
Hexokinase-2-Linked Glycolytic Overload and Unscheduled Glycolysis—Driver of Insulin Resistance and Development of Vascular Complications of Diabetes




Abstract
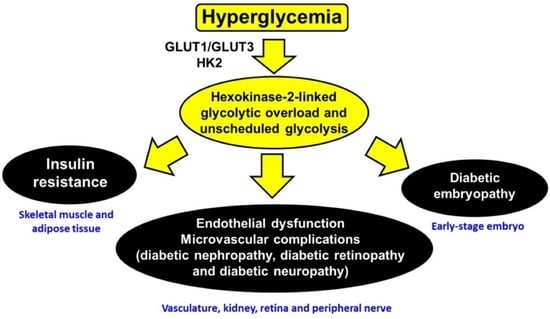
:
1. Introduction: Human Hexokinase-2—Overview of Molecular Characteristics and Subcellular and Tissue Expression
2. Hexokinase-2-Linked Glycolytic Overload and Unscheduled Glycolysis
- (i)
- (ii)
- (iii)
- (iv)
- (v)
3. Hexokinase-2-Linked Glycolytic Overload versus Oxidative Stress as an Initiator of Metabolic Dysfunction in Hyperglycemia
4. Evidence of Hexokinase-2 Linked Glycolytic Overload Occurring at Sites of Vascular Complications of Diabetes
5. Evidence for Hexokinase-2 Linked Unscheduled Glycolysis in Insulin Resistance and the Development of Type 2 Diabetes
- a.
- Overexpression of GLUT1 in skeletal-muscle-induced impairment of insulin-responsive glucose uptake
- b.
- Partial knockdown of hexokinase-2 (HK2 (−/+)) in mice improved glucose tolerance in the late stage of glucose challenge
- c.
- Overexpression of hexokinase-2 in skeletal muscle of mice impaired uptake of glucose on a high fat diet in hyperinsulinemic euglycemic clamp studies
- d.
- Downstream metabolic signaling in skeletal muscle and adipose tissue in insulin resistance resembles HK2-linked unscheduled glycolysis
6. How May Glyoxalase 1 Inducer, trans-Resveratrol, and Hesperetin Prevent the Development of Vascular Complications of Diabetes and Correct Insulin Resistance in Skeletal Muscle and Adipose Tissue?
7. Concluding Remarks
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Traut, T. Hexokinase. In Allosteric Regulatory Enzymes; Springer: Boston, MA, USA, 2008; pp. 179–198. [Google Scholar]
- Xia, H.-G.; Najafov, A.; Geng, J.; Galan-Acosta, L.; Han, X.; Guo, Y.; Shan, B.; Zhang, Y.; Norberg, E.; Zhang, T.; et al. Degradation of HK2 by chaperone-mediated autophagy promotes metabolic catastrophe and cell death. J. Cell Biol. 2015, 210, 705–716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- John, S.; Weiss, J.N.; Ribalet, B. Subcellular Localization of Hexokinases I and II Directs the Metabolic Fate of Glucose. PLoS ONE 2011, 6, e17674. [Google Scholar] [CrossRef] [Green Version]
- Ritov, V.B.; Kelley, D.E. Hexokinase Isozyme Distribution in Human Skeletal Muscle. Diabetes 2001, 50, 1253–1262. [Google Scholar] [CrossRef] [Green Version]
- Bryan, N.; Raisch, K.P. Identification of a mitochondrial-binding site on the N-terminal end of hexokinase II. Biosci. Rep. 2015, 35, e00205. [Google Scholar] [CrossRef]
- Pedersen, P.L. Voltage dependent anion channels (VDACs): A brief introduction with a focus on the outer mitochondrial compartment’s roles together with hexokinase-2 in the “Warburg effect” in cancer. J. Bioenerg. Biomembr. 2008, 40, 123–126. [Google Scholar] [CrossRef] [PubMed]
- Wolf, A.; Agnihotri, S.; Munoz, D.; Guha, A. Developmental profile and regulation of the glycolytic enzyme hexokinase 2 in normal brain and glioblastoma multiforme. Neurobiol. Dis. 2011, 44, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Hogan, A.; Heyner, S.; Charron, M.J.; Copeland, N.G.; Gilbert, D.J.; Jenkins, N.A.; Thorens, B.; Schultz, G.A. Glucose transporter gene expression in early mouse embryos. Development 1991, 113, 363–372. [Google Scholar] [CrossRef]
- Yu, L.; Chen, X.; Sun, X.; Wang, L.; Chen, S. The Glycolytic Switch in Tumors: How Many Players Are Involved? J. Cancer 2017, 8, 3430–3440. [Google Scholar] [CrossRef] [PubMed]
- Mitsuishi, Y.; Taguchi, K.; Kawatani, Y.; Shibata, T.; Nukiwa, T.; Aburatani, H.; Yamamoto, M.; Motohashi, H. Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell 2012, 22, 66–79. [Google Scholar] [CrossRef] [Green Version]
- Rempel, A.; Mathupala, S.P.; Pedersen, P.L. Glucose catabolism in cancer cells: Regulation of the type II hexokinase promoter by glucose and cyclic AMP. FEBS Lett. 1996, 385, 233–237. [Google Scholar] [CrossRef] [Green Version]
- Mathupala, S.P.; Rempel, A.; Pedersen, P.L. Glucose Catabolism in Cancer Cells: Isolation, sequence, and activity of the promoter for type II hexokinase. J. Biol. Chem. 1995, 270, 16918–16925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irshad, Z.; Xue, M.; Ashour, A.; Larkin, J.R.; Thornalley, P.J.; Rabbani, N. Activation of the unfolded protein response in high glucose treated endothelial cells is mediated by methylglyoxal. Sci. Rep. 2019, 9, 7889. [Google Scholar] [CrossRef] [Green Version]
- Laakso, M.; Malkki, M.; Kekäläinen, P.; Kuusisto, J.; Deeb, S.S. Polymorphisms of the human hexokinase II gene: Lack of association with NIDDM and insulin resistance. Diabetologia 1995, 38, 617–622. [Google Scholar] [CrossRef] [PubMed]
- Lehto, M.; Huang, X.; Davis, E.M.; Le Beau, M.M.; Laurila, E.; Eriksson, K.F.; Bell, G.I.; Groop, L. Human hexokinase II gene: Exon-intron organization, mutation screening in NIDDM, and its relationship to muscle hexokinase activity. Diabetologia 1995, 38, 1466–1474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ardehali, H.; Tiller, G.E.; Printz, R.L.; Mochizuki, H.; Prochazka, M.; Granner, D.K. A novel (TA)n polymorphism in the hexokinase II gene: Application to noninsulin-dependent diabetes mellitus in the Pima Indians. Hum. Genet. 1996, 97, 482–485. [Google Scholar] [CrossRef] [PubMed]
