
Splenic Architecture and Function Requires Tight Control of Transmembrane TNF Expression



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
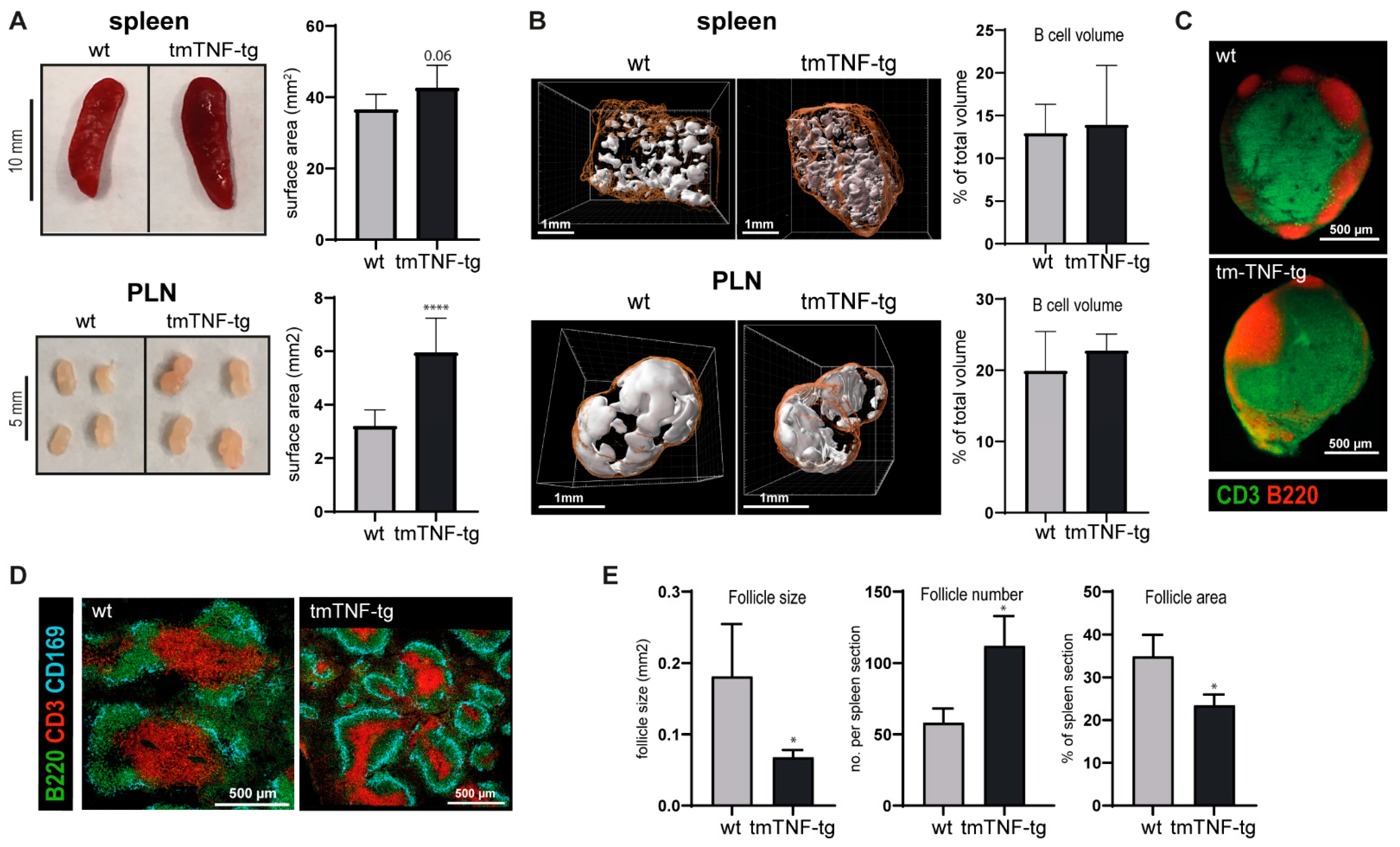
2.1. tmTNF Overexpression Leads to Enlarged SLOs and Aberrant Splenic Lymphoid Follicle Structures
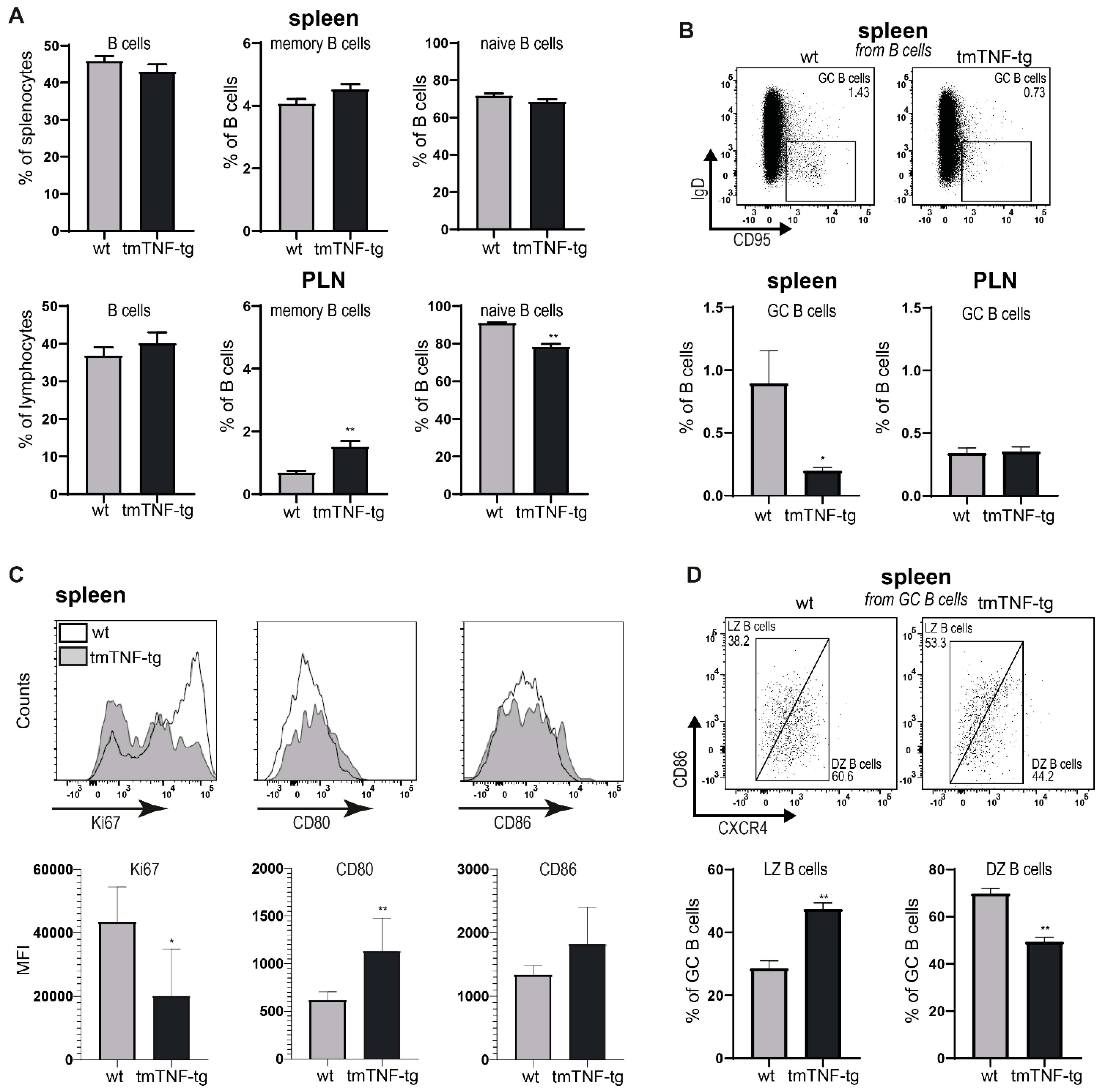
2.2. tmTNF Overexpression Reduces GC B Cell Formation in the Spleen but Not in PLN
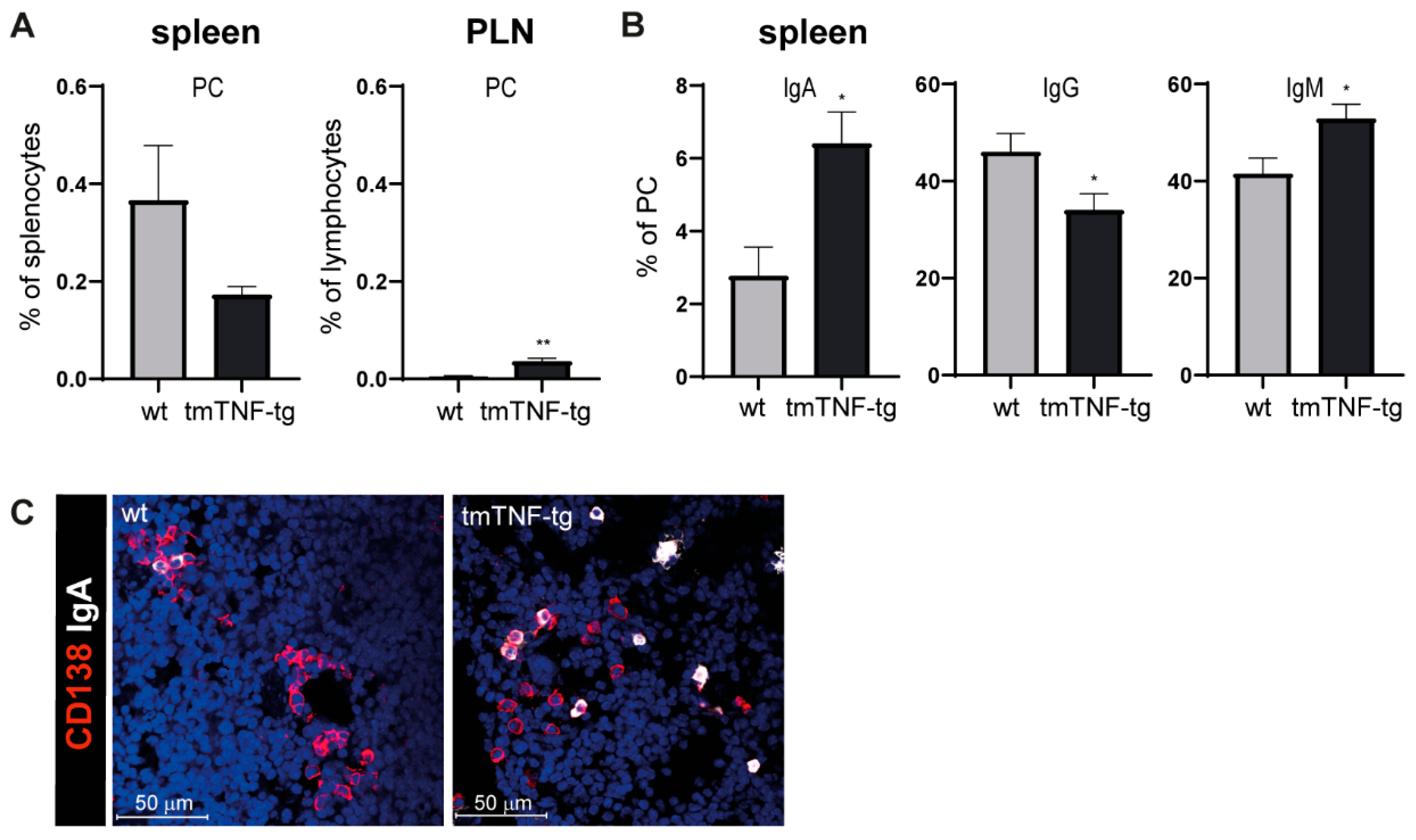
2.3. tmTNF Overexpression Results in Increased IgA and IgM Switching
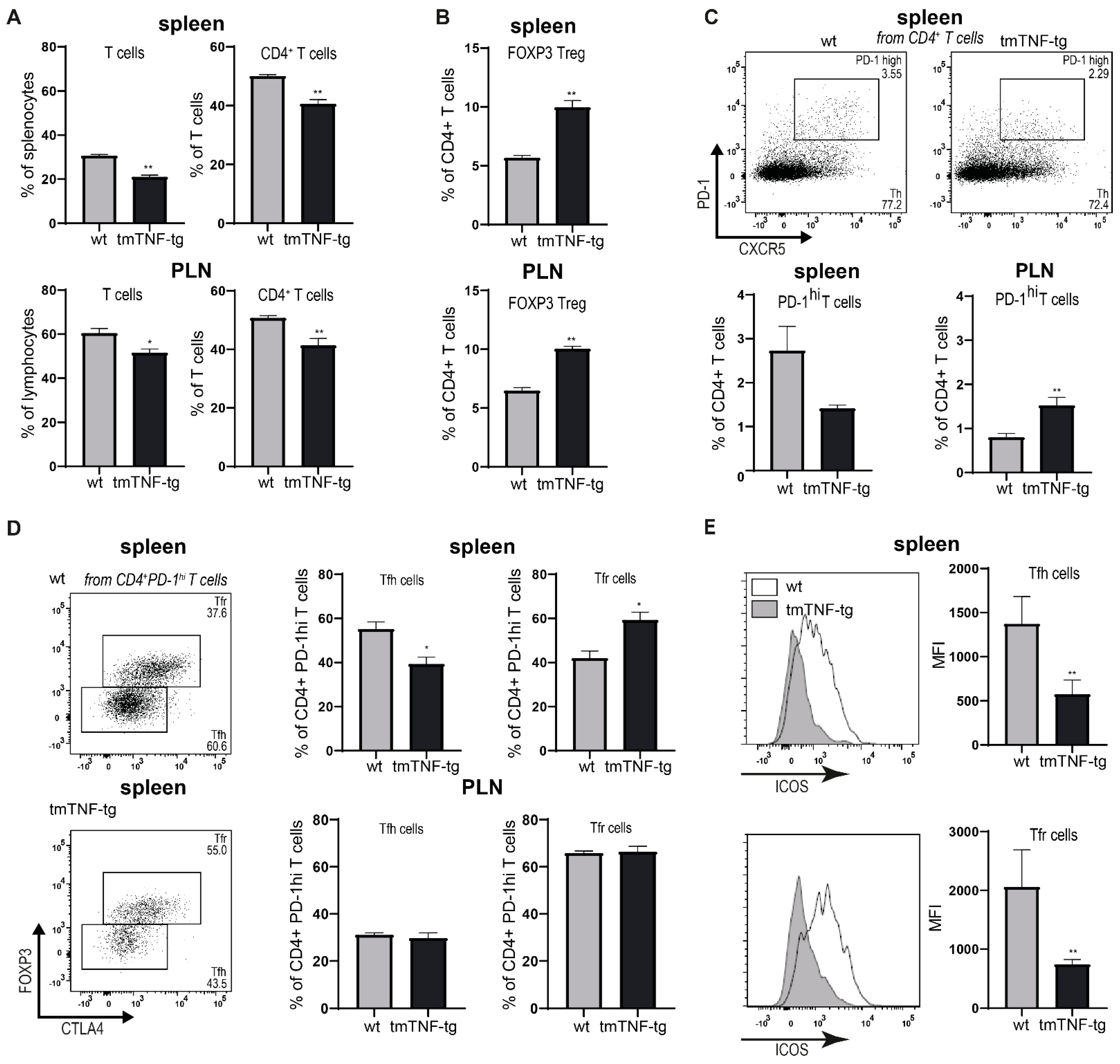
2.4. tmTNF Overexpression Affects GC-Associated T cells in Spleen
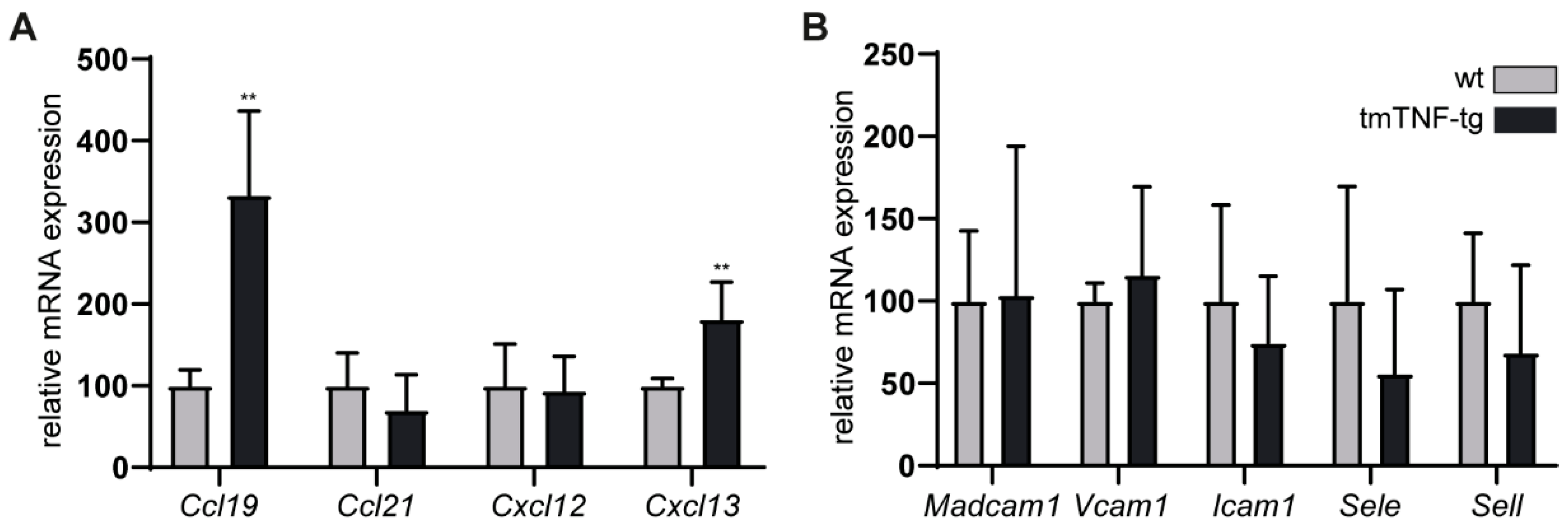
2.5. The Effects of tmTNF Overexpression on Chemokine and Adhesion Molecule Expression in the Spleen
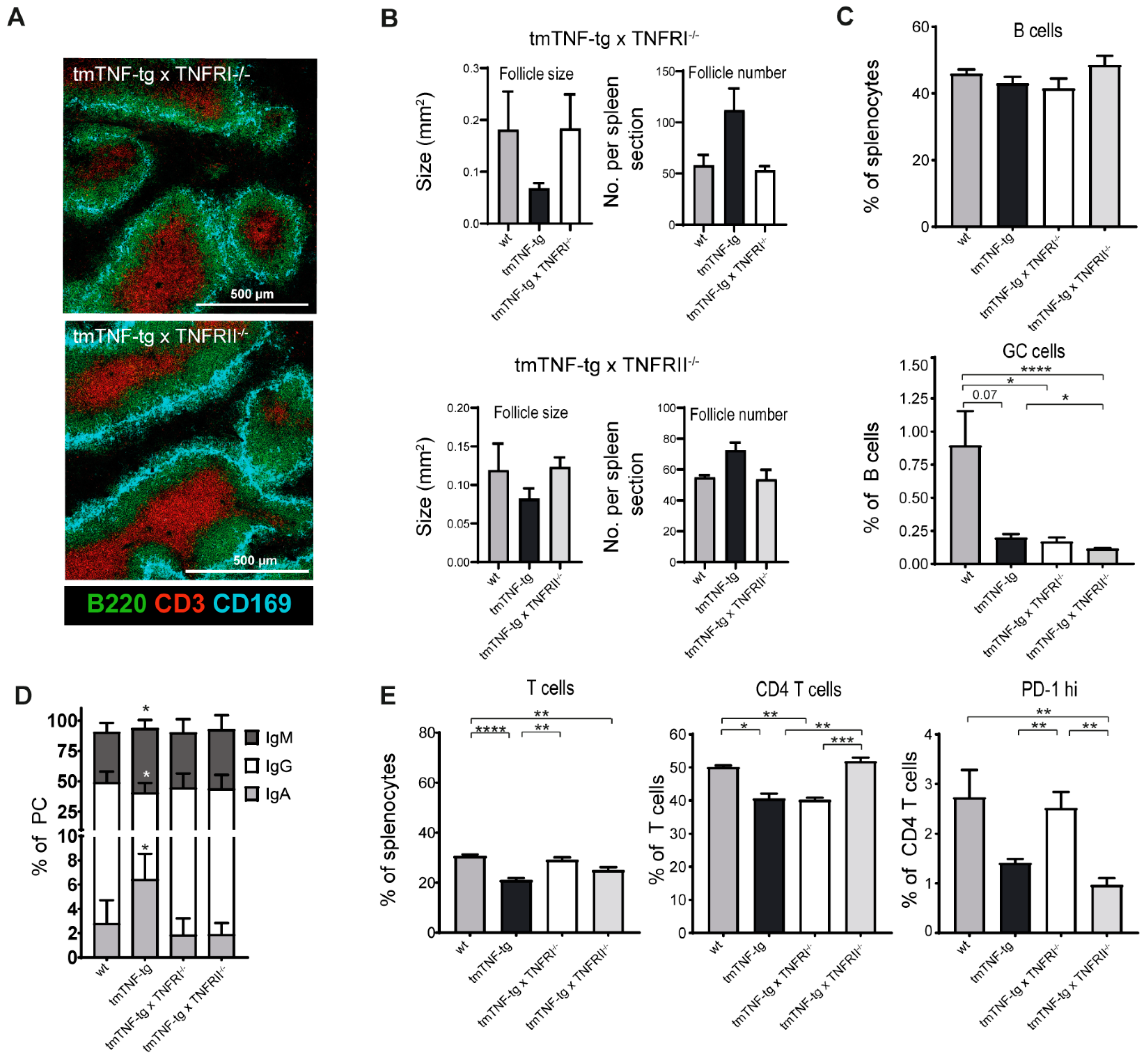
2.6. Splenic Defects Caused by tmTNF Overexpression Are Mediated via TNFRI and TNFRII Signaling
3. Discussion
4. Materials and Methods
4.1. Mice
4.2. Sample Processing
4.3. Flow Cytometry
4.4. Histology
4.5. Immunofluorescence Staining
4.6. RNA Isolation and Quantitative RT-PCR
4.7. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Randall, T.D.; Carragher, D.M.; Rangel-Moreno, J. Development of Secondary Lymphoid Organs. Annu. Rev. Immunol. 2008, 26, 627–650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stebegg, M.; Kumar, S.; Silva-Cayetano, A.; Fonseca, V.R.; Linterman, M.A.; Graca, L. Regulation of the Germinal Center Response. Front. Immunol. 2018, 9, 2469. [Google Scholar] [CrossRef] [Green Version]
- Bradley, J. TNF-mediated inflammatory disease. J. Pathol. 2008, 214, 149–160. [Google Scholar] [CrossRef]
- Holbrook, J.; Lara-Reyna, S.; Jarosz-Griffiths, H.; McDermott, M.F. Tumour necrosis factor signalling in health and disease. F1000Research 2019, 8, 111. [Google Scholar] [CrossRef]
