
SALL4 Oncogenic Function in Cancers: Mechanisms and Therapeutic Relevance


Abstract
:1. Introduction
2. Mechanisms of Action of SALL4 in Tumors
2.1. SALL4 Activates the Wnt/β-Catenin Signaling Pathway
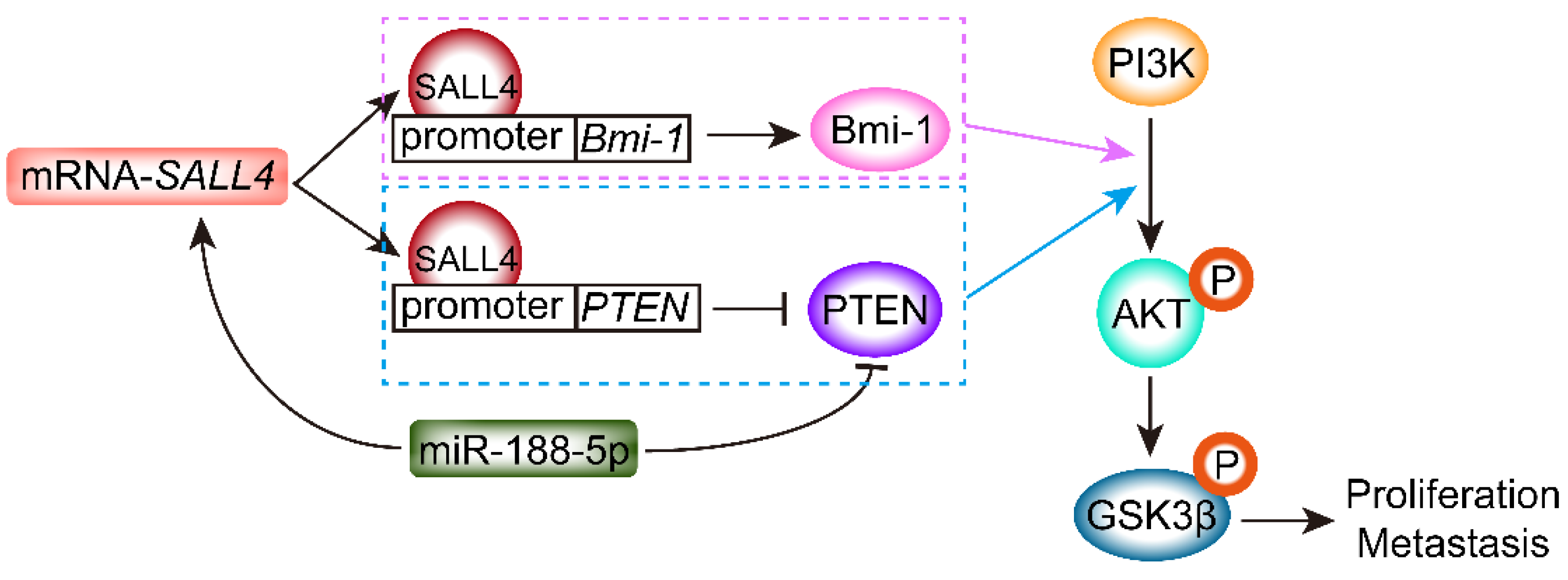
2.2. SALL4 Inhibits the Expression of PTEN and Activates the PI3K/AKT Signaling Pathway
2.3. SALL4 Activates the Notch Signaling Pathway
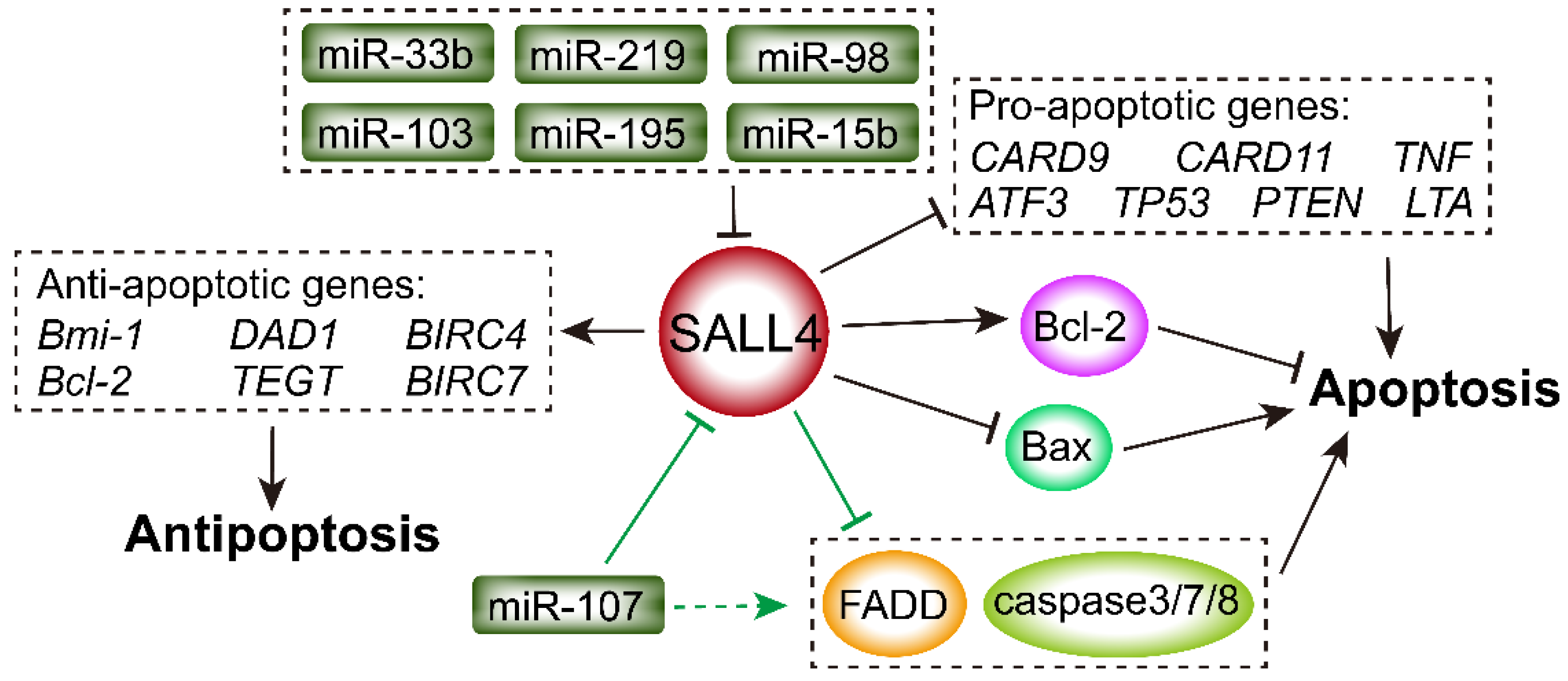
2.4. SALL4 Regulates Expression of Bcl-2 and Bax
2.5. SALL4 Inhibits Caspase-Related and Death-Receptor Pathways
2.6. MiRNAs Regulate the Interaction of SALL4 and TNF Family
2.7. SALL4 Regulates Gene Expression through Epigenetic Mechanisms
2.8. SALL4 Induces Mitochondrial Oxidative Phosphorylation during Tumorigenesis
2.9. Brief Summary of Mechanisms of SALL4 in Cancers
3. Function of SALL4 in Tumors
3.1. Lung Cancer
3.2. Hepatocellular Carcinoma
3.3. Breast Cancer
3.4. Gastric Cancer
3.5. Colorectal Cancer
3.6. Osteosarcoma
3.7. Normal Hematopoietic Function and Leukemia
3.8. Ovarian-Related Diseases
3.9. Glioma
4. Analysis and Prospects in the Future
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Herbst, R.S.; Morgensztern, D.; Boshoff, C. The biology and management of non-small cell lung cancer. Nature 2018, 553, 446–454. [Google Scholar] [CrossRef] [PubMed]
- Ma, T.; Shi, S.; Jiang, H.; Chen, X.; Xu, D.; Ding, X.; Zhang, H.; Xi, Y. A pan-cancer study of spalt-like transcription factors 1/2/3/4 as therapeutic targets. Arch. Biochem. Biophys. 2021, 711, 109016. [Google Scholar] [CrossRef] [PubMed]
- Misawa, K.; Mochizuki, D.; Imai, A.; Misawa, Y.; Endo, S.; Mima, M.; Kawasaki, H.; Carey, T.E.; Kanazawa, T. Epigenetic silencing of SALL3 is an independent predictor of poor survival in head and neck cancer. Clin. Epigenetics 2017, 9, 64. [Google Scholar] [CrossRef] [PubMed]
- Misawa, K.; Misawa, Y.; Imai, A.; Mochizuki, D.; Endo, S.; Mima, M.; Ishikawa, R.; Kawasaki, H.; Yamatodani, T.; Kanazawa, T. Epigenetic modification of SALL1 as a novel biomarker for the prognosis of early stage head and neck cancer. J. Cancer 2018, 9, 941–949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hermosilla, V.E.; Hepp, M.I.; Escobar, D.; Farkas, C.; Riffo, E.N.; Castro, A.F.; Pincheira, R. Developmental SALL2 transcription factor: A new player in cancer. Carcinogenesis 2017, 38, 680–690. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Yuan, X.; Zhu, W.; Qian, H.; Xu, W. SALL4: An emerging cancer biomarker and target. Cancer Lett. 2015, 357, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Imai, A.; Mochizuki, D.; Misawa, Y.; Nakagawa, T.; Endo, S.; Mima, M.; Yamada, S.; Kawasaki, H.; Kanazawa, T.; Misawa, K. SALL2 Is a Novel Prognostic Methylation Marker in Patients with Oral Squamous Carcinomas: Associations with SALL1 and SALL3 Methylation Status. DNA Cell Biol. 2019, 38, 678–687. [Google Scholar] [CrossRef]
- Wei, X.; Zhang, S.; Cao, D.; Zhao, M.; Zhang, Q.; Zhao, J.; Yang, T.; Pei, M.; Wang, L.; Li, Y.; et al. Aberrant Hypermethylation of SALL3 with HPV Involvement Contributes to the Carcinogenesis of Cervical Cancer. PLoS ONE 2015, 10, e0145700. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Wei, X.; Yang, T.; Zhao, M.Y.; Pei, M.L.; Yang, X.F. Correlation between Methylation of SALL3 and Cervical Cancer. Zhongguo Yi Xue Ke Xue Yuan Xue Bao. Acta Acad. Med. Sin. 2019, 41, 609–614. [Google Scholar] [CrossRef]
- Chen, M.; Li, L.; Zheng, P.S. SALL4 promotes the tumorigenicity of cervical cancer cells through activation of the Wnt/β-catenin pathway via CTNNB1. Cancer Sci. 2019, 110, 2794–2805. [Google Scholar] [CrossRef] [Green Version]
- Chi, D.; Zhang, W.; Jia, Y.; Cong, D.; Hu, S. Spalt-Like Transcription Factor 1 (SALL1) Gene Expression Inhibits Cell Proliferation and Cell Migration of Human Glioma Cells Through the Wnt/β-Catenin Signaling Pathway. Med. Sci. Monit. Basic Res. 2019, 25, 128–138. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, C.; Fukuda, N.; Matsumoto, T.; Higuchi, T.; Ueno, T.; Matsumoto, K. Zinc-finger transcriptional factor Sall1 induces angiogenesis by activation of the gene for VEGF-A. Hypertens. Res. Off. J. Jpn. Soc. Hypertens. 2010, 33, 143–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sung, C.K.; Yim, H. Roles of SALL2 in tumorigenesis. Arch. Pharmacal. Res. 2017, 40, 146–151. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Zhang, W.; Zhou, Q.; Zhao, T.; Song, Y.; Chai, L.; Li, Y. Low-expression of microRNA-107 inhibits cell apoptosis in glioma by upregulation of SALL4. Int. J. Biochem. Cell Biol. 2013, 45, 1962–1973. [Google Scholar] [CrossRef] [PubMed]
- Du, W.; Ni, L.; Liu, B.; Wei, Y.; Lv, Y.; Qiang, S.; Dong, J.; Liu, X. Upregulation of SALL4 by EGFR activation regulates the stemness of CD44-positive lung cancer. Oncogenesis 2018, 7, 36. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Yao, F.; Mao, X.; Li, W.; Chen, H. Effect of SALL4 on the Proliferation, Invasion and Apoptosis of Breast Cancer Cells. Technol. Cancer Res. Treat. 2020, 19, 1533033820980074. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, N.; Li, M.Y.; Du, M.F. Long non-coding RNA ZEB2-AS1 regulates osteosarcoma progression by acting as a molecular sponge of miR-107 to modulate SALL4 expression. Am. J. Transl. Res. 2021, 13, 1140–1154. [Google Scholar]
- Diener, J.; Baggiolini, A.; Pernebrink, M.; Dalcher, D.; Lerra, L.; Cheng, P.F.; Varum, S.; Hausel, J.; Stierli, S.; Treier, M.; et al. Epigenetic control of melanoma cell invasiveness by the stem cell factor SALL4. Nat. Commun. 2021, 12, 5056. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Huang, J.; Ma, Q.; Liu, G. Association between quantitative parameters of CEUS and Sall4/Wnt/beta-catenin signaling in patients with hepatocellular carcinoma. Cancer Manag Res. 2019, 11, 3339–3347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Zhang, P.; Shao, M.; Zang, X.; Zhang, J.; Mao, F.; Qian, H.; Xu, W. SALL4 activates TGF-β/SMAD signaling pathway to induce EMT and promote gastric cancer metastasis. Cancer Manag Res. 2018, 10, 4459–4470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Wang, X.; Liu, Y.; Hu, Y.; Li, Z.; Li, Z.; Bu, Z.; Wu, X.; Zhang, L.; Ji, J. Up-Regulation of SALL4 Is Associated With Survival and Progression via Putative WNT Pathway in Gastric Cancer. Front. Cell Dev. Biol. 2021, 9, 600344. [Google Scholar] [CrossRef] [PubMed]
- Al-Baradie, R.; Yamada, K.; St Hilaire, C.; Chan, W.M.; Andrews, C.; McIntosh, N.; Nakano, M.; Martonyi, E.J.; Raymond, W.R.; Okumura, S.; et al. Duane radial ray syndrome (Okihiro syndrome) maps to 20q13 and results from mutations in SALL4, a new member of the SAL family. Am. J. Hum. Genet. 2002, 71, 1195–1199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uez, N.; Lickert, H.; Kohlhase, J.; de Angelis, M.H.; Kühn, R.; Wurst, W.; Floss, T. Sall4 isoforms act during proximal-distal and anterior-posterior axis formation in the mouse embryo. Genesis 2008, 46, 463–477. [Google Scholar] [CrossRef] [PubMed]