- Ashour, A.; Xue, M.; Al-Motawa, M.; Thornalley, P.J.; Rabbani, N. Glycolytic overload-driven dysfunction of periodontal ligament fibroblasts in high glucose concentration, corrected by glyoxalase 1 inducer. BMJ Open Diabetes Res. Care 2020, 8, e001458. [Google Scholar] [CrossRef] [PubMed]
- Viticchiè, G.; Agostini, M.; Lena, A.M.; Mancini, M.; Zhou, H.; Zolla, L.; Dinsdale, D.; Saintigny, G.; Melino, G.; Candi, E. p63 supports aerobic respiration through hexokinase II. Proc. Natl. Acad. Sci. USA 2015, 112, 11577–11582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishikawa, T.; Edelstein, D.; Liang Du, X.; Yamagishi, S.; Matsumura, T.; Kaneda, Y.; Yorek, M.A.; Beede, D.; Oates, P.J.; Hammes, H.P.; et al. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemia damage. Nature 2000, 404, 787–790. [Google Scholar] [CrossRef]
- Xue, M.; Qian, Q.; Adaikalakoteswari, A.; Rabbani, N.; Babaei-Jadidi, R.; Thornalley, P.J. Activation of NF-E2-Related Factor-2 Reverses Biochemical Dysfunction of Endothelial Cells Induced by Hyperglycemia Linked to Vascular Disease. Diabetes 2008, 57, 2809–2817. [Google Scholar] [CrossRef] [Green Version]
- Quijano, C.; Castro, L.; Peluffo, G.; Valez, V.; Radi, R. Enhanced mitochondrial superoxide in hyperglycemic endothelial cells: Direct measurements and formation of hydrogen peroxide and peroxynitrite. AJP—Heart Circ. Physiol. 2007, 293, H3404–H3414. [Google Scholar] [CrossRef] [Green Version]
- Noguchi, R.; Kubota, H.; Yugi, K.; Toyoshima, Y.; Komori, Y.; Soga, T.; Kuroda, S. The selective control of glycolysis, gluconeogenesis and glycogenesis by temporal insulin patterns. Mol. Syst. Biol. 2013, 9, 664. [Google Scholar] [CrossRef]
- Printz, R.L.; Koch, S.; Potter, L.R.; O’Doherty, R.M.; Tiesinga, J.J.; Moritz, S.; Granner, D.K. Hexokinase II mRNA and gene structure, regulation by insulin, and evolution. J. Biol. Chem. 1993, 268, 5209–5219. [Google Scholar] [CrossRef]
- Osawa, H.; Sutherland, C.; Robey, R.B.; Printz, R.L.; Granner, D.K. Analysis of the Signaling Pathway Involved in the Regulation of Hexokinase II Gene Transcription by Insulin. J. Biol. Chem. 1996, 271, 16690–16694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Probst, I.; Unthan-Fechner, K. Activation of glycolysis by insulin with a sequential increase of the 6-phosphofructo-2-kinase activity, fructose-2,6-bisphosphate level and pyruvate kinase activity in cultured rat hepatocytes. Eur. J. Biochem. 1985, 153, 347–353. [Google Scholar] [CrossRef] [PubMed]
- Nasrin, N.; Ercolani, L.; Denaro, M.; Kong, X.F.; Kang, I.; Alexander, M. An insulin response element in the glyceraldehyde-3-phosphate dehydrogenase gene binds a nuclear protein induced by insulin in cultured cells and by nutritional manipulations in vivo. Proc. Natl. Acad. Sci. USA 1990, 87, 5273–5277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sans, C.L.; Satterwhite, D.J.; Stoltzman, C.A.; Breen, K.T.; Ayer, D.E. MondoA-Mlx Heterodimers Are Candidate Sensors of Cellular Energy Status: Mitochondrial Localization and Direct Regulation of Glycolysis. Mol. Cell. Biol. 2006, 26, 4863–4871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeong, Y.S.; Kim, D.; Lee, Y.S.; Kim, H.J.; Han, J.Y.; Im, S.S.; Chong, H.K.; Kwon, J.K.; Cho, Y.H.; Kim, W.K.; et al. Integrated Expression Profiling and Genome-Wide Analysis of ChREBP Targets Reveals the Dual Role for ChREBP in Glucose-Regulated Gene Expression. PLoS ONE 2011, 6, e22544. [Google Scholar]
- Ma, L.; Robinson, L.N.; Towle, H.C. ChREBP•Mlx Is the Principal Mediator of Glucose-induced Gene Expression in the Liver. J. Biol. Chem. 2006, 281, 28721–28730. [Google Scholar] [CrossRef] [Green Version]
- Arden, C.; Tudhope, S.J.; Petrie, J.L.; Al-Oanzi, Z.H.; Cullen, K.S.; Lange, A.J.; Towle, H.C.; Agius, L. Fructose 2,6-bisphosphate is essential for glucose-regulated gene transcription of glucose-6-phosphatase and other ChREBP target genes in hepatocytes. Biochem. J. 2012, 443, 111–123. [Google Scholar] [CrossRef] [Green Version]
- Rabbani, N.; Thornalley, P.J. Hexokinase-2 Glycolytic Overload in Diabetes and Ischemia–Reperfusion Injury. Trends Endocrinol. Metab. 2019, 30, 419–431. [Google Scholar] [CrossRef] [Green Version]
- Brownlee, M. Biochemistry and molecular cell biology of diabetic complications. Nature 2001, 414, 813–820. [Google Scholar] [CrossRef] [PubMed]
- Hink, U.; Li, H.; Mollnau, H.; Oelze, M.; Matheis, E.; Hartmann, M.; Skatchkov, M.; Thaiss, F.; Stahl, R.A.K.; Warnholtz, A.; et al. Mechanisms underlying endothelial dysfunction in diabetes mellitus. Circ. Res. 2001, 88, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Guzik, T.J.; Mussa, S.; Gastaldi, D.; Sadowski, J.; Ratnatunga, C.; Pillai, R.; Channon, K.M. Mechanisms of Increased Vascular Superoxide Production in Human Diabetes Mellitus: Role of NAD(P)H Oxidase and Endothelial Nitric Oxide Synthase. Circulation 2002, 105, 1656–1662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandes, R.P.; Kreuzer, J. Vascular NADPH oxidases: Molecular mechanisms of activation. Cardiovasc. Res. 2005, 65, 16–27. [Google Scholar] [CrossRef]
