ovariectomized (OVX) and estradiol-supplemented ovariectomized female rats [152,154] | Immobilization (IMO) | (1) The reduction of LV contractility and the increase of heart rate in response to emotional stress were attenuated by supplement of estradiol in the ovariectomized rats. (2) Emotional stress and a surge of catecholamine upregulated heme oxygenase-1 (HO-1) | |
| Cynomolgus monkeys [192] | Intravenous infusion of epinephrine overdose | LV dysfunction with apical ballooning and wall motion abnormalities | |
| Rabbits [193] | Vagal stimulation | The cardiac lesions related to ventricular arrhythmias were involved in the basal portion, mitral valve, and papillary muscles but not the apex | |
| Mice [185,195,206] | A single dose injection of isoprenaline | (1) Lipotoxicity was closely related to catecholamine-induced myocardial dysfunction, including neurogenic stunning, metabolic stunning, and electrophysiological stunning (2) ISO reduced GLS and circumferential (GCS) strains of males and females | |
| Rats [190,194,197,198,199,200,201,202,203,205] | Isoprenaline (ISO) | (1) TTS rats had significantly lower left ventricular end-diastolic pressure and significantly better estimates of cardiac function. (2) Its apical perfusion was not impaired in the early stage of TTS (3) ISO significantly increased the levels of reactive oxygen species (ROS) in the setting of TTS (4) Early treatment with isoflurane could reduce LV dyskinesia and improve the survival rate of experimental TTS (5) GPER, azelnidipine, Tempol and amlodipine also played a protective role for TTS | |
| Rats [204] | Epinephrine | GPER played a protective role against TTS | |
| hiPSC-CMs models | hiPSC-CMs models [14] | Isoprenaline | Estradiol had protective effects against catecholamine excess and hence reduction in estrogen level may increase the risk of acquired long QT syndrome in TTC |
| hiPSC-CMs models [204] | Epinephrine | Knockdown of GPER by siRNA abolished E2 effects on increasing ICa-L and action potential duration in the stress state | |
| hiPSC-CMs models [95] | Epinephrine | High concentrations of epinephrine inhibited the depolarization rate in hiPSC-CMs, the duration of action potentials and induced arrhythmia events while the effect of epinephrine was attenuated by alpha-adrenergic receptor blockers-phentolamine | |
| TTS-iPSC-CMs [132] | The β-adrenergic signaling, including cAMP response and cAMP-dependent PKA activity, was increased in TTS-iPSC-CMs | ||
| Other cells model | H9C2 [205] | Isoproterenol | Pretreatment with Tempolcould reduce the production of reactive oxygen species and the deposition of lipid droplets and protect mitochondrial function by reducing mitochondrial swelling |
| Computational model [227] | Three potential dominant mechanisms are related to the effects of β-adrenergic stimulation |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fan, X.; Yang, G.; Kowitz, J.; Akin, I.; Zhou, X.; El-Battrawy, I. Takotsubo Syndrome: Translational Implications and Pathomechanisms. Int. J. Mol. Sci. 2022, 23, 1951. https://doi.org/10.3390/ijms23041951
Fan X, Yang G, Kowitz J, Akin I, Zhou X, El-Battrawy I. Takotsubo Syndrome: Translational Implications and Pathomechanisms. International Journal of Molecular Sciences. 2022; 23(4):1951. https://doi.org/10.3390/ijms23041951
Chicago/Turabian StyleFan, Xuehui, Guoqiang Yang, Jacqueline Kowitz, Ibrahim Akin, Xiaobo Zhou, and Ibrahim El-Battrawy. 2022. "Takotsubo Syndrome: Translational Implications and Pathomechanisms" International Journal of Molecular Sciences 23, no. 4: 1951. https://doi.org/10.3390/ijms23041951
APA StyleFan, X., Yang, G., Kowitz, J., Akin, I., Zhou, X., & El-Battrawy, I. (2022). Takotsubo Syndrome: Translational Implications and Pathomechanisms. International Journal of Molecular Sciences, 23(4), 1951. https://doi.org/10.3390/ijms23041951