- Horiuchi, T.; Mitoma, H.; Harashima, S.-I.; Tsukamoto, H.; Shimoda, T. Transmembrane TNF-: Structure, function and interaction with anti-TNF agents. Rheumatology 2010, 49, 1215–1228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Körner, H.; Cook, M.; Riminton, D.S.; Lemckert, F.A.; Hoek, R.M.; Ledermann, B.; Köntgen, F.; Groth, B.F.D.S.; Sedgwick, J.D. Distinct roles for lymphotoxin-α and tumor necrosis factor in organogenesis and spatial organization of lymphoid tissue. Eur. J. Immunol. 1997, 27, 2600–2609. [Google Scholar] [CrossRef] [PubMed]
- Pasparakis, M.; Alexopoulou, L.; Grell, M.; Pfizenmaier, K.; Bluethmann, H.; Kollias, G. Peyer’s patch organogenesis is intact yet formation of B lymphocyte follicles is defective in peripheral lymphoid organs of mice deficient for tumor necrosis factor and its 55-kDa receptor. Proc. Natl. Acad. Sci. USA 1997, 94, 6319–6323. [Google Scholar] [CrossRef] [Green Version]
- Jeucken, K.C.M.; Koning, J.J.; van Hamburg, J.P.; Mebius, R.E.; Tas, S.W. A Straightforward Method for 3D Visualization of B Cell Clusters and High Endothelial Venules in Lymph Nodes Highlights Differential Roles of TNFRI and -II. Front. Immunol. 2021, 12, 2492. [Google Scholar] [CrossRef]
- Alexopoulou, L.; Kranidioti, K.; Xanthoulea, S.; Denis, M.; Kotanidou, A.; Douni, E.; Blackshear, P.J.; Kontoyiannis, D.L.; Kollias, G. Transmembrane TNF protects mutant mice against intracellular bacterial infections, chronic inflammation and autoimmunity. Eur. J. Immunol. 2006, 36, 2768–2780. [Google Scholar] [CrossRef]
- Ruuls, S.R.; Hoek, R.M.; Ngo, V.; McNeil, T.; Lucian, A.L.; Janatpour, M.J.; Körner, H.; Scheerens, H.; Hessel, E.M.; Cyster, J.G.; et al. Membrane-Bound TNF Supports Secondary Lymphoid Organ Structure but Is Subservient to Secreted TNF in Driving Autoimmune Inflammation. Immunity 2001, 15, 533–543. [Google Scholar] [CrossRef] [Green Version]