- Lonn, E.; Yusuf, S.; Hoogwerf, B.; Pogue, J.; Yi, Q.L.; Zinman, B.; Bosch, J.; Dagenais, G.; Mann, J.F.E.; Gerstein, H.C. Effects of vitamin E on cardiovascular and microvascular outcomes in high-risk patients with diabetes—Results of the HOPE study and MICRO-HOPE substudy. Diabetes Care 2002, 25, 1919–1927. [Google Scholar] [CrossRef] [Green Version]
- Han, T.; Bai, J.; Liu, W.; Hu, Y. A systematic review and meta-analysis of α-lipoic acid in the treatment of diabetic peripheral neuropathy. Eur. J. Endocrinol. 2012, 167, 465–471. [Google Scholar] [CrossRef] [Green Version]
- Karachalias, N.; Babaei-Jadidi, R.; Rabbani, N.; Thornalley, P.J. Increased protein damage in renal glomeruli, retina, nerve, plasma and urine and its prevention by thiamine and benfotiamine therapy in a rat model of diabetes. Diabetologia 2010, 53, 1506–1516. [Google Scholar] [CrossRef] [Green Version]
- Feldman, E.L.; Callaghan, B.C.; Pop-Busui, R.; Zochodne, D.W.; Wright, D.E.; Bennett, D.L.; Bril, V.; Russell, J.W.; Viswanathan, V. Diabetic neuropathy. Nat. Rev. Dis. Primers 2019, 5, 41. [Google Scholar] [CrossRef]
- Gardiner, N.J.; Wang, Z.; Luke, C.; Gott, A.; Price, S.A.; Fernyhough, P. Expression of hexokinase isoforms in the dorsal root ganglion of the adult rat and effect of experimental diabetes. Brain Res. 2007, 1175 (Suppl. C), 143–154. [Google Scholar] [CrossRef]
- Kim, E.S.; Isoda, F.; Kurland, I.; Mobbs, C.V. Glucose-Induced Metabolic Memory in Schwann Cells: Prevention by PPAR Agonists. Endocrinology 2013, 154, 3054–3066. [Google Scholar] [CrossRef]
- Pan, S.; Chan, J.R. Regulation and dysregulation of axon infrastructure by myelinating glia. J. Cell Biol. 2017, 216, 3903–3916. [Google Scholar] [CrossRef]
- Xue, M.; Rabbani, N.; Momiji, H.; Imbasi, P.; Anwar, M.M.; Kitteringham, N.R.; Park, B.K.; Souma, T.; Moriguchi, T.; Yamamoto, M.; et al. Transcriptional control of glyoxalase 1 by Nrf2 provides a stress responsive defence against dicarbonyl glycation. Biochem. J. 2012, 443, 213–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, M.; Weickert, M.O.; Qureshi, S.; Ngianga-Bakwin, K.; Anwar, A.; Waldron, M.; Shafie, A.; Messenger, D.; Fowler, M.; Jenkins, G.; et al. Improved glycemic control and vascular function in overweight and obese subjects by glyoxalase 1 inducer formulation. Diabetes 2016, 65, 2282–2294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iori, E.; Millioni, R.; Puricelli, L.; Arrigoni, G.; Lenzini, L.; Trevisan, R.; James, P.; Rossi, G.P.; Pinna, L.A.; Tessari, P. Glycolytic enzyme expression and pyruvate kinase activity in cultured fibroblasts from type 1 diabetic patients with and without nephropathy. Biochim. Biophys. Acta (BBA)—Mol. Basis Dis. 2008, 1782, 627–633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isoe, T.; Makino, Y.; Mizumoto, K.; Sakagami, H.; Fujita, Y.; Honjo, J.; Takiyama, Y.; Itoh, H.; Haneda, M. High glucose activates HIF-1-mediated signal transduction in glomerular mesangial cells through a carbohydrate response element binding protein. Kidney Intern. 2010, 78, 48–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beemer, F.A.; Vlug, A.M.C.; Rijksen, G.; Hamburg, A.; Staal, G.E.J. Characterization of Some Glycolytic Enzymes from Human Retina and Retinoblastoma. Cancer Res. 1982, 42, 4228–4232. [Google Scholar] [PubMed]
- Magnani, P.; Cherian, P.V.; Gould, G.W.; Greene, D.A.; Sima, A.A.F.; Brosius, F.C., III. Glucose transporters in rat peripheral nerve: Paradonal expression of GLUT1 and GLUT3. Metabolism 1996, 45, 1466–1473. [Google Scholar] [CrossRef]
- Echave, P.; Machado-da-Silva, G.; Arkell, R.S.; Duchen, M.R.; Jacobson, J.; Mitter, R.; Lloyd, A.C. Extracellular growth factors and mitogens cooperate to drive mitochondrial biogenesis. J. Cell Sci. 2009, 122, 4516–4525. [Google Scholar] [CrossRef] [Green Version]
- Segev, H.; Fishman, B.; Schulman, R.; Itskovitz-Eldor, J. The Expression of the Class 1 Glucose Transporter Isoforms in Human Embryonic Stem Cells, and the Potential Use of GLUT2 as a Marker for Pancreatic Progenitor Enrichment. Stem Cells Dev. 2012, 21, 1653–1661. [Google Scholar] [CrossRef]
- Johnson, M.T.; Mahmood, S.; Patel, M.S. Intermediary Metabolism and Energetics during Murine Early Embryogenesis. J. Biol. Chem. 2003, 278, 31457–31460. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Håkan Borg, L.A.; Eriksson, U.J. Altered mitochondrial morphology of rat embryos in diabetic pregnancy. Anat. Rec. 1995, 241, 255–267. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, U.J.; Wentzel, P.; Minhas, H.S.; Thornalley, P.J. Teratogenicity of 3-deoxyglucosone and diabetic embryopathy. Diabetes 1998, 47, 1960–1966. [Google Scholar] [CrossRef]
- Horal, M.; Zhang, Z.; Stanton, R.; Virkamäki, A.; Loeken, M.R. Activation of the hexosamine pathway causes oxidative stress and abnormal embryo gene expression: Involvement in diabetic teratogenesis. Birth Defects Res. Part A Clin. Mol. Teratol. 2004, 70, 519–527. [Google Scholar] [CrossRef]