- Pantier, R.; Chhatbar, K.; Quante, T.; Skourti-Stathaki, K.; Cholewa-Waclaw, J.; Alston, G.; Alexander-Howden, B.; Lee, H.Y.; Cook, A.G.; Spruijt, C.G.; et al. SALL4 controls cell fate in response to DNA base composition. Mol. Cell 2021, 81, 845–858 e848. [Google Scholar] [CrossRef] [PubMed]
- Kong, N.R.; Bassal, M.A.; Tan, H.K.; Kurland, J.V.; Yong, K.J.; Young, J.J.; Yang, Y.; Li, F.; Lee, J.D.; Liu, Y.; et al. Zinc Finger Protein SALL4 Functions through an AT-Rich Motif to Regulate Gene Expression. Cell Rep. 2021, 34, 108574. [Google Scholar] [CrossRef] [PubMed]
- Shao, M.; Zhang, J.; Zhang, J.; Shi, H.; Zhang, Y.; Ji, R.; Mao, F.; Qian, H.; Xu, W.; Zhang, X. SALL4 promotes gastric cancer progression via hexokinase II mediated glycolysis. Cancer Cell Int. 2020, 20, 188. [Google Scholar] [CrossRef]
- Li, A.; Jiao, Y.; Yong, K.J.; Wang, F.; Gao, C.; Yan, B.; Srivastava, S.; Lim, G.S.; Tang, P.; Yang, H.; et al. SALL4 is a new target in endometrial cancer. Oncogene 2015, 34, 63–72. [Google Scholar] [CrossRef] [Green Version]
- Zeng, S.S.; Yamashita, T.; Kondo, M.; Nio, K.; Hayashi, T.; Hara, Y.; Nomura, Y.; Yoshida, M.; Hayashi, T.; Oishi, N.; et al. The transcription factor SALL4 regulates stemness of EpCAM-positive hepatocellular carcinoma. J. Hepatol. 2014, 60, 127–134. [Google Scholar] [CrossRef] [Green Version]
- Yang, J. SALL4 as a transcriptional and epigenetic regulator in normal and leukemic hematopoiesis. Biomark. Res. 2018, 6, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yong, K.J.; Chai, L.; Tenen, D.G. Oncofetal gene SALL4 in aggressive hepatocellular carcinoma. N. Engl. J. Med. 2013, 369, 1171–1172. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Zhong, N.; Li, X.; Chen, M.B. TRIB3 promotes lung cancer progression by activating β-catenin signaling. Eur. J. Pharmacol. 2019, 863, 172697. [Google Scholar] [CrossRef] [PubMed]
- Hua, F.; Shang, S.; Yang, Y.W.; Zhang, H.Z.; Xu, T.L.; Yu, J.J.; Zhou, D.D.; Cui, B.; Li, K.; Lv, X.X.; et al. TRIB3 Interacts With β-Catenin and TCF4 to Increase Stem Cell Features of Colorectal Cancer Stem Cells and Tumorigenesis. Gastroenterology 2019, 156, 708–721.e715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Y.; Cui, W.; Yang, J.; Qu, J.; Di, C.; Amin, H.M.; Lai, R.; Ritz, J.; Krause, D.S.; Chai, L. SALL4, a novel oncogene, is constitutively expressed in human acute myeloid leukemia (AML) and induces AML in transgenic mice. Blood 2006, 108, 2726–2735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reya, T.; Clevers, H. Wnt signalling in stem cells and cancer. Nature 2005, 434, 843–850. [Google Scholar] [CrossRef]
- Wang, Y.; Krivtsov, A.V.; Sinha, A.U.; North, T.E.; Goessling, W.; Feng, Z.; Zon, L.I.; Armstrong, S.A. The Wnt/beta-catenin pathway is required for the development of leukemia stem cells in AML. Science 2010, 327, 1650–1653. [Google Scholar] [CrossRef] [Green Version]
- Heidel, F.H.; Mar, B.G.; Armstrong, S.A. Self-renewal related signaling in myeloid leukemia stem cells. Int. J. Hematol. 2011, 94, 109–117. [Google Scholar] [CrossRef] [Green Version]
- Ng, S.W.; Mitchell, A.; Kennedy, J.A.; Chen, W.C.; McLeod, J.; Ibrahimova, N.; Arruda, A.; Popescu, A.; Gupta, V.; Schimmer, A.D.; et al. A 17-gene stemness score for rapid determination of risk in acute leukaemia. Nature 2016, 540, 433–437. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Huang, F.; Deng, G.; Nie, W.; Huang, W.; Xu, H.; Zheng, S.; Yi, Z.; Wan, T. Knockdown of Sall4 inhibits intrahepatic cholangiocarcinoma cell migration and invasion in ICC-9810 cells. OncoTargets Ther. 2016, 9, 5297–5305. [Google Scholar] [CrossRef] [Green Version]
- Hao, L.; Zhao, Y.; Wang, Z.; Yin, H.; Zhang, X.; He, T.; Song, S.; Sun, S.; Wang, B.; Li, Z.; et al. Expression and clinical significance of SALL4 and β-catenin in colorectal cancer. J. Mol. Histol. 2016, 47, 117–128. [Google Scholar] [CrossRef]
- Liu, S.; Ma, X.; Ai, Q.; Huang, Q.; Shi, T.; Zhu, M.; Wang, B.; Zhang, X. NOTCH1 functions as an oncogene by regulating the PTEN/PI3K/AKT pathway in clear cell renal cell carcinoma. Urol. Oncol. 2013, 31, 938–948. [Google Scholar] [CrossRef]
- Ji, Y.; Zheng, M.; Ye, S.; Chen, J.; Chen, Y. PTEN and Ki67 expression is associated with clinicopathologic features of non-small cell lung cancer. J. Biomed. Res. 2014, 28, 462–467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, P.C.; Lin, J.K.; Lin, H.H.; Lan, Y.T.; Lin, C.C.; Yang, S.H.; Chen, W.S.; Liang, W.Y.; Jiang, J.K.; Chang, S.C. A comprehensive analysis of phosphatase and tensin homolog deleted on chromosome 10 (PTEN) loss in colorectal cancer. World J. Surg. Oncol. 2015, 13, 186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dasari, V.R.; Kaur, K.; Velpula, K.K.; Gujrati, M.; Fassett, D.; Klopfenstein, J.D.; Dinh, D.H.; Rao, J.S. Upregulation of PTEN in glioma cells by cord blood mesenchymal stem cells inhibits migration via downregulation of the PI3K/Akt pathway. PLoS ONE 2010, 5, e10350. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.W.; Kayani, M.A.; Shabbir, G.; Ali, S.M.; Shinwari, W.U.; Mahjabeen, I. Expression of PTEN and its correlation with proliferation marker Ki-67 in head and neck cancer. Int. J. Biol. Markers 2016, 31, e193–203. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.L.; Mu, G.G.; Ding, Q.S.; Li, Y.X.; Shi, Y.B.; Dai, J.F.; Yu, H.G. Phosphatase and Tensin Homolog (PTEN) Represses Colon Cancer Progression through Inhibiting Paxillin Transcription via PI3K/AKT/NF-κB Pathway. J. Biol. Chem. 2015, 290, 15018–15029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, G.; Liu, C.T. Knockdown of SALL4 overcomes cisplatin-resistance through AKT/mTOR signaling in lung cancer cells. Int. J. Clin. Exp. Pathol. 2018, 11, 634–641. [Google Scholar] [PubMed]
- Nicoletti, N.F.; Erig, T.C.; Zanin, R.F.; Pereira, T.C.; Bogo, M.R.; Campos, M.M.; Morrone, F.B. Mechanisms involved in kinin-induced glioma cells proliferation: The role of ERK1/2 and PI3K/Akt pathways. J. Neuro-Oncol. 2014, 120, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Yang, Q.; Yu, J.; Li, X.; Yu, S.; Zhang, X. SPOCK1 promotes the proliferation, migration and invasion of glioma cells through PI3K/AKT and Wnt/β-catenin signaling pathways. Oncol. Rep. 2016, 35, 3566–3576. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Jeong, H.W.; Kong, N.; Yang, Y.; Carroll, J.; Luo, H.R.; Silberstein, L.E.; Yupoma; Chai, L. Stem cell factor SALL4 represses the transcriptions of PTEN and SALL1 through an epigenetic repressor complex. PLoS ONE 2009, 4, e5577. [Google Scholar] [CrossRef]