- Kaaij, M.H.; Van Tok, M.N.; Blijdorp, I.C.; Ambarus, C.A.; Stock, M.; Pots, D.; Knaup, V.L.; Armaka, M.; Christodoulou-Vafeiadou, E.; Van Melsen, T.K.; et al. Transmembrane TNF drives osteoproliferative joint inflammation reminiscent of human spondyloarthritis. J. Exp. Med. 2020, 217, e20200288. [Google Scholar] [CrossRef] [PubMed]
- Alexopoulou, L.; Pasparakis, M.; Kollias, G. A murine transmembrane tumor necrosis factor (TNF) transgene induces arthritis by cooperative p55/p75 TNF receptor signaling. Eur. J. Immunol. 1997, 27, 2588–2592. [Google Scholar] [CrossRef] [PubMed]
- Campbell, I.K.; O’Donnell, K.; Lawlor, K.; Wicks, I.P. Severe inflammatory arthritis and lymphadenopathy in the absence of TNF. J. Clin. Investig. 2001, 107, 1519–1527. [Google Scholar] [CrossRef] [PubMed]
- Grabowska, J.; Lopez-Venegas, M.; Affandi, A.J.; Haan, J.D. CD169+ Macrophages Capture and Dendritic Cells Instruct: The Interplay of the Gatekeeper and the General of the Immune System. Front. Immunol. 2018, 9, 2472. [Google Scholar] [CrossRef] [Green Version]
- Kaaij, M.H.; Rip, J.; Jeucken, K.C.M.; Kan, Y.Y.; van Rooijen, C.C.N.; Saris, J.; Pots, D.; Frey, S.; Grootjans, J.; Schett, G.; et al. Overexpression of Transmembrane TNF Drives Development of Ectopic Lymphoid Structures in the Bone Marrow and B Cell Lineage Alterations in Experimental Spondyloarthritis. J. Immunol. 2021, 207, 2337–2346. [Google Scholar] [CrossRef]
- Crotty, S. T Follicular Helper Cell Biology: A Decade of Discovery and Diseases. Immunity 2019, 50, 1132–1148. [Google Scholar] [CrossRef]
- Deng, J.; Wei, Y.; Fonseca, V.R.; Graca, L.; Yu, D. T follicular helper cells and T follicular regulatory cells in rheumatic diseases. Nat. Rev. Rheumatol. 2019, 15, 475–490. [Google Scholar] [CrossRef]
- Liu, D.; Xu, H.; Shih, C.; Wan, Z.; Ma, X.; Ma, W.; Luo, D.; Qi, H. T–B-cell entanglement and ICOSL-driven feed-forward regulation of germinal centre reaction. Nature 2015, 517, 214–218. [Google Scholar] [CrossRef]
