
New and Emerging Targeted Therapies for Advanced Breast Cancer


Abstract
:1. Introduction
2. Targeted Therapies for Endocrine Therapy Resistance