- Liu, C.; Wu, H.; Li, Y.; Shen, L.; Yu, R.; Yin, H.; Sun, T.; Sun, C.; Zhou, Y.; Du, Z. SALL4 suppresses PTEN expression to promote glioma cell proliferation via PI3K/AKT signaling pathway. J. Neuro-Oncol. 2017, 135, 263–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Chai, L.; Liu, F.; Fink, L.M.; Lin, P.; Silberstein, L.E.; Amin, H.M.; Ward, D.C.; Ma, Y. Bmi-1 is a target gene for SALL4 in hematopoietic and leukemic cells. Proc. Natl. Acad. Sci. USA 2007, 104, 10494–10499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Qiu, R.; Gong, Z.; Zhao, X.; Wang, T.; Zhou, L.; Lu, W.; Shen, B.; Zhu, W.; Xu, W. miR-188-5p emerges as an oncomiRNA to promote gastric cancer cell proliferation and migration via upregulation of SALL4. J. Cell Biochem. 2019, 120, 15027–15037. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Kohashi, K.; Yoshizumi, T.; Okumura, Y.; Tanaka, Y.; Shimokawa, M.; Iwasaki, T.; Aishima, S.; Maehara, Y.; Oda, Y. Coexpression of SALL4 with HDAC1 and/or HDAC2 is associated with underexpression of PTEN and poor prognosis in patients with hepatocellular carcinoma. Hum. Pathol. 2017, 64, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Shen, Q.; Liu, S.; Hu, J.; Chen, S.; Yang, L.; Li, B.; Wu, X.; Ma, Y.; Yang, J.; Ma, Y.; et al. The differential expression pattern of the BMI-1, SALL4 and ABCA3 genes in myeloid leukemia. Cancer Cell Int. 2012, 12, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, J.; Tang, Q.; Gao, Y.; Zhang, W.; Zhao, Z.; Yang, F.; Hu, X.; Zhang, D.; Wang, Y.; Zhang, H.; et al. VHL mutation-mediated SALL4 overexpression promotes tumorigenesis and vascularization of clear cell renal cell carcinoma via Akt/GSK-3β signaling. J. Exp. Clin. Cancer Res. CR 2020, 39, 104. [Google Scholar] [CrossRef] [PubMed]
- Oishi, N.; Yamashita, T.; Kaneko, S. Molecular biology of liver cancer stem cells. Liver Cancer 2014, 3, 71–84. [Google Scholar] [CrossRef]
- Forghanifard, M.M.; Kasebi, P.; Abbaszadegan, M.R. SOX2/SALL4 stemness axis modulates Notch signaling genes to maintain self-renewal capacity of esophageal squamous cell carcinoma. Mol. Cell. Biochem. 2021, 476, 921–929. [Google Scholar] [CrossRef] [PubMed]
- Park, J.T.; Chen, X.; Tropè, C.G.; Davidson, B.; Shih Ie, M.; Wang, T.L. Notch3 overexpression is related to the recurrence of ovarian cancer and confers resistance to carboplatin. Am. J. Pathol. 2010, 177, 1087–1094. [Google Scholar] [CrossRef]
- Qi, H.; Pei, D. The magic of four: Induction of pluripotent stem cells from somatic cells by Oct4, Sox2, Myc and Klf4. Cell Res. 2007, 17, 578–580. [Google Scholar] [CrossRef] [Green Version]
- Forghanifard, M.M.; Ardalan Khales, S.; Javdani-Mallak, A.; Rad, A.; Farshchian, M.; Abbaszadegan, M.R. Stemness state regulators SALL4 and SOX2 are involved in progression and invasiveness of esophageal squamous cell carcinoma. Med. Oncol. 2014, 31, 922. [Google Scholar] [CrossRef] [PubMed]
- Yin, T.; Li, L. The stem cell niches in bone. J. Clin. Investig. 2006, 116, 1195–1201. [Google Scholar] [CrossRef] [PubMed]
- Lévesque, J.P.; Helwani, F.M.; Winkler, I.G. The endosteal ’osteoblastic’ niche and its role in hematopoietic stem cell homing and mobilization. Leukemia 2010, 24, 1979–1992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kode, A.; Manavalan, J.S.; Mosialou, I.; Bhagat, G.; Rathinam, C.V.; Luo, N.; Khiabanian, H.; Lee, A.; Murty, V.V.; Friedman, R.; et al. Leukaemogenesis induced by an activating β-catenin mutation in osteoblasts. Nature 2014, 506, 240–244. [Google Scholar] [CrossRef] [Green Version]
- Weber, J.M.; Calvi, L.M. Notch signaling and the bone marrow hematopoietic stem cell niche. Bone 2010, 46, 281–285. [Google Scholar] [CrossRef] [Green Version]
- Pajcini, K.V.; Speck, N.A.; Pear, W.S. Notch signaling in mammalian hematopoietic stem cells. Leukemia 2011, 25, 1525–1532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, J.; Cui, L.; Xu, A.; Yin, X.; Li, F.; Gao, J. MEIS1 inhibits clear cell renal cell carcinoma cells proliferation and in vitro invasion or migration. BMC Cancer 2017, 17, 176. [Google Scholar] [CrossRef] [Green Version]
- Song, F.; Wang, H.; Wang, Y. Myeloid ecotropic viral integration site 1 inhibits cell proliferation, invasion or migration in human gastric cancer. Oncotarget 2017, 8, 90050–90060. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Liu, L.; Gao, H.; Pinnamaneni, J.P.; Sanagasetti, D.; Singh, V.P.; Wang, K.; Mathison, M.; Zhang, Q.; Chen, F.; et al. The stem cell factor SALL4 is an essential transcriptional regulator in mixed lineage leukemia-rearranged leukemogenesis. J. Hematol. Oncol. 2017, 10, 159. [Google Scholar] [CrossRef] [Green Version]
- Cillo, C.; Cantile, M.; Faiella, A.; Boncinelli, E. Homeobox genes in normal and malignant cells. J. Cell. Physiol. 2001, 188, 161–169. [Google Scholar] [CrossRef]
- Seifert, A.; Werheid, D.F.; Knapp, S.M.; Tobiasch, E. Role of Hox genes in stem cell differentiation. World J. Stem Cells 2015, 7, 583–595. [Google Scholar] [CrossRef]
- Mahmoudian, R.A.; Forghanifard, M.M. Crosstalk between MEIS1 and markers of different cell signaling pathways in esophageal squamous cell carcinoma. Mol. Biol. Rep. 2020, 47, 3439–3448. [Google Scholar] [CrossRef] [PubMed]
- Chipuk, J.E.; Moldoveanu, T.; Llambi, F.; Parsons, M.J.; Green, D.R. The BCL-2 family reunion. Mol. Cell 2010, 37, 299–310. [Google Scholar] [CrossRef]
- Czabotar, P.E.; Lessene, G.; Strasser, A.; Adams, J.M. Control of apoptosis by the BCL-2 protein family: Implications for physiology and therapy. Nat. Rev. Mol. Cell Biol. 2014, 15, 49–63. [Google Scholar] [CrossRef] [PubMed]
- Akl, H.; Vervloessem, T.; Kiviluoto, S.; Bittremieux, M.; Parys, J.B.; De Smedt, H.; Bultynck, G. A dual role for the anti-apoptotic Bcl-2 protein in cancer: Mitochondria versus endoplasmic reticulum. Biochim. Biophys. Acta 2014, 1843, 2240–2252. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Gao, C.; Lu, J.; Tatetsu, H.; Williams, D.A.; Müller, L.U.; Cui, W.; Chai, L. Leukemic survival factor SALL4 contributes to defective DNA damage repair. Oncogene 2016, 35, 6087–6095. [Google Scholar] [CrossRef] [Green Version]
- Liu, K.F.; Shan, Y.X. Effects of siRNA-mediated silencing of Sal-like 4 expression on proliferation and apoptosis of prostate cancer C4-2 cells. Genet. Mol. Res. GMR 2016, 15. [Google Scholar] [CrossRef]
- Hesari, A.; Anoshiravani, A.A.; Talebi, S.; Noruzi, S.; Mohammadi, R.; Salarinia, R.; Zare, R.; Ghasemi, F. Knockdown of sal-like 4 expression by small interfering RNA induces apoptosis in breast cancer cells. J. Cell Biochem. 2019, 120, 9392–9399. [Google Scholar] [CrossRef] [PubMed]
- Hesari, A.; Rajab, S.; Rezaei, M.; Basam, M.; Golmohamadi, S.; Ghasemi, F. Knockdown of Sal-like 4 expression by siRNA induces apoptosis in colorectal cancer. J. Cell Biochem. 2019. [Google Scholar] [CrossRef]
- Nie, X.; Guo, E.; Wu, C.; Liu, D.; Sun, W.; Zhang, L.; Long, G.; Mei, Q.; Wu, K.; Xiong, H.; et al. SALL4 induces radioresistance in nasopharyngeal carcinoma via the ATM/Chk2/p53 pathway. Cancer Med. 2019, 8, 1779–1792. [Google Scholar] [CrossRef]