- Alexopoulou, L.; Pasparakis, M.; Kollias, G. Complementation of Lymphotoxin α Knockout Mice with Tumor Necrosis Factor–expressing Transgenes Rectifies Defective Splenic Structure and Function. J. Exp. Med. 1998, 188, 745–754. [Google Scholar] [CrossRef] [Green Version]
- Edwards, C.K., 3rd; Bendele, A.M.; Reznikov, L.I.; Fantuzzi, G.; Chlipala, E.S.; Li, L.; Moldawer, L.L.; Mountz, J.D.; Li, Y.Y.; Dinarello, C.A. Soluble human p55 and p75 tumor necrosis factor receptors reverse spontaneous arthritis in transgenic mice expressing transmembrane tumor necrosis factor alpha. Arthritis Rheum 2006, 54, 2872–2885. [Google Scholar] [CrossRef]
- Pasparakis, M.; Alexopoulou, L.; Episkopou, V.; Kollias, G. Immune and inflammatory responses in TNF alpha-deficient mice: A critical requirement for TNF alpha in the formation of primary B cell follicles, follicular dendritic cell networks and germinal centers, and in the maturation of the humoral immune response. J. Exp. Med. 1996, 184, 1397–1411. [Google Scholar] [CrossRef] [PubMed]
- Marino, M.W.; Dunn, A.; Grail, D.; Inglese, M.; Noguchi, Y.; Richards, E.; Jungbluth, A.; Wada, H.; Moore, M.; Williamson, B.; et al. Characterization of tumor necrosis factor-deficient mice. Proc. Natl. Acad. Sci. USA 1997, 94, 8093–8098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peschon, J.J.; Slack, J.L.; Reddy, P.; Stocking, K.L.; Sunnarborg, S.W.; Lee, D.C.; Russell, W.E.; Castner, B.J.; Johnson, R.S.; Fitzner, J.N.; et al. An Essential Role for Ectodomain Shedding in Mammalian Development. Science 1998, 282, 1281–1284. [Google Scholar] [CrossRef] [PubMed]
- Campbell, I.K.; Roberts, L.J.; Wicks, I.P. Molecular targets in immune-mediated diseases: The case of tumour necrosis factor and rheumatoid arthritis. Immunol. Cell Biol. 2003, 81, 354–366. [Google Scholar] [CrossRef]