- Hiramatsu, Y.; Sekiguchi, N.; Hayashi, M.; Isshiki, K.; Yokota, T.; King, G.L.; Loeken, M.R. Diacylglycerol Production and Protein Kinase C Activity Are Increased in a Mouse Model of Diabetic Embryopathy. Diabetes 2002, 51, 2804–2810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singleton, J.R.; Smith, A.G.; Russell, J.W.; Feldman, E.L. Microvascular Complications of Impaired Glucose Tolerance. Diabetes 2003, 52, 2867–2873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva, C.M.; Arnegard, M.E.; Maric-Bilkan, C. Dysglycemia in Pregnancy and Maternal/Fetal Outcomes. J. Women’s Health 2021, 30, 187–193. [Google Scholar] [CrossRef]
- Lefort, N.; Glancy, B.; Bowen, B.; Willis, W.T.; Bailowitz, Z.; De Filippis, E.A.; Brophy, C.; Meyer, C.; Højlund, K.; Yi, Z.; et al. Increased Reactive Oxygen Species Production and Lower Abundance of Complex I Subunits and Carnitine Palmitoyltransferase 1B Protein Despite Normal Mitochondrial Respiration in Insulin-Resistant Human Skeletal Muscle. Diabetes 2010, 59, 2444–2452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yki-Järvinen, H.; Daniels, M.C.; Virkamäki, A.; Mäkimattila, S.; DeFronzo, R.A.; McClain, D. Increased Glutamine: Fructose-6-Phosphate Amidotransferase Activity in Skeletal Muscle of Patients With NIDDM. Diabetes 1996, 45, 302–307. [Google Scholar] [CrossRef]
- Li, M.; Vienberg, S.G.; Bezy, O.; O’Neill, B.T.; Kahn, C.R. Role of PKCδ in Insulin Sensitivity and Skeletal Muscle Metabolism. Diabetes 2015, 64, 4023–4032. [Google Scholar] [CrossRef] [Green Version]
- Mey, J.T.; Blackburn, B.K.; Miranda, E.R.; Chaves, A.B.; Briller, J.; Bonini, M.G.; Haus, J.M. Dicarbonyl stress and glyoxalase enzyme system regulation in human skeletal muscle. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2018, 314, R181–R190. [Google Scholar] [CrossRef]
- Pendergrass, M.; Koval, J.; Vogt, C.; Yki-Jarvinen, H.; Iozzo, P.; Pipek, R.; Ardehali, H.; Printz, R.; Granner, D.; DeFronzo, R.A. Insulin-Induced hexokinase II expression is reduced in obesity and NIDDM. Diabetes 1998, 47, 387–394. [Google Scholar] [CrossRef]
- Lavis, V.R. Hexokinase isozymes of normal human subcutaneous adipose tissue. Metabolism 1978, 27, 1101–1108. [Google Scholar] [CrossRef]
- Masania, J.; Malczewska-Malec, M.; Razny, U.; Goralska, J.; Zdzienicka, A.; Kiec-Wilk, B.; Gruca, A.; Stancel-Mozwillo, J.; Dembinska-Kiec, A.; Rabbani, N.; et al. Dicarbonyl stress in clinical obesity. Glycoconj. J. 2016, 33, 581–589. [Google Scholar] [CrossRef] [Green Version]
- Fazakerley, D.J.; Minard, A.Y.; Krycer, J.R.; Thomas, K.C.; Stöckli, J.; Harney, D.J.; Burchfield, J.G.; Maghzal, G.J.; Caldwell, S.T.; Hartley, R.C.; et al. Mitochondrial oxidative stress causes insulin resistance without disrupting oxidative phosphorylation. J. Biol. Chem. 2018, 293, 7315–7328. [Google Scholar] [CrossRef] [Green Version]
- Brandon, A.E.; Liao, B.M.; Diakanastasis, B.; Parker, B.L.; Raddatz, K.; McManus, S.A.; O’Reilly, L.; Kimber, E.; van der Kraan, A.G.; Hancock, D.; et al. Protein Kinase C Epsilon Deletion in Adipose Tissue, but Not in Liver, Improves Glucose Tolerance. Cell Metab. 2019, 29, 183–191.e7. [Google Scholar] [CrossRef] [Green Version]
- Morton, N.M.; Nelson, Y.B.; Michailidou, Z.; Di Rollo, E.M.; Ramage, L.; Hadoke, P.W.F.; Seckl, J.R.; Bunger, L.; Horvat, S.; Kenyon, C.J.; et al. A stratified transcriptomics analysis of polygenic fat and lean mouse adipose tissues identifies novel candidate obesity genes. PLoS ONE 2011, 6, e23944. [Google Scholar] [CrossRef]
- Komolafe, O.A.; Ofusori, D.A.; Adewole, O.S.; Ayoka, A.O.; Bejide, R. Histological and Histochemical Studies of the Aorta and Pulmonary Trunk in STZ-induced Diabetic Wistar Rats Treated with Momordica charantia. Int. J. Morphol. 2013, 31, 716–723. [Google Scholar] [CrossRef] [Green Version]
- Kang, J.; Dai, X.-S.; Yu, T.-B.; Wen, B.; Yang, Z.-W. Glycogen accumulation in renal tubules, a key morphological change in the diabetic rat kidney. Acta Diabetol. 2005, 42, 110–116. [Google Scholar] [CrossRef]
- Geoffrion, M.; Du, X.; Irshad, Z.; Vanderhyden, B.C.; Courville, K.; Sui, G.; D’Agati, V.D.; Ott-Braschi, S.; Rabbani, N.; Thornalley, P.J.; et al. Differential effects of glyoxalase 1 overexpression on diabetic atherosclerosis and renal dysfunction in streptozotocin-treated, apolipoprotein E-deficient mice. Physiol. Rep. 2014, 2, e12043. [Google Scholar] [CrossRef]
- Fantus, I.G.; Goldberg, H.J.; Whiteside, C.I.; Topic, D. The Hexosamine Biosynthesis Pathway. In The Diabetic Kidney; Cortes, P., Mogensen, C.E., Eds.; Humana Press: Totowa, NJ, USA, 2006; pp. 117–133. [Google Scholar]
- Kiritoshi, S.; Nishikawa, T.; Sonoda, K.; Kukidome, D.; Senokuchi, T.; Matsuo, T.; Matsumura, T.; Tokunaga, H.; Brownlee, M.; Araki, E. Reactive oxygen species from mitochondria Induce cyclooxygenase-2 gene expression in human mesangial cells: Potential role in diabetic nephropathy. Diabetes 2003, 52, 2570–2577. [Google Scholar] [CrossRef]
- Shiba, T.; Inoguchi, T.; Sportsman, J.R.; Heath, W.F.; Bursell, S.; King, G.L. Correlation of diacylglycerol level and protein kinase C activity in rat retina to retinal circulation. Am. J. Physiol. 1993, 265, E783–E793. [Google Scholar] [CrossRef]