- Yang, J.; Chai, L.; Gao, C.; Fowles, T.C.; Alipio, Z.; Dang, H.; Xu, D.; Fink, L.M.; Ward, D.C.; Ma, Y. SALL4 is a key regulator of survival and apoptosis in human leukemic cells. Blood 2008, 112, 805–813. [Google Scholar] [CrossRef] [Green Version]
- Ueno, S.; Lu, J.; He, J.; Li, A.; Zhang, X.; Ritz, J.; Silberstein, L.E.; Chai, L. Aberrant expression of SALL4 in acute B cell lymphoblastic leukemia: Mechanism, function, and implication for a potential novel therapeutic target. Exp. Hematol. 2014, 42, 307–316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, J.; Deng, R.; Zhang, P.; Wu, C.; Wu, K.; Shi, L.; Liu, X.; Bai, J.; Deng, M.; Shuai, X.; et al. miR-219-5p plays a tumor suppressive role in colon cancer by targeting oncogene Sall4. Oncol. Rep. 2015, 34, 1923–1932. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Zou, B.; Liu, L.; Cui, K.; Gao, J.; Yuan, S.; Cong, N. MicroRNA-98 acts as a tumor suppressor in hepatocellular carcinoma via targeting SALL4. Oncotarget 2016, 7, 74059–74073. [Google Scholar] [CrossRef]
- Tian, Q.; Xiao, Y.; Wu, Y.; Liu, Y.; Song, Z.; Gao, W.; Zhang, J.; Yang, J.; Zhang, Y.; Guo, T.; et al. MicroRNA-33b suppresses the proliferation and metastasis of hepatocellular carcinoma cells through the inhibition of Sal-like protein 4 expression. Int. J. Mol. Med. 2016, 38, 1587–1595. [Google Scholar] [CrossRef] [Green Version]
- Jiang, B.; Li, M.; Ji, F.; Nie, Y. MicroRNA-219 exerts a tumor suppressive role in glioma via targeting Sal-like protein 4. Exp. Ther. Med. 2017, 14, 6213–6221. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.P.; Zhang, N.N.; Ren, X.Q.; He, J.; Li, Y. miR-103/miR-195/miR-15b Regulate SALL4 and Inhibit Proliferation and Migration in Glioma. Molecules 2018, 23, 2938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, C.; Han, Q.; Xu, D.; Zheng, B.; Zhao, X.; Zhang, J. SALL4-mediated upregulation of exosomal miR-146a-5p drives T-cell exhaustion by M2 tumor-associated macrophages in HCC. Oncoimmunology 2019, 8, 1601479. [Google Scholar] [CrossRef] [PubMed]
- Naugler, W.E.; Sakurai, T.; Kim, S.; Maeda, S.; Kim, K.; Elsharkawy, A.M.; Karin, M. Gender disparity in liver cancer due to sex differences in MyD88-dependent IL-6 production. Science 2007, 317, 121–124. [Google Scholar] [CrossRef] [Green Version]
- Scharping, N.E.; Delgoffe, G.M. Tumor Microenvironment Metabolism: A New Checkpoint for Anti-Tumor Immunity. Vaccines 2016, 4, 46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kao, C.; Oestreich, K.J.; Paley, M.A.; Crawford, A.; Angelosanto, J.M.; Ali, M.A.; Intlekofer, A.M.; Boss, J.M.; Reiner, S.L.; Weinmann, A.S.; et al. Transcription factor T-bet represses expression of the inhibitory receptor PD-1 and sustains virus-specific CD8+ T cell responses during chronic infection. Nat. Immunol. 2011, 12, 663–671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, B.; Wang, Y.; Tan, X.; Ke, K.; Zheng, X.; Wang, F.; Lan, S.; Liao, N.; Cai, Z.; Shi, Y.; et al. Inflammatory Micro-environment Contributes to Stemness Properties and Metastatic Potential of HCC via the NF-κB/miR-497/SALL4 Axis. Mol. Oncolytics 2019, 15, 79–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, A.; Yang, Y.; Gao, C.; Lu, J.; Jeong, H.W.; Liu, B.H.; Tang, P.; Yao, X.; Neuberg, D.; Huang, G.; et al. A SALL4/MLL/HOXA9 pathway in murine and human myeloid leukemogenesis. J. Clin. Investig. 2013, 123, 4195–4207. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Corsello, T.R.; Ma, Y. Stem cell gene SALL4 suppresses transcription through recruitment of DNA methyltransferases. J. Biol. Chem. 2012, 287, 1996–2005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.; Souto, J.; Liao, W.; Jiang, Y.; Li, Y.; Nishinakamura, R.; Huang, S.; Rosengart, T.; Yang, V.W.; Schuster, M.; et al. Histone lysine-specific demethylase 1 (LSD1) protein is involved in Sal-like protein 4 (SALL4)-mediated transcriptional repression in hematopoietic stem cells. J. Biol. Chem. 2013, 288, 34719–34728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Chai, L.; Fowles, T.C.; Alipio, Z.; Xu, D.; Fink, L.M.; Ward, D.C.; Ma, Y. Genome-wide analysis reveals Sall4 to be a major regulator of pluripotency in murine-embryonic stem cells. Proc. Natl. Acad. Sci. USA 2008, 105, 19756–19761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, C.; Dimitrov, T.; Yong, K.J.; Tatetsu, H.; Jeong, H.W.; Luo, H.R.; Bradner, J.E.; Tenen, D.G.; Chai, L. Targeting transcription factor SALL4 in acute myeloid leukemia by interrupting its interaction with an epigenetic complex. Blood 2013, 121, 1413–1421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dou, Y.; Milne, T.A.; Ruthenburg, A.J.; Lee, S.; Lee, J.W.; Verdine, G.L.; Allis, C.D.; Roeder, R.G. Regulation of MLL1 H3K4 methyltransferase activity by its core components. Nat. Struct. Mol. Biol. 2006, 13, 713–719. [Google Scholar] [CrossRef]
- Campos-Sanchez, E.; Deleyto-Seldas, N.; Dominguez, V.; Carrillo-de-Santa-Pau, E.; Ura, K.; Rocha, P.P.; Kim, J.; Aljoufi, A.; Esteve-Codina, A.; Dabad, M.; et al. Wolf-Hirschhorn Syndrome Candidate 1 Is Necessary for Correct Hematopoietic and B Cell Development. Cell Rep. 2017, 19, 1586–1601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nimura, K.; Ura, K.; Shiratori, H.; Ikawa, M.; Okabe, M.; Schwartz, R.J.; Kaneda, Y. A histone H3 lysine 36 trimethyltransferase links Nkx2-5 to Wolf-Hirschhorn syndrome. Nature 2009, 460, 287–291. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, A.T.; Zhang, Y. The diverse functions of Dot1 and H3K79 methylation. Genes Dev. 2011, 25, 1345–1358. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Liu, L.; Leung, L.H.; Cooney, A.J.; Chen, C.; Rosengart, T.K.; Ma, Y.; Yang, J. Knockdown of SALL4 Protein Enhances All-trans Retinoic Acid-induced Cellular Differentiation in Acute Myeloid Leukemia Cells. J. Biol. Chem. 2015, 290, 10599–10609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wouters, B.J.; Delwel, R. Epigenetics and approaches to targeted epigenetic therapy in acute myeloid leukemia. Blood 2016, 127, 42–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallipoli, P.; Giotopoulos, G.; Huntly, B.J. Epigenetic regulators as promising therapeutic targets in acute myeloid leukemia. Ther. Adv. Hematol. 2015, 6, 103–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernt, K.M.; Armstrong, S.A. Targeting epigenetic programs in MLL-rearranged leukemias. Hematol. Am. Soc. Hematology. Educ. Program 2011, 2011, 354–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saygin, C.; Carraway, H.E. Emerging therapies for acute myeloid leukemia. J. Hematol. Oncol. 2017, 10, 93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, Y.; Wu, F.; Wu, J. Targeting histone methylation for cancer therapy: Enzymes, inhibitors, biological activity and perspectives. J. Hematol. Oncol. 2016, 9, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shain, A.H.; Bastian, B.C. From melanocytes to melanomas. Nat. Rev. Cancer 2016, 16, 345–358. [Google Scholar] [CrossRef]