- Gearing, A.J.H.; Beckett, P.; Christodoulou, M.; Churchill, M.; Clements, J.; Davidson, A.H.; Drummond, A.H.; Galloway, W.A.; Gilbert, R.; Gordon, J.L.; et al. Processing of tumour necrosis factor-α precursor by metalloproteinases. Nature 1994, 370, 555–557. [Google Scholar] [CrossRef]
- Corneth, O.B.J.; Reijmers, R.; Mus, A.M.C.; Asmawidjaja, P.S.; Van Hamburg, J.P.; Papazian, N.; Siegers, J.Y.; Mourcin, F.; Amin, R.; Tarte, K.; et al. Loss of IL-22 inhibits autoantibody formation in collagen-induced arthritis in mice. Eur. J. Immunol. 2016, 46, 1404–1414. [Google Scholar] [CrossRef]
- Moschovakis, G.L.; Bubke, A.; Friedrichsen, M.; Falk, C.S.; Feederle, R.; Förster, R. T cell specific Cxcr5 deficiency prevents rheumatoid arthritis. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef]
- Yang, S.; Wang, J.; Brand, D.; Zheng, S.G. Role of TNF–TNF Receptor 2 Signal in Regulatory T Cells and Its Therapeutic Implications. Front. Immunol. 2018, 9, 784. [Google Scholar] [CrossRef] [Green Version]
- Salomon, B.L.; Leclerc, M.; Tosello, J.; Ronin, E.; Piaggio, E.; Cohen, J.L. Tumor Necrosis Factor α and Regulatory T Cells in Oncoimmunology. Front. Immunol. 2018, 9, 444. [Google Scholar] [CrossRef]
- Pedros, C.; Zhang, Y.; Hu, J.K.; Choi, Y.S.; Canonigo-Balancio, A.J.; Yates, J.R., 3rd; Altman, A.; Crotty, S.; Kong, K.F. A TRAF-like motif of the inducible costimulator ICOS controls development of germinal center TFH cells via the kinase TBK1. Nat. Immunol. 2016, 17, 825–833. [Google Scholar] [CrossRef]
- Xu, H.; Li, X.; Liu, D.; Li, J.; Zhang, X.; Chen, X.; Hou, S.; Peng, L.; Xu, C.; Liu, W.; et al. Follicular T-helper cell recruitment governed by bystander B cells and ICOS-driven motility. Nature 2013, 496, 523–527. [Google Scholar] [CrossRef] [PubMed]