- Powell, H.C.; Rosoff, J.; Myers, R.R. Microangiopathy in human diabetic neuropathy. Acta Neuropathol. 1985, 68, 295–305. [Google Scholar] [CrossRef]
- Bierhaus, A.; Fleming, T.; Stoyanov, S.; Leffler, A.; Babes, A.; Neacsu, C.; Sauer, S.K.; Eberhardt, M.; Schnolzer, M.; Lasischka, F.; et al. Methylglyoxal modification of Nav1.8 facilitates nociceptive neuron firing and causes hyperalgesia in diabetic neuropathy. Nat. Med. 2012, 18, 926–933. [Google Scholar] [CrossRef]
- Zhang, L.; Yu, C.; Vasquez, F.E.; Galeva, N.; Onyango, I.; Swerdlow, R.H.; Dobrowsky, R.T. Hyperglycemia alters the schwann cell mitochondrial proteome and decreases coupled respiration in the absence of superoxide production. J. Proteome Res. 2010, 9, 458–471. [Google Scholar] [CrossRef] [Green Version]
- Russell, J.W.; Berent-Spillson, A.; Vincent, A.M.; Freimann, C.L.; Sullivan, K.A.; Feldman, E.L. Oxidative injury and neuropathy in diabetes and impaired glucose tolerance. Neurobiol. Dis. 2008, 30, 420–429. [Google Scholar] [CrossRef] [Green Version]
- Duran-Jimenez, B.; Dobler, D.; Moffatt, S.; Rabbani, N.; Streuli, C.H.; Thornalley, P.J.; Tomlinson, D.R.; Gardiner, N.J. Advanced Glycation Endproducts in extracellular matrix proteins contribute to the failure of sensory nerve regeneration in diabetes. Diabetes 2009, 58, 2893–2903. [Google Scholar] [CrossRef] [Green Version]
- Schellini, S.A.; Gregorio, E.A.; Spadella, C.T.; Machado, J.L.M.; Demoraessilva, M.A. Muller cells and diabetic-retinopathy. Braz. J. Med. Biol. Res. 1995, 28, 977–980. [Google Scholar]
- Yao, D.; Taguchi, T.; Matsumura, T.; Pestell, R.; Edelstein, D.; Giardino, I.; Suske, G.; Rabbani, N.; Thornalley, P.J.; Sarthy, V.P.; et al. High glucose increases angiopoietin-2 transcription in microvascular endothelial cells through methylglyoxal modification of mSin3A. J. Biol. Chem. 2007, 282, 31038–31045. [Google Scholar] [CrossRef] [Green Version]
- Tien, T.; Zhang, J.; Muto, T.; Kim, D.; Sarthy, V.P.; Roy, S. High Glucose Induces Mitochondrial Dysfunction in Retinal Müller Cells: Implications for Diabetic Retinopathy. Investig. Ophthalmol. Vis. Sci. 2017, 58, 2915–2921. [Google Scholar] [CrossRef]
- Semba, R.D.; Huang, H.; Lutty, G.A.; Van Eyk, J.E.; Hart, G.W. The role of O-GlcNAc signaling in the pathogenesis of diabetic retinopathy. Proteom. Clin. Appl. 2014, 8, 218–231. [Google Scholar] [CrossRef] [Green Version]
- Ellington, S.K.L. Effects of excess glucose on mammalian post-implantation embryos. Int. J. Dev. BioI. 1997, 41, 299–306. [Google Scholar]
- Nannipieri, M.; Lanfranchi, A.; Santerini, D.; Catalano, C.; van de Werve, G.; Ferrannini, E. Influence of Long-Term Diabetes on Renal Glycogen Metabolism in the Rat. Nephron 2001, 87, 50–57. [Google Scholar] [CrossRef]
- Salceda, R.; Coffe, V.M.; Hernández-Berrones, J. Glycogen Levels in the Normal and Diabetic Rat Retina. Investig. Ophthalmol. Vis. Sci. 2003, 44, 3879. [Google Scholar]
- Yagihashi, S.; Matsunaga, M. Ultrastructural pathology of peripheral nerves in patients with diabetic neuropathy. Tohoku J. Exp. Med. 1979, 129, 357–366. [Google Scholar] [CrossRef] [Green Version]
- Moore, S.A.; Peterson, R.G.; Felten, D.L.; O’Connor, B.L. Glycogen accumulation in tibial nerves of experimentally diabetic and aging control rats. J. Neurol. Sci. 1981, 52, 289–303. [Google Scholar] [CrossRef]
- Ishibashi, F.; Kojima, R.; Taniguchi, M.; Kosaka, A.; Uetake, H.; Tavakoli, M. The Expanded Bead Size of Corneal C-Nerve Fibers Visualized by Corneal Confocal Microscopy Is Associated with Slow Conduction Velocity of the Peripheral Nerves in Patients with Type 2 Diabetes Mellitus. J. Diabetes Res. 2016, 2016, 3653459. [Google Scholar] [CrossRef] [Green Version]
- Lillioja, S.; Mott, D.M.; Howard, B.V.; Bennett, P.H.; Yki-Järvinen, H.; Freymond, D.; Nyomba, B.L.; Zurlo, F.; Swinburn, B.; Bogardus, C. Impaired glucose tolerance as a disorder of insulin action. Longitudinal and cross-sectional studies in Pima Indians. N. Engl. J. Med. 1988, 318, 1217–1225. [Google Scholar] [CrossRef]
- Warram, J.H.; Martin, B.C.; Krolewski, A.S.; Soeldner, J.S.; Kahn, C.R. Slow glucose removal rate and hyperinsulinemia precede the development of type II diabetes in the offspring of diabetic parents. Ann. Intern. Med. 1990, 113, 909–915. [Google Scholar] [CrossRef]
- DeFronzo, R.A.; Tripathy, D. Skeletal muscle insulin resistance is the primary defect in type 2 diabetes. Diabetes Care 2009, 32 (Suppl. S2), S157–S163. [Google Scholar] [CrossRef] [Green Version]
- DeFronzo, R.A.; Hendler, R.; Simonson, D. Insulin Resistance is a Prominent Feature of Insulin-dependent Diabetes. Diabetes 1982, 31, 795–801. [Google Scholar] [CrossRef]
- Vogt, C.; Ardehali, H.; Iozzo, P.; Yki-Järvinen, H.; Koval, J.; Maezono, K.; Pendergrass, M.; Printz, R.; Granner, D.; DeFronzo, R.; et al. Regulation of hexokinase II expression in human skeletal muscle in vivo. Metab.—Clin. Exp. 2000, 49, 814–818. [Google Scholar] [CrossRef]
- Vogt, C.; Yki-Jarvinen, H.; Iozzo, P.; Pipek, R.; Pendergrass, M.; Koval, J.; Ardehali, H.; Printz, R.; Granner, D.; DeFronzo, R.; et al. Effects of Insulin on Subcellular Localization of Hexokinase II in Human Skeletal Muscle in Vivo. J. Clin. Endocrinol. Metab. 1998, 83, 230–234. [Google Scholar] [CrossRef] [PubMed]