- Rice, K.L.; Hormaeche, I.; Licht, J.D. Epigenetic regulation of normal and malignant hematopoiesis. Oncogene 2007, 26, 6697–6714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goyama, S.; Kitamura, T. Epigenetics in normal and malignant hematopoiesis: An overview and update 2017. Cancer Sci. 2017, 108, 553–562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, L.W.; Sun, Q.Y.; Tan, K.T.; Chien, W.; Mayakonda, A.; Yeoh, A.E.J.; Kawamata, N.; Nagata, Y.; Xiao, J.F.; Loh, X.Y.; et al. Mutational Landscape of Pediatric Acute Lymphoblastic Leukemia. Cancer Res. 2017, 77, 390–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, Z.; Yao, Y.; Zhou, C.; Chen, F.; Wu, F.; Wei, L.; Liu, W.; Dong, S.; Redell, M.; Mo, Q.; et al. Pharmacological inhibition of LSD1 for the treatment of MLL-rearranged leukemia. J. Hematol. Oncol. 2016, 9, 24. [Google Scholar] [CrossRef] [Green Version]
- Aguila, J.R.; Liao, W.; Yang, J.; Avila, C.; Hagag, N.; Senzel, L.; Ma, Y. SALL4 is a robust stimulator for the expansion of hematopoietic stem cells. Blood 2011, 118, 576–585. [Google Scholar] [CrossRef] [Green Version]
- Shen, B.; Zhang, Y.; Dai, W.; Ma, Y.; Jiang, Y. Ex-vivo expansion of nonhuman primate CD34(+) cells by stem cell factor Sall4B. Stem Cell Res. Ther. 2016, 7, 152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mossahebi-Mohammadi, M.; Atashi, A.; Kaviani, S.; Soleimani, M. Efficient Expansion of SALL4-Transduced Umbilical Cord Blood Derived CD133+Hematopoietic Stem Cells. Acta Med. Iran. 2017, 55, 290–296. [Google Scholar] [PubMed]
- Milanovich, S.; Peterson, J.; Allred, J.; Stelloh, C.; Rajasekaran, K.; Fisher, J.; Duncan, S.A.; Malarkannan, S.; Rao, S. Sall4 overexpression blocks murine hematopoiesis in a dose-dependent manner. Exp. Hematol. 2015, 43, 53–64.e51-58. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Tam, W.L.; Tong, G.Q.; Wu, Q.; Chan, H.Y.; Soh, B.S.; Lou, Y.; Yang, J.; Ma, Y.; Chai, L.; et al. Sall4 modulates embryonic stem cell pluripotency and early embryonic development by the transcriptional regulation of Pou5f1. Nat. Cell Biol. 2006, 8, 1114–1123. [Google Scholar] [CrossRef] [PubMed]
- Misawa, K.; Misawa, Y.; Mima, M.; Yamada, S.; Imai, A.; Mochizuki, D.; Nakagawa, T.; Kurokawa, T.; Endo, S.; Kawasaki, H.; et al. Overexpression of Sal-like protein 4 in head and neck cancer: Epigenetic effects and clinical correlations. Cell. Oncol. (Dordr. ) 2020, 43, 631–641. [Google Scholar] [CrossRef]
- Tan, J.L.; Li, F.; Yeo, J.Z.; Yong, K.J.; Bassal, M.A.; Ng, G.H.; Lee, M.Y.; Leong, C.Y.; Tan, H.K.; Wu, C.S.; et al. New High-Throughput Screening Identifies Compounds That Reduce Viability Specifically in Liver Cancer Cells That Express High Levels of SALL4 by Inhibiting Oxidative Phosphorylation. Gastroenterology 2019, 157, 1615–1629.e1617. [Google Scholar] [CrossRef] [Green Version]
- Xie, J.; Ruan, S.; Zhu, Z.; Wang, M.; Cao, Y.; Ou, M.; Yu, P.; Shi, J. Database mining analysis revealed the role of the putative H(+)/sugar transporter solute carrier family 45 in skin cutaneous melanoma. Channels 2021, 15, 496–506. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.N.; Halling-Brown, M.D.; Tym, J.E.; Workman, P.; Al-Lazikani, B. Objective assessment of cancer genes for drug discovery. Nat. Rev. Drug Discov. 2013, 12, 35–50. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Thorne, N.; McKew, J.C. Phenotypic screens as a renewed approach for drug discovery. Drug Discov. Today 2013, 18, 1067–1073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilding, J.L.; Bodmer, W.F. Cancer cell lines for drug discovery and development. Cancer Res. 2014, 74, 2377–2384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, C.; Lan, P.; Han, Q.; Huang, M.; Zhang, Z.; Xu, G.; Song, J.; Wang, J.; Wei, H.; Zhang, J.; et al. Oncofetal gene SALL4 reactivation by hepatitis B virus counteracts miR-200c in PD-L1-induced T cell exhaustion. Nat. Commun. 2018, 9, 1241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirsch, F.R.; Scagliotti, G.V.; Mulshine, J.L.; Kwon, R.; Curran, W.J., Jr.; Wu, Y.L.; Paz-Ares, L. Lung cancer: Current therapies and new targeted treatments. Lancet (Lond. Engl. ) 2017, 389, 299–311. [Google Scholar] [CrossRef]
- Jemal, A.; Siegel, R.; Xu, J.; Ward, E. Cancer statistics, 2010. CA A Cancer J. Clin. 2010, 60, 277–300. [Google Scholar] [CrossRef] [PubMed]
- DeSantis, C.E.; Lin, C.C.; Mariotto, A.B.; Siegel, R.L.; Stein, K.D.; Kramer, J.L.; Alteri, R.; Robbins, A.S.; Jemal, A. Cancer treatment and survivorship statistics, 2014. CA A Cancer J. Clin. 2014, 64, 252–271. [Google Scholar] [CrossRef] [PubMed]
- Jemal, A.; Bray, F.; Center, M.M.; Ferlay, J.; Ward, E.; Forman, D. Global cancer statistics. CA A Cancer J. Clin. 2011, 61, 69–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez-Canales, J.; Parra-Cuentas, E.; Wistuba, I.I. Diagnosis and Molecular Classification of Lung Cancer. Cancer Treat. Res. 2016, 170, 25–46. [Google Scholar] [CrossRef]
- Bagchi, S.; Yuan, R.; Engleman, E.G. Immune Checkpoint Inhibitors for the Treatment of Cancer: Clinical Impact and Mechanisms of Response and Resistance. Annu. Rev. Pathol. 2021, 16, 223–249. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, D.; Kuribayashi, K.; Tanaka, M.; Watanabe, N. Overexpression of SALL4 in lung cancer and its importance in cell proliferation. Oncol. Rep. 2011, 26, 965–970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Zhang, Y.; Tao, X.; You, Q.; Tao, Z.; Zhang, Y.; He, Z.; Ou, J. Knockdown of SALL4 inhibits the proliferation, migration, and invasion of human lung cancer cells in vivo and in vitro. Ann. Transl. Med. 2020, 8, 1678. [Google Scholar] [CrossRef] [PubMed]
- Jia, X.; Qian, R.; Zhang, B.; Zhao, S. The expression of SALL4 is significantly associated with EGFR, but not KRAS or EML4-ALK mutations in lung cancer. J. Thorac. Dis. 2016, 8, 2682–2688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yong, K.J.; Li, A.; Ou, W.B.; Hong, C.K.; Zhao, W.; Wang, F.; Tatetsu, H.; Yan, B.; Qi, L.; Fletcher, J.A.; et al. Targeting SALL4 by entinostat in lung cancer. Oncotarget 2016, 7, 75425–75440. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Sauer, M.A.; Hussein, S.G.; Yang, J.; Tenen, D.G.; Chai, L. SALL4 and microRNA: The Role of Let-7. Genes 2021, 12, 1301. [Google Scholar] [CrossRef]
- Xia, H.; Niu, Q.; Ding, Y.; Zhang, Z.; Yuan, J.; Jin, W. Long noncoding HOXA11-AS knockdown suppresses the progression of non-small cell lung cancer by regulating miR-3619-5p/SALL4 axis. J. Mol. Histol. 2021, 52, 729–740. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef]
- Sun, X.; Zhuo, X.B.; Hu, Y.P.; Zheng, X.; Zhao, Q.J. A novel matrine derivative WM622 inhibits hepatocellular carcinoma by inhibiting PI3K/AKT signaling pathways. Mol. Cell. Biochem. 2018, 449, 47–54. [Google Scholar] [CrossRef]