- Tumanov, A.V.; Grivennikov, S.I.; Kruglov, A.; Shebzukhov, Y.V.; Koroleva, E.P.; Piao, Y.; Cui, C.-Y.; Kuprash, D.; Nedospasov, S.A. Cellular source and molecular form of TNF specify its distinct functions in organization of secondary lymphoid organs. Blood 2010, 116, 3456–3464. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.-X.; Huang, G.; Matsumoto, M.; Molina, H.; Chaplin, D.D. Independent signals regulate development of primary and secondary follicle structure in spleen and mesenteric lymph node. Proc. Natl. Acad. Sci. USA 1997, 94, 5739–5743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaminski, D.A.; Stavnezer, J. Enhanced IgA Class Switching in Marginal Zone and B1 B Cells Relative to Follicular/B2 B Cells. J. Immunol. 2006, 177, 6025–6029. [Google Scholar] [CrossRef]
- Cerutti, A.; Cols, M.; Puga, I. Marginal zone B cells: Virtues of innate-like antibody-producing lymphocytes. Nat. Rev. Immunol. 2013, 13, 118–132. [Google Scholar] [CrossRef] [Green Version]
- Duan, Z.; Zheng, H.; Liu, H.; Li, M.; Tang, M.; Weng, X.; Yi, W.; Bode, A.M.; Cao, Y. AID expression increased by TNF-alpha is associated with class switch recombination of Igalpha gene in cancers. Cell Mol. Immunol. 2016, 13, 484–491. [Google Scholar] [CrossRef] [Green Version]
- Jin, J.; Xiao, Y.; Chang, J.H.; Yu, J.; Hu, H.; Starr, R.; Brittain, G.C.; Chang, M.; Cheng, X.; Sun, S.C. The kinase TBK1 controls IgA class switching by negatively regulating noncanonical NF-kappaB signaling. Nat. Immunol. 2012, 13, 1101–1109. [Google Scholar] [CrossRef] [Green Version]
- Tucker, E.; O’Donnell, K.; Fuchsberger, M.; Hilton, A.A.; Metcalf, D.; Greig, K.; Sims, N.A.; Quinn, J.M.; Alexander, W.S.; Hilton, D.J.; et al. A novel mutation in the Nfkb2 gene generates an NF-kappa B2 “super repressor”. J. Immunol. 2007, 179, 7514–7522. [Google Scholar] [CrossRef] [Green Version]
- Schulz, O.; Hammerschmidt, S.I.; Moschovakis, G.L.; Förster, R. Chemokines and Chemokine Receptors in Lymphoid Tissue Dynamics. Annu. Rev. Immunol. 2016, 34, 203–242. [Google Scholar] [CrossRef]