- Roberts, D.J.; Tan-Sah, V.P.; Smith, J.M.; Miyamoto, S. Akt phosphorylates HK-II at Thr-473 and increases mitochondrial HK-II association to protect cardiomyocytes. J. Biol. Chem. 2013, 288, 23798–23806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vestergaard, H.; Bjørbaek, C.; Hansen, T.; Larsen, F.S.; Granner, D.K.; Pedersen, O. Impaired activity and gene expression of hexokinase II in muscle from non-insulin-dependent diabetes mellitus patients. J. Clin. Investig. 1995, 96, 2639–2645. [Google Scholar] [CrossRef] [PubMed]
- DeFronzo, R.A.; Jacot, E.; Jequier, E.; Maeder, E.; Wahren, J.; Felber, J.P. The Effect of Insulin on the Disposal of Intravenous Glucose: Results from Indirect Calorimetry and Hepatic and Femoral Venous Catheterization. Diabetes 1981, 30, 1000–1007. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, O.; Bak, J.F.; Andersen, P.H.; Lund, S.; Moller, D.E.; Flier, J.S.; Kahn, B.B. Evidence against altered expression of GLUT1 or GLUT4 in skeletal muscle of patients with obesity or NIDDM. Diabetes 1990, 39, 865–870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciaraldi, T.P.; Mudaliar, S.; Barzin, A.; Macievic, J.A.; Edelman, S.V.; Park, K.S.; Henry, R.R. Skeletal Muscle GLUT1 Transporter Protein Expression and Basal Leg Glucose Uptake Are Reduced in Type 2 Diabetes. J. Clin. Endocrinol. Metab. 2005, 90, 352–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marshall, B.A.; Ren, J.M.; Johnson, D.W.; Gibbs, E.M.; Lillquist, J.S.; Soeller, W.C.; Holloszy, J.O.; Mueckler, M. Germline manipulation of glucose homeostasis via alteration of glucose transporter levels in skeletal muscle. J. Biol. Chem. 1993, 268, 18442–18445. [Google Scholar] [CrossRef]
- Gulve, E.A.; Ren, J.M.; Marshall, B.A.; Gao, J.; Hansen, P.A.; Holloszy, J.O.; Mueckler, M. Glucose transport activity in skeletal muscles from transgenic mice overexpressing GLUT1. Increased basal transport is associated with a defective response to diverse stimuli that activate GLUT4. J. Biol. Chem. 1994, 269, 18366–18370. [Google Scholar] [CrossRef]
- Buse, M.G.; Robinson, K.A.; Marshall, B.A.; Hresko, R.C.; Mueckler, M.M. Enhanced O-GlcNAc protein modification is associated with insulin resistance in GLUT1-overexpressing muscles. Am. J. Physiol. Endocrinol. Metab. 2002, 283, E241–E250. [Google Scholar] [CrossRef] [Green Version]
- Fazakerley, D.J.; Krycer, J.R.; Kearney, A.L.; Hocking, S.L.; James, D.E. Muscle and adipose tissue insulin resistance: Malady without mechanism? J. Lipid Res. 2019, 60, 1720–1732. [Google Scholar] [CrossRef] [PubMed]
- Hansen, P.A.; Gulve, E.A.; Marshall, B.A.; Gao, J.; Pessin, J.E.; Holloszy, J.O.; Mueckler, M. Skeletal muscle glucose transport and metabolism are enhanced in transgenic mice overexpressing the Glut4 glucose transporter. J. Biol. Chem. 1995, 270, 1679–1684. [Google Scholar] [CrossRef]
- Heikkinen, S.; Pietilä, M.; Halmekytö, M.; Suppola, S.; Pirinen, E.; Deeb, S.S.; Jänne, J.; Laakso, M. Hexokinase II-deficient mice: Prenatal death of homozygotes without disturbances in glucose tolerance in heterozygotes. J. Biol. Chem. 1999, 274, 22517–22523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, P.Y.; Jensen, J.; Printz, R.L.; Granner, D.K.; Ivy, J.L.; Moller, D.E. Overexpression of hexokinase II in transgenic mice. Evidence that increased phosphorylation augments muscle glucose uptake. J. Biol. Chem. 1996, 271, 14834–14839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fueger, P.T.; Bracy, D.P.; Malabanan, C.M.; Pencek, R.R.; Granner, D.K.; Wasserman, D.H. Hexokinase II overexpression improves exercise-stimulated but not insulin-stimulated muscle glucose uptake in high-fat-fed C57BL/6J mice. Diabetes 2004, 53, 306–314. [Google Scholar] [CrossRef] [Green Version]
- Hansen, P.A.; Marshall, B.A.; Chen, M.; Holloszy, J.O.; Mueckler, M. Transgenic Overexpression of Hexokinase II in Skeletal Muscle Does Not Increase Glucose Disposal in Wild-type or Glut1-overexpressing Mice. J. Biol. Chem. 2000, 275, 22381–22386. [Google Scholar] [CrossRef] [Green Version]
- Thiebaud, D.; Jacot, E.; Defronzo, R.A.; Maeder, E.; Jequier, E.; Felber, J.-P. The Effect of Graded Doses of Insulin on Total Glucose Uptake, Glucose Oxidation, and Glucose Storage in Man. Diabetes 1982, 31, 957–963. [Google Scholar] [CrossRef]
- Ceperuelo-Mallafré, V.; Ejarque, M.; Serena, C.; Duran, X.; Montori-Grau, M.; Rodríguez, M.A.; Yanes, O.; Núñez-Roa, C.; Roche, K.; Puthanveetil, P.; et al. Adipose tissue glycogen accumulation is associated with obesity-linked inflammation in humans. Mol. Metab. 2016, 5, 5–18. [Google Scholar] [CrossRef]
- Ebeling, P.; Koistinen, H.A.; Koivisto, V.A. Insulin-independent glucose transport regulates insulin sensitivity. FEBS Lett. 1998, 436, 301–303. [Google Scholar] [CrossRef]
- Stoltzman, C.A.; Peterson, C.W.; Breen, K.T.; Muoio, D.M.; Billin, A.N.; Ayer, D.E. Glucose sensing by MondoA:Mlx complexes: A role for hexokinases and direct regulation of thioredoxin-interacting protein expression. Proc. Natl. Acad. Sci. USA 2008, 105, 6912–6917. [Google Scholar] [CrossRef] [Green Version]