- Beck, M.; Schirmacher, P.; Singer, S. Alterations of the nuclear transport system in hepatocellular carcinoma—New basis for therapeutic strategies. J. Hepatol. 2017, 67, 1051–1061. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Liang, N.; Yang, T.; Li, Y.; Li, J.; Huang, Q.; Wu, C.; Sun, L.; Zhou, X.; Cheng, X.; et al. DNMT1-mediated methylation of BEX1 regulates stemness and tumorigenicity in liver cancer. J. Hepatol. 2021. [Google Scholar] [CrossRef]
- Yong, K.J.; Gao, C.; Lim, J.S.; Yan, B.; Yang, H.; Dimitrov, T.; Kawasaki, A.; Ong, C.W.; Wong, K.F.; Lee, S.; et al. Oncofetal gene SALL4 in aggressive hepatocellular carcinoma. N. Engl. J. Med. 2013, 368, 2266–2276. [Google Scholar] [CrossRef] [Green Version]
- Fitzmorris, P.; Shoreibah, M.; Anand, B.S.; Singal, A.K. Management of hepatocellular carcinoma. J. Cancer Res. Clin. Oncol. 2015, 141, 861–876. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.S.; Liu, J.B.; Lin, L.; Zhang, H.; Wu, J.J.; Shi, Y.; Jia, C.Y.; Zhang, D.D.; Yu, F.; Wang, H.M.; et al. Exosomal microRNA-15a from mesenchymal stem cells impedes hepatocellular carcinoma progression via downregulation of SALL4. Cell Death Discov. 2021, 7, 224. [Google Scholar] [CrossRef] [PubMed]
- Fan, H.; Cui, Z.; Zhang, H.; Mani, S.K.; Diab, A.; Lefrancois, L.; Fares, N.; Merle, P.; Andrisani, O. DNA demethylation induces SALL4 gene re-expression in subgroups of hepatocellular carcinoma associated with Hepatitis B or C virus infection. Oncogene 2017, 36, 2435–2445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, M.; Sun, L.; Lai, J.Z.; Shi, H.; Mei, K.; He, X.; Jin, X.; Lai, J.; Cao, D. Expression of RNA-binding protein LIN28 in classic gastric hepatoid carcinomas, gastric fetal type gastrointestinal adenocarcinomas, and hepatocellular carcinomas: An immunohistochemical study with comparison to SALL4, alpha-fetoprotein, glypican-3, and Hep Par1. Pathol. Res. Pract. 2018, 214, 1707–1712. [Google Scholar] [CrossRef]
- Nakra, T.; Roy, M.; Yadav, R.; Agarwala, S.; Jassim, M.; Khanna, G.; Das, P.; Jain, D.; Mathur, S.R.; Iyer, V.K. Cytomorphology of hepatoblastoma with histological correlation and role of SALL4 immunocytochemistry in its diagnosis, subtyping, and prognostication. Cancer Cytopathol. 2020, 128, 190–200. [Google Scholar] [CrossRef]
- Azamjah, N.; Soltan-Zadeh, Y.; Zayeri, F. Global Trend of Breast Cancer Mortality Rate: A 25-Year Study. Asian Pac. J. Cancer Prev. 2019, 20, 2015–2020. [Google Scholar] [CrossRef]
- Parrella, P. Epigenetic Signatures in Breast Cancer: Clinical Perspective. Breast Care 2010, 5, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.; DeSantis, C.; Virgo, K.; Stein, K.; Mariotto, A.; Smith, T.; Cooper, D.; Gansler, T.; Lerro, C.; Fedewa, S.; et al. Cancer treatment and survivorship statistics, 2012. CA A Cancer J. Clin. 2012, 62, 220–241. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.K.; Singh, S.; Lillard, J.W., Jr.; Singh, R. Drug delivery approaches for breast cancer. Int. J. Nanomed. 2017, 12, 6205–6218. [Google Scholar] [CrossRef] [Green Version]
- Xue, H.Y.; Wong, H.L. Targeting megalin to enhance delivery of anti-clusterin small-interfering RNA nanomedicine to chemo-treated breast cancer. Eur. J. Pharm. Biopharm. Off. J. Arb. Fur Pharm. Verfahr. E.V 2012, 81, 24–32. [Google Scholar] [CrossRef]
- Chen, Y.Y.; Li, Z.Z.; Ye, Y.Y.; Xu, F.; Niu, R.J.; Zhang, H.C.; Zhang, Y.J.; Liu, Y.B.; Han, B.S. Knockdown of SALL4 inhibits the proliferation and reverses the resistance of MCF-7/ADR cells to doxorubicin hydrochloride. BMC Mol. Biol. 2016, 17, 6. [Google Scholar] [CrossRef] [Green Version]
- Noruzi, S.; Vatanchian, M.; Azimian, A.; Niroomand, A.; Salarinia, R.; Oroojalian, F. Silencing SALL-4 Gene by Transfecting Small Interfering RNA with Targeted Aminoglycoside-Carboxyalkyl Polyethylenimine Nano-Polyplexes Reduced Migration of MCF-7 Breast Cancer Cells. Avicenna J. Med. Biotechnol. 2021, 13, 2–8. [Google Scholar] [CrossRef]
- Chen, T.; Tsang, J.Y.S.; Su, X.C.; Li, P.; Sun, W.Q.; Wong, I.L.K.; Choy, K.Y.; Yang, Q.; Tse, G.M.K.; Chan, T.H.; et al. SALL4 promotes tumor progression in breast cancer by targeting EMT. Mol. Carcinog. 2020, 59, 1209–1226. [Google Scholar] [CrossRef] [PubMed]
- Boustan, A.; Mosaffa, F.; Jahangiri, R.; Heidarian-Miri, H.; Dahmardeh-Ghalehno, A.; Jamialahmadi, K. Role of SALL4 and Nodal in the prognosis and tamoxifen resistance of estrogen receptor-positive breast cancer. Mol. Biol. Res. Commun. 2021, 10, 109–119. [Google Scholar] [CrossRef]
- Itou, J.; Li, W.; Ito, S.; Tanaka, S.; Matsumoto, Y.; Sato, F.; Toi, M. Sal-like 4 protein levels in breast cancer cells are post-translationally down-regulated by tripartite motif-containing 21. J. Biol. Chem. 2018, 293, 6556–6564. [Google Scholar] [CrossRef] [Green Version]
- Lo, P.K.; Wolfson, B.; Zhou, Q. Cancer stem cells and early stage basal-like breast cancer. World J. Obstet. Gynecol. 2016, 5, 150–161. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Zuo, X.; Wei, D. Concise Review: Emerging Role of CD44 in Cancer Stem Cells: A Promising Biomarker and Therapeutic Target. Stem Cells Transl. Med. 2015, 4, 1033–1043. [Google Scholar] [CrossRef]
- Louderbough, J.M.; Schroeder, J.A. Understanding the dual nature of CD44 in breast cancer progression. Mol. Cancer Res. MCR 2011, 9, 1573–1586. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, Y.; Itou, J.; Sato, F.; Toi, M. SALL4—KHDRBS3 network enhances stemness by modulating CD44 splicing in basal-like breast cancer. Cancer Med. 2018, 7, 454–462. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA A Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kudou, K.; Saeki, H.; Nakashima, Y.; Sasaki, S.; Jogo, T.; Hirose, K.; Hu, Q.; Tsuda, Y.; Kimura, K.; Nakanishi, R.; et al. Postoperative development of sarcopenia is a strong predictor of a poor prognosis in patients with adenocarcinoma of the esophagogastric junction and upper gastric cancer. Am. J. Surg. 2019, 217, 757–763. [Google Scholar] [CrossRef] [PubMed]
- Araki, I.; Washio, M.; Yamashita, K.; Hosoda, K.; Ema, A.; Mieno, H.; Moriya, H.; Katada, N.; Kikuchi, S.; Watanabe, M. Robust vascular invasion concurrent with intense EGFR immunostaining can predict recurrence in patients with stage IB node-negative gastric cancer. Surg. Today 2018, 48, 478–485. [Google Scholar] [CrossRef]
- Cats, A.; Jansen, E.P.M.; van Grieken, N.C.T.; Sikorska, K.; Lind, P.; Nordsmark, M.; Meershoek-Klein Kranenbarg, E.; Boot, H.; Trip, A.K.; Swellengrebel, H.A.M.; et al. Chemotherapy versus chemoradiotherapy after surgery and preoperative chemotherapy for resectable gastric cancer (CRITICS): An international, open-label, randomised phase 3 trial. Lancet. Oncol. 2018, 19, 616–628. [Google Scholar] [CrossRef]