- Yang, S.; Xie, C.; Chen, Y.; Wang, J.; Chen, X.; Lu, Z.; June, R.R.; Zheng, S.G. Differential roles of TNFalpha-TNFR1 and TNFalpha-TNFR2 in the differentiation and function of CD4(+)Foxp3(+) induced Treg cells in vitro and in vivo periphery in autoimmune diseases. Cell Death Dis. 2019, 10, 27. [Google Scholar] [CrossRef]
- Naudé, P.J.W.; Boer, J.A.D.; Luiten, P.G.M.; Eisel, U.L.M. Tumor necrosis factor receptor cross-talk. FEBS J. 2011, 278, 888–898. [Google Scholar] [CrossRef] [PubMed]
- Le Hir, M.; Bluethmann, H.; Kosco-Vilbois, M.H.; Müller, M.; di Padova, F.; Moore, M.; Ryffel, B.; Eugster, H.P. Differentiation of follicular dendritic cells and full antibody responses require tumor necrosis factor receptor-1 signaling. J. Exp. Med. 1996, 183, 2367–2372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peschon, J.J.; Torrance, D.S.; Stocking, K.L.; Glaccum, M.B.; Otten, C.; Willis, C.R.; Charrier, K.; Morrissey, P.J.; Ware, C.B.; Mohler, K.M. TNF receptor-deficient mice reveal divergent roles for p55 and p75 in several models of inflammation. J. Immunol. 1998, 160, 943–952. [Google Scholar]
- MacEwan, D.J. TNF receptor subtype signalling: Differences and cellular consequences. Cell. Signal. 2002, 14, 477–492. [Google Scholar] [CrossRef]
- Lata, M.; Hettinghouse, A.S.; Liu, C. Targeting tumor necrosis factor receptors in ankylosing spondylitis. Ann. N. Y. Acad. Sci. 2018, 1442, 5–16. [Google Scholar] [CrossRef]
- Van Hauwermeiren, F.; Vandenbroucke, R.E.; Libert, C. Treatment of TNF mediated diseases by selective inhibition of soluble TNF or TNFR1. Cytokine Growth Factor Rev. 2011, 22, 311–319. [Google Scholar] [CrossRef]
- Salomon, B.L. Insights into the biology and therapeutic implications of TNF and regulatory T cells. Nat. Rev. Rheumatol. 2021, 17, 487–504. [Google Scholar] [CrossRef]
- Medler, J.; Wajant, H. Tumor necrosis factor receptor-2 (TNFR2): An overview of an emerging drug target. Expert Opin. Ther. Targets 2019, 23, 295–307. [Google Scholar] [CrossRef]