- Ahn, B.; Soundarapandian, M.M.; Sessions, H.; Peddibhotla, S.; Roth, G.P.; Li, J.-L.; Sugarman, E.; Koo, A.; Malany, S.; Wang, M.; et al. MondoA coordinately regulates skeletal myocyte lipid homeostasis and insulin signaling. J. Clin. Investig. 2016, 126, 3567–3579. [Google Scholar] [CrossRef] [PubMed]
- Ahn, B.; Wan, S.; Jaiswal, N.; Vega, R.B.; Ayer, D.E.; Titchenell, P.M.; Han, X.; Won, K.J.; Kelly, D.P. MondoA drives muscle lipid accumulation and insulin resistance. JCI Insight 2019, 5, e129119. [Google Scholar] [CrossRef] [PubMed]
- Mozaffary, A.; Asgari, S.; Tohidi, M.; Kazempour-Ardebili, S.; Azizi, F.; Hadaegh, F. Change in fasting plasma glucose and incident type 2 diabetes mellitus: Results from a prospective cohort study. BMJ Open 2016, 6, e010889. [Google Scholar] [CrossRef] [Green Version]
- Bock, G.; Chittilapilly, E.; Basu, R.; Toffolo, G.; Cobelli, C.; Chandramouli, V.; Landau, B.R.; Rizza, R.A. Contribution of Hepatic and Extrahepatic Insulin Resistance to the Pathogenesis of Impaired Fasting Glucose: Role of Increased Rates of Gluconeogenesis. Diabetes 2007, 56, 1703–1711. [Google Scholar] [CrossRef] [Green Version]
- Kaul, K.; Apostolopoulou, M.; Roden, M. Insulin resistance in type 1 diabetes mellitus. Metabolism 2015, 64, 1629–1639. [Google Scholar] [CrossRef] [PubMed]
- Pereira, R.I.; Snell-Bergeon, J.K.; Erickson, C.; Schauer, I.E.; Bergman, B.C.; Rewers, M.; Maahs, D.M. Adiponectin Dysregulation and Insulin Resistance in Type 1 Diabetes. J. Clin. Endocrinol. Metab. 2012, 97, E642–E647. [Google Scholar] [CrossRef] [PubMed]
- Rabbani, N.; Xue, M.; Thornalley, P.J. Dicarbonyl stress, protein glycation and the unfolded protein response. Glycoconj. J. 2021, 38, 331–334. [Google Scholar] [CrossRef] [PubMed]
- Rabbani, N.; Xue, M.; Thornalley, P.J. Methylglyoxal-induced dicarbonyl stress in aging and disease: First steps towards glyoxalase 1-based treatments. Clin. Sci. 2016, 130, 1677–1696. [Google Scholar] [CrossRef]
- Lerner, A.G.; Upton, J.-P.; Praveen, P.V.K.; Ghosh, R.; Nakagawa, Y.; Igbaria, A.; Shen, S.; Nguyen, V.; Backes, B.J.; Heiman, M.; et al. IRE1α induces thioredoxin-interacting protein to activate the NLRP3 inflammasome and promote programmed cell death under irremediable ER stress. Cell Metab. 2012, 16, 250–264. [Google Scholar] [CrossRef] [Green Version]
- Waldhart, A.N.; Dykstra, H.; Peck, A.S.; Boguslawski, E.A.; Madaj, Z.B.; Wen, J.; Veldkamp, K.; Hollowell, M.; Zheng, B.; Cantley, L.C.; et al. Phosphorylation of TXNIP by AKT Mediates Acute Influx of Glucose in Response to Insulin. Cell Rep. 2017, 19, 2005–2013. [Google Scholar] [CrossRef] [Green Version]
- Oslowski, C.M.; Hara, T.; O’Sullivan-Murphy, B.; Kanekura, K.; Lu, S.; Hara, M.; Ishigaki, S.; Zhu, L.J.; Hayashi, E.; Hui, S.T.; et al. Thioredoxin-interacting protein mediates ER stress-induced β cell death through initiation of the inflammasome. Cell Metab. 2012, 16, 265–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jo, S.H.; Kim, M.Y.; Park, J.M.; Kim, T.H.; Ahn, Y.H. Txnip contributes to impaired glucose tolerance by upregulating the expression of genes involved in hepatic gluconeogenesis in mice. Diabetologia 2013, 56, 2723–2732. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Reddy, M.A.; Miao, F.; Shanmugam, N.; Yee, J.-K.; Hawkins, D.; Ren, B.; Natarajan, R. Role of the Histone H3 Lysine 4 Methyltransferase, SET7/9, in the Regulation of NF-κB-dependent Inflammatory Genes: Relevance to Diabetes and Inflammation. J. Biol. Chem. 2008, 283, 26771–26781. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Guo, Y.; Zeng, W.; Huang, L.; Pang, Q.; Nie, L.; Mu, J.; Yuan, F.; Feng, B. ER stress triggers MCP-1 expression through SET7/9-induced histone methylation in the kidneys of db/db mice. Am. J. Physiol.—Ren. Physiol. 2014, 306, F916–F925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hotamisligil, G.S. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell 2010, 140, 900–917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hotamisligil, G.S. Mechanisms of TNF-alpha-induced insulin resistance. Exp. Clin. Endocrinol. Diabetes 1999, 107, 119–125. [Google Scholar] [CrossRef]
- Rabbani, N.; Xue, M.; Weickert, M.O.; Thornalley, P.J. Reversal of Insulin Resistance in Overweight and Obese Subjects by trans-Resveratrol and Hesperetin Combination—Link to Dysglycemia, Blood Pressure, Dyslipidemia, and Low-Grade Inflammation. Nutrients 2021, 13, 2374. [Google Scholar] [CrossRef]
- Parikh, H.; Carlsson, E.; Chutkow, W.A.; Johansson, L.E.; Storgaard, H.; Poulsen, P.; Saxena, R.; Ladd, C.; Schulze, P.C.; Mazzini, M.J.; et al. TXNIP Regulates Peripheral Glucose Metabolism in Humans. PLoS Med. 2007, 4, e158. [Google Scholar] [CrossRef] [Green Version]
- Uruno, A.; Yagishita, Y.; Katsuoka, F.; Kitajima, Y.; Nunomiya, A.; Nagatomi, R.; Pi, J.; Biswal, S.S.; Yamamoto, M. Nrf2-Mediated Regulation of Skeletal Muscle Glycogen Metabolism. Mol. Cell. Biol. 2016, 36, 1655–1672. [Google Scholar] [CrossRef] [Green Version]