- Zhang, L.; Xu, Z.; Xu, X.; Zhang, B.; Wu, H.; Wang, M.; Zhang, X.; Yang, T.; Cai, J.; Yan, Y.; et al. SALL4, a novel marker for human gastric carcinogenesis and metastasis. Oncogene 2014, 33, 5491–5500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yanagihara, N.; Kobayashi, D.; Kuribayashi, K.; Tanaka, M.; Hasegawa, T.; Watanabe, N. Significance of SALL4 as a drug-resistant factor in lung cancer. Int. J. Oncol. 2015, 46, 1527–1534. [Google Scholar] [CrossRef] [Green Version]
- Pan, L.; Liang, W.; Gu, J.; Zang, X.; Huang, Z.; Shi, H.; Chen, J.; Fu, M.; Zhang, P.; Xiao, X.; et al. Long noncoding RNA DANCR is activated by SALL4 and promotes the proliferation and invasion of gastric cancer cells. Oncotarget 2018, 9, 1915–1930. [Google Scholar] [CrossRef] [Green Version]
- Kretz, M.; Webster, D.E.; Flockhart, R.J.; Lee, C.S.; Zehnder, A.; Lopez-Pajares, V.; Qu, K.; Zheng, G.X.; Chow, J.; Kim, G.E.; et al. Suppression of progenitor differentiation requires the long noncoding RNA ANCR. Genes Dev. 2012, 26, 338–343. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Zhang, M.; Liang, L.; Li, J.; Chen, Y.X. Over-expression of lncRNA DANCR is associated with advanced tumor progression and poor prognosis in patients with colorectal cancer. Int. J. Clin. Exp. Pathol. 2015, 8, 11480–11484. [Google Scholar]
- Yuan, X.; Zhang, X.; Zhang, W.; Liang, W.; Zhang, P.; Shi, H.; Zhang, B.; Shao, M.; Yan, Y.; Qian, H.; et al. SALL4 promotes gastric cancer progression through activating CD44 expression. Oncogenesis 2016, 5, e268. [Google Scholar] [CrossRef]
- Jiang, X.; Wang, Z. miR-16 targets SALL4 to repress the proliferation and migration of gastric cancer. Oncol. Lett. 2018, 16, 3005–3012. [Google Scholar] [CrossRef]
- Sato, T.; Stange, D.E.; Ferrante, M.; Vries, R.G.; Van Es, J.H.; Van den Brink, S.; Van Houdt, W.J.; Pronk, A.; Van Gorp, J.; Siersema, P.D.; et al. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett’s epithelium. Gastroenterology 2011, 141, 1762–1772. [Google Scholar] [CrossRef] [PubMed]
- Wolpin, B.M.; Mayer, R.J. Systemic treatment of colorectal cancer. Gastroenterology 2008, 134, 1296–1310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forghanifard, M.M.; Moghbeli, M.; Raeisossadati, R.; Tavassoli, A.; Mallak, A.J.; Boroumand-Noughabi, S.; Abbaszadegan, M.R. Role of SALL4 in the progression and metastasis of colorectal cancer. J. Biomed. Sci. 2013, 20, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, J.; Deng, R.; Wu, C.; Zhang, P.; Wu, K.; Shi, L.; Liu, X.; Bai, J.; Deng, M.; Gao, J.; et al. Inhibition of SALL4 suppresses carcinogenesis of colorectal cancer via regulating Gli1 expression. Int. J. Clin. Exp. Pathol. 2015, 8, 10092–10101. [Google Scholar] [PubMed]
- Ardalan Khales, S.; Abbaszadegan, M.R.; Abdollahi, A.; Raeisossadati, R.; Tousi, M.F.; Forghanifard, M.M. SALL4 as a new biomarker for early colorectal cancers. J. Cancer Res. Clin. Oncol. 2015, 141, 229–235. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Zheng, R.; Baade, P.D.; Zhang, S.; Zeng, H.; Bray, F.; Jemal, A.; Yu, X.Q.; He, J. Cancer statistics in China, 2015. CA A Cancer J. Clin. 2016, 66, 115–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bacci, G.; Briccoli, A.; Rocca, M.; Ferrari, S.; Donati, D.; Longhi, A.; Bertoni, F.; Bacchini, P.; Giacomini, S.; Forni, C.; et al. Neoadjuvant chemotherapy for osteosarcoma of the extremities with metastases at presentation: Recent experience at the Rizzoli Institute in 57 patients treated with cisplatin, doxorubicin, and a high dose of methotrexate and ifosfamide. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2003, 14, 1126–1134. [Google Scholar] [CrossRef]
- Kager, L.; Zoubek, A.; Pötschger, U.; Kastner, U.; Flege, S.; Kempf-Bielack, B.; Branscheid, D.; Kotz, R.; Salzer-Kuntschik, M.; Winkelmann, W.; et al. Primary metastatic osteosarcoma: Presentation and outcome of patients treated on neoadjuvant Cooperative Osteosarcoma Study Group protocols. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2003, 21, 2011–2018. [Google Scholar] [CrossRef] [PubMed]
- Meyers, P.A.; Heller, G.; Healey, J.H.; Huvos, A.; Applewhite, A.; Sun, M.; LaQuaglia, M. Osteogenic sarcoma with clinically detectable metastasis at initial presentation. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 1993, 11, 449–453. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Jiang, F.; Wang, X.; Li, G. Knockdown of SALL4 Inhibits Proliferation, Migration, and Invasion in Osteosarcoma Cells. Oncol. Res. 2017, 25, 763–771. [Google Scholar] [CrossRef]
- Wang, F.; Lv, H.; Zhao, B.; Zhou, L.; Wang, S.; Luo, J.; Liu, J.; Shang, P. Iron and leukemia: New insights for future treatments. J. Exp. Clin. Cancer Res. CR 2019, 38, 406. [Google Scholar] [CrossRef] [Green Version]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA A Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [Green Version]
- Farawela, H.M.; Zawam, H.M.; Al-Wakeel, H.A.; El-Nagdy, M.H.; El-Refaey, F.A.; Abdel-Rahman, H.A. Expression pattern and prognostic implication of SALL4 gene in myeloid leukemias: A case-control study. Scand. J. Clin. Lab. Invest. 2019, 79, 65–70. [Google Scholar] [CrossRef]
- Ibraheem, F.M.; Badawy, R.; Ayoub, M.A.; Hassan, N.M.; Mostafa, M.N. SALL4 Gene Expression in Acute Myeloid Leukemia. Asian Pac. J. Cancer Prev. 2019, 20, 3121–3127. [Google Scholar] [CrossRef]
- Sanz, M.A.; Iacoboni, G.; Montesinos, P.; Venditti, A. Emerging strategies for the treatment of older patients with acute myeloid leukemia. Ann. Hematol. 2016, 95, 1583–1593. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, R.C.; Rego, E. Management of APL in developing countries: Epidemiology, challenges and opportunities for international collaboration. Hematol. Am. Soc. Hematology. Educ. Program. 2006, 162–168. [Google Scholar] [CrossRef] [Green Version]
- Sheikhrezaei, Z.; Heydari, P.; Farsinezhad, A.; Fatemi, A.; Khanamani Falahati-Pour, S.; Darakhshan, S.; Noroozi Karimabad, M.; Darekordi, A.; Khorramdelazad, H.; Hassanshahi, G. A New Indole Derivative Decreased SALL4 Gene Expression in Acute Promyelocytic Leukemia Cell Line (NB4). Iran. Biomed. J. 2018, 22, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Swelem, R.S.; Elneely, D.A.; Shehata, A.A.R. The Study of SALL4 Gene and BMI-1 Gene Expression in Acute Myeloid Leukemia Patients. Lab. Med. 2020, 51, 265–270. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Kong, N.R.; Chai, L. The role of stem cell factor SALL4 in leukemogenesis. Crit. Rev. Oncog. 2011, 16, 117–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, J.; Ma, Y.; Kong, N.; Alipio, Z.; Gao, C.; Krause, D.S.; Silberstein, L.E.; Chai, L. Dissecting the role of SALL4, a newly identified stem cell factor, in chronic myelogenous leukemia. Leukemia 2011, 25, 1211–1213. [Google Scholar] [CrossRef] [PubMed]
- American Cancer Society Cancer Statistics 2021 Report. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2021, 62, 12n.