- Ticha, O.; Slanina, P.; Moos, L.; Stichova, J.; Vlkova, M.; Bekeredjian-Ding, I. TNFR2 expression is a hallmark of human memory B cells with suppressive function. Eur. J. Immunol. 2021, 51, 1195–1205. [Google Scholar] [CrossRef]
- Udalova, I.; Monaco, C.; Nanchahal, J.; Feldmann, M. Anti-TNF therapy. Microbiol. Spectrum. 2016, 4, 4. [Google Scholar] [CrossRef] [Green Version]
- Atreya, R.; Neumann, H.; Neufert, C.; Waldner, M.J.; Billmeier, U.; Zopf, Y.; Willma, M.; App, C.; Munster, T.; Kessler, H.; et al. In vivo imaging using fluorescent antibodies to tumor necrosis factor predicts therapeutic response in Crohn’s disease. Nat. Med. 2014, 20, 313–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jeucken, K.C.M.; Kaaij, M.H.; Rip, J.; van Rooijen, C.C.N.; Kan, Y.Y.; Corneth, O.B.J.; van Hamburg, J.P.; Tas, S.W. Splenic Architecture and Function Requires Tight Control of Transmembrane TNF Expression. Int. J. Mol. Sci. 2022, 23, 2229. https://doi.org/10.3390/ijms23042229
Jeucken KCM, Kaaij MH, Rip J, van Rooijen CCN, Kan YY, Corneth OBJ, van Hamburg JP, Tas SW. Splenic Architecture and Function Requires Tight Control of Transmembrane TNF Expression. International Journal of Molecular Sciences. 2022; 23(4):2229. https://doi.org/10.3390/ijms23042229
Chicago/Turabian StyleJeucken, Kim C. M., Merlijn H. Kaaij, Jasper Rip, Charlotte C. N. van Rooijen, Yik Y. Kan, Odilia B. J. Corneth, Jan Piet van Hamburg, and Sander W. Tas. 2022. "Splenic Architecture and Function Requires Tight Control of Transmembrane TNF Expression" International Journal of Molecular Sciences 23, no. 4: 2229. https://doi.org/10.3390/ijms23042229
APA StyleJeucken, K. C. M., Kaaij, M. H., Rip, J., van Rooijen, C. C. N., Kan, Y. Y., Corneth, O. B. J., van Hamburg, J. P., & Tas, S. W. (2022). Splenic Architecture and Function Requires Tight Control of Transmembrane TNF Expression. International Journal of Molecular Sciences, 23(4), 2229. https://doi.org/10.3390/ijms23042229