- Park, S.-J.; Ahmad, F.; Philp, A.; Baar, K.; Williams, T.; Luo, H.; Ke, H.; Rehmann, H.; Taussig, R.; Brown, A.L.; et al. Resveratrol Ameliorates Aging-Related Metabolic Phenotypes by Inhibiting cAMP Phosphodiesterases. Cell 2012, 148, 421–433. [Google Scholar] [CrossRef] [Green Version]
- Hwang, S.-L.; Lin, J.-A.; Shih, P.-H.; Yeh, C.-T.; Yen, G.-C. Pro-Cellular survival and neuroprotection of citrus flavonoid: The actions of hesperetin in PC12 cells. Food Funct. 2012, 3, 1082–1090. [Google Scholar] [CrossRef] [PubMed]
- Xue, M.; Momiji, H.; Rabbani, N.; Barker, G.; Bretschneider, T.; Shmygol, A.; Rand, D.A.; Thornalley, P.J. Frequency modulated translocational oscillations of Nrf2 mediate the ARE cytoprotective transcriptional response. Antioxid. Redox Signal. 2015, 23, 613–629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, M.; Momiji, H.; Rabbani, N.; Bretschneider, T.; Rand, D.A.; Thornalley, P.J. Frequency modulated translocational oscillations of Nrf2, a transcription factor functioning like a wireless sensor. Biochem. Soc. Trans. 2015, 43, 669–673. [Google Scholar] [CrossRef] [PubMed]
- Gerhart-Hines, Z.; Dominy, J.E.; Blättler, S.M.; Jedrychowski, M.P.; Banks, A.S.; Lim, J.-H.; Chim, H.; Gygi, S.P.; Puigserver, P. The cAMP/PKA pathway rapidly activates SIRT1 to promote fatty acid oxidation independently of changes in NAD(+). Mol. Cell 2011, 44, 851–863. [Google Scholar] [CrossRef] [Green Version]
- Steinberg, G.R. Cellular Energy Sensing and Metabolism—Implications for Treating Diabetes: The 2017 Outstanding Scientific Achievement Award Lecture. Diabetes 2018, 67, 169–179. [Google Scholar] [CrossRef] [Green Version]
- Rena, G.; Hardie, D.G.; Pearson, E.R. The mechanisms of action of metformin. Diabetologia 2017, 60, 1577–1585. [Google Scholar] [CrossRef] [Green Version]
- Martin-Montalvo, A.; Mercken, E.M.; Mitchell, S.J.; Palacios, H.H.; Mote, P.L.; Scheibye-Knudsen, M.; Gomes, A.P.; Ward, T.M.; Minor, R.K.; Blouin, M.J.; et al. Metformin improves healthspan and lifespan in mice. Nat. Commun. 2013, 4, 2192. [Google Scholar] [CrossRef]
- Graham, G.G.; Punt, J.; Arora, M.; Day, R.O.; Doogue, M.P.; Duong, J.; Furlong, T.J.; Greenfield, J.R.; Greenup, L.C.; Kirkpatrick, C.M.; et al. Clinical Pharmacokinetics of Metformin. Clin. Pharmacokinet. 2011, 50, 81–98. [Google Scholar] [CrossRef]
- Stumvoll, M.; Nurjhan, N.; Perriello, G.; Dailey, G.; Gerich, J.E. Metabolic Effects of Metformin in Non-Insulin-Dependent Diabetes Mellitus. N. Engl. J. Med. 1995, 333, 550–554. [Google Scholar] [CrossRef]
- Zhou, G.; Myers, R.; Li, Y.; Chen, Y.; Shen, X.; Fenyk-Melody, J.; Wu, M.; Ventre, J.; Doebber, T.; Fujii, N.; et al. Role of AMP-activated protein kinase in mechanism of metformin action. J. Clin. Investig. 2001, 108, 1167–1174. [Google Scholar] [CrossRef]
- Del Prato, S. In search of normoglycaemia in diabetes: Controlling postprandial glucose. Int. J. Obes. 2002, 26, S9–S17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beisswenger, P.J.; Howell, S.K.; Touchette, A.; Lal, S.; Szwergold, B.S. Metformin reduces systemic methylglyoxal levels in type 2 diabetes. Diabetes 1999, 48, 198–202. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pathogenesis | Tissue/Cell Type | Indications | References |
|---|---|---|---|
| Insulin resistance (skeletal muscle) | Skeletal muscle myocytes |
| [58,59,60,61,62] |
| Insulin resistance (adipose tissue) | Adipose tissue, insulin-resistant 3T3-L1 adipocytes in vitro |
| [63,64,65,66,67] |
| Diabetic endothelial dysfunction | Endothelial cells |
| [13,19,20,68] |
| Diabetic nephropathy | Renal mesangial, cells, podocytes, and tubular epithelial cells |
| [46,69,70,71,72,73] |
| Diabetic neuropathy | Schwann cells (also dorsal root ganglia and sciatic nerve) |
| [41,74,75,76,77,78] |
| Diabetic retinopathy | Muller cells, endothelial cells and pericytes (also intact retina) |
| [47,73,79,80,81,82] |
| Diabetic embryopathy | Early-stage embryo (typically rat embryo, day 9–11 gestation) |
| [7,52,53,54,55,83] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rabbani, N.; Xue, M.; Thornalley, P.J. Hexokinase-2-Linked Glycolytic Overload and Unscheduled Glycolysis—Driver of Insulin Resistance and Development of Vascular Complications of Diabetes. Int. J. Mol. Sci. 2022, 23, 2165. https://doi.org/10.3390/ijms23042165
Rabbani N, Xue M, Thornalley PJ. Hexokinase-2-Linked Glycolytic Overload and Unscheduled Glycolysis—Driver of Insulin Resistance and Development of Vascular Complications of Diabetes. International Journal of Molecular Sciences. 2022; 23(4):2165. https://doi.org/10.3390/ijms23042165
Chicago/Turabian StyleRabbani, Naila, Mingzhan Xue, and Paul J. Thornalley. 2022. "Hexokinase-2-Linked Glycolytic Overload and Unscheduled Glycolysis—Driver of Insulin Resistance and Development of Vascular Complications of Diabetes" International Journal of Molecular Sciences 23, no. 4: 2165. https://doi.org/10.3390/ijms23042165
APA StyleRabbani, N., Xue, M., & Thornalley, P. J. (2022). Hexokinase-2-Linked Glycolytic Overload and Unscheduled Glycolysis—Driver of Insulin Resistance and Development of Vascular Complications of Diabetes. International Journal of Molecular Sciences, 23(4), 2165. https://doi.org/10.3390/ijms23042165