- Peres, L.C.; Cushing-Haugen, K.L.; Anglesio, M.; Wicklund, K.; Bentley, R.; Berchuck, A.; Kelemen, L.E.; Nazeran, T.M.; Gilks, C.B.; Harris, H.R.; et al. Histotype classification of ovarian carcinoma: A comparison of approaches. Gynecol. Oncol. 2018, 151, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Desai, A.; Xu, J.; Aysola, K.; Qin, Y.; Okoli, C.; Hariprasad, R.; Chinemerem, U.; Gates, C.; Reddy, A.; Danner, O.; et al. Epithelial ovarian cancer: An overview. World J. Transl. Med. 2014, 3, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Ueland, F.R. A Perspective on Ovarian Cancer Biomarkers: Past, Present and Yet-To-Come. Diagnostic 2017, 7, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muinao, T.; Deka Boruah, H.P.; Pal, M. Multi-biomarker panel signature as the key to diagnosis of ovarian cancer. Heliyon 2019, 5, e02826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharbatoghli, M.; Shamshiripour, P.; Fattahi, F.; Kalantari, E.; Habibi Shams, Z.; Panahi, M.; Totonchi, M.; Asadi-Lari, Z.; Madjd, Z.; Saeednejad Zanjani, L. Co-expression of cancer stem cell markers, SALL4/ALDH1A1, is associated with tumor aggressiveness and poor survival in patients with serous ovarian carcinoma. J. Ovarian Res. 2022, 15, 17. [Google Scholar] [CrossRef]
- Yang, M.; Xie, X.; Ding, Y. SALL4 is a marker of poor prognosis in serous ovarian carcinoma promoting invasion and metastasis. Oncol. Rep. 2016, 35, 1796–1806. [Google Scholar] [CrossRef]
- Penumatsa, K.; Edassery, S.L.; Barua, A.; Bradaric, M.J.; Luborsky, J.L. Differential expression of aldehyde dehydrogenase 1a1 (ALDH1) in normal ovary and serous ovarian tumors. J. Ovarian Res. 2010, 3, 28. [Google Scholar] [CrossRef] [Green Version]
- Atabiekov, I.; Hobeika, E.; Sheikh, U.; El Andaloussi, A.; Al-Hendy, A. The Role of Gene Therapy in Premature Ovarian Insufficiency Management. Biomedicines 2018, 6, 102. [Google Scholar] [CrossRef] [Green Version]
- Webber, L.; Davies, M.; Anderson, R.; Bartlett, J.; Braat, D.; Cartwright, B.; Cifkova, R.; de Muinck Keizer-Schrama, S.; Hogervorst, E.; Janse, F.; et al. ESHRE Guideline: Management of women with premature ovarian insufficiency. Hum. Reprod. 2016, 31, 926–937. [Google Scholar] [CrossRef] [Green Version]
- Sweetman, D.; Münsterberg, A. The vertebrate spalt genes in development and disease. Dev. Biol. 2006, 293, 285–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Li, D.; Cai, B.; Chen, Q.; Li, C.; Wu, Y.; Jin, L.; Wang, X.; Zhang, X.; Zhang, F. Whole-exome sequencing reveals SALL4 variants in premature ovarian insufficiency: An update on genotype-phenotype correlations. Hum. Genet. 2019, 138, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2015. CA A Cancer J. Clin. 2015, 65, 5–29. [Google Scholar] [CrossRef] [PubMed]
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012. CA A Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [Green Version]
- Ludwig, K.; Kornblum, H.I. Molecular markers in glioma. J. Neuro-Oncol. 2017, 134, 505–512. [Google Scholar] [CrossRef]
- Jia, Z.; Wang, K.; Wang, G.; Zhang, A.; Pu, P. MiR-30a-5p antisense oligonucleotide suppresses glioma cell growth by targeting SEPT7. PLoS ONE 2013, 8, e55008. [Google Scholar] [CrossRef]
- An, L.; Liu, Y.; Wu, A.; Guan, Y. microRNA-124 inhibits migration and invasion by down-regulating ROCK1 in glioma. PLoS ONE 2013, 8, e69478. [Google Scholar] [CrossRef]
- Zhou, Y.; Peng, Y.; Liu, M.; Jiang, Y. MicroRNA-181b Inhibits Cellular Proliferation and Invasion of Glioma Cells via Targeting Sal-Like Protein 4. Oncol. Res. 2017, 25, 947–957. [Google Scholar] [CrossRef]
- Xia, Z.; Qiu, D.; Deng, J.; Jiao, X.; Yang, R.; Sun, Z.; Wan, X.; Li, J. Methylation-induced downregulation and tumor-suppressive role of microRNA-98 in glioma through targeting Sal-like protein 4. Int. J. Mol. Med. 2018, 41, 2651–2659. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Yan, Y.; Jiang, Y.; Qian, J.; Jiang, L.; Hu, G.; Lu, Y.; Luo, C. Knockdown of SALL4 expression using RNA interference induces cell cycle arrest, enhances early apoptosis, inhibits invasion and increases chemosensitivity to temozolomide in U251 glioma cells. Oncol. Lett. 2017, 14, 4263–4269. [Google Scholar] [CrossRef]
- Di, C.; Sun, J.; Zhang, H.; Zhou, P.; Kong, J. High expression of SALL4 is associated with poor prognosis in squamous cell carcinoma of the uterine cervix. Int. J. Clin. Exp. Pathol. 2018, 11, 1391–1398. [Google Scholar] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SALL4 Family Member | Function in Tumors | Mechanism | References |
|---|---|---|---|
| SALL1 | Anti-proliferative effects in glioma | Inhibit Wnt/β-catenin signaling | [11] |
| Induce angiogenesis | Active the VEGF-A gene | [12] | |
| SALL2 | Tumor suppressor | Inhibit c-Myc gene | [13] |
| SALL3 | Promote tumorigenesis | SALL3 promoter hypermethylation | [3] |
| SALL4 | Oncogene in glioma | Induce epithelial–mesenchymal transition | [14] |
| Promote progression in lung cancer | Activate EGFR by ERK1/2 signaling | [15] | |
| Promote proliferation, migration, and invasion in breast cancer | Activate Wnt/β-catenin signaling | [16] | |
| Promote growth and invasion in osteosarcoma | Sponge miR-107 | [17] | |
| Promote invasiveness in melanoma | Interact with the HDAC 2 and bind to invasiveness genes | [18] | |
| Promote development in hepatocellular carcinoma | Activate Wnt/β-catenin signaling | [19] | |
| Promote metastasis in gastric cancer | Activate the TGF-β/SMAD signaling | [20] |
| Abbreviation | Full Name |
|---|---|
| ALDH1 | aldehyde dehydrogenase 1 |
| ALL | acute lymphoblastic leukemia |
| AML | acute myeloid leukemia |
| APL | acute promyelocytic leukemia |
| AXIN2 | axis inhibition protein |
| B-ALL | B-cell lymphoblastic leukemia |
| Bax | Bcl-like-protein 4 |
| BC | breast cancer |
| Bcl-2 | B-cell lymphoma 2 |
| BM | bone marrow |
| Bmi-1 | B cell-specific Moloney murine leukemia virus integration site 1 |
| ccRCC | clear cell renal cell carcinoma |
| CEA | carcinoembryonic antigen |
| CLL | chronic lymphoblastic leukemia |
| CML | chronic myeloid leukemia |
| CRC | colorectal cancer |
| CTLA-4 | cytotoxic-T-lymphocyte-antigen-4 |
| DFS | disease-free survival |
| DLL1 | delta-like 1 |
| DNMTs | DNA methyltransferases |
| DNMT3A | DNA methyltransferase 3 alpha |
| DOT1 L | disruptor of telomeric silencing 1-like |
| DSS | disease-specific survival |
| EMT | epithelial–mesenchymal transition |
| EOC | epithelial ovarian carcinoma |
| ERK1/2 | extracellular signal-regulated kinase 1/2 |
| ESCC | esophageal squamous cell carcinoma |
| ESCs | embryonic stem cells |
| GBM | glioblastoma multiforme |
| GC | gastric cancer |
| GHCs | gastric hepatoid carcinomas |
| HB | hepatoblastoma |
| HCC | hepatocellular carcinoma |
| HDAC | high histone deacetylase |
| HE4 | human epididymis protein 4 |
| HEY2 | hairy/enhancer of split related to YRPW motif family member 2 |
| HK-2 | exokinase II |
| HNSCC | neck squamous cell carcinoma |
| HOXA9 | homeobox A9 |
| HOXA11-AS | homeobox A11 antisense |
| HPC | hematopoietic progenitor cell |
| HSC | hematopoietic stem cell |
| ICC | intrahepatic cholangiocarcinoma |
| IFN-γ | TNF-α and interferon-γ |
| IL-2 | interleukin-2 |
| KDMs | histone 3 lysine 9-specific demethylases |
| LSD1 | lysine-specific demethylase 1 |
| MAML1 | mastermind-like transcriptional coactivator 1 |
| MBD2 | methyl-CpG-binding domain 2 protein |
| MDS | myelodysplastic syndrome |
| MEIS1 | myeloid ecotropic viral insertion site 1 |
| miRNA/miR | microRNA |
| MLL | mixed lineage leukemia |
| NF-κB | transcription factor nuclear factor κB |
| NPC | nasopharyngeal carcinoma |
| NSCLC | non-small cell lung cancer |
| NuRD | nucleosome remodeling deacetylase |
| OC | ovarian cancer |
| OCT4 | octamer-binding transcription factor 4 |
| OS | osteosarcoma |
| OXPHOS | oxidative phosphorylation |
| PD-1 | programmed death ligand 1 |
| PFS | progression-free survival |
| POI | premature ovarian insufficiency |
| POLII | RNA polymerase II |
| PRC | polycomb repressive complex |
| PTEN | phosphatase and tension homolog |
| ROMA | risk of ovarian malignancy algorithm |
| SALLs | spalt-like transcription factors |
| SALL4TSS | SALL4 transcriptional start site TSS |
| SCLC | small cell lung cancer |
| SOC | serous ovarian carcinoma |
| SOX2 | sex-determining region Y (SRY)-Box 2 |
| STAT3 | activator of transcription 3 |
| TAMR | tamoxifen-resistant |
| TNF-α | tumor necrosis factor alpha |
| TNM | tumor node metastasis |
| TRIB3 | tribbles pseudokinase 3 |
| TRIM21 | tripartite motif-containing 21 |
| UTR | untranslated region |
| WHSC1 | Wolf–Hirschhorn syndrome candidate gene-1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, B.; Xu, L.; Bi, W.; Ou, W.-B. SALL4 Oncogenic Function in Cancers: Mechanisms and Therapeutic Relevance. Int. J. Mol. Sci. 2022, 23, 2053. https://doi.org/10.3390/ijms23042053
Sun B, Xu L, Bi W, Ou W-B. SALL4 Oncogenic Function in Cancers: Mechanisms and Therapeutic Relevance. International Journal of Molecular Sciences. 2022; 23(4):2053. https://doi.org/10.3390/ijms23042053
Chicago/Turabian StyleSun, Boshu, Liangliang Xu, Wenhui Bi, and Wen-Bin Ou. 2022. "SALL4 Oncogenic Function in Cancers: Mechanisms and Therapeutic Relevance" International Journal of Molecular Sciences 23, no. 4: 2053. https://doi.org/10.3390/ijms23042053
APA StyleSun, B., Xu, L., Bi, W., & Ou, W. -B. (2022). SALL4 Oncogenic Function in Cancers: Mechanisms and Therapeutic Relevance. International Journal of Molecular Sciences, 23(4), 2053. https://doi.org/10.3390/ijms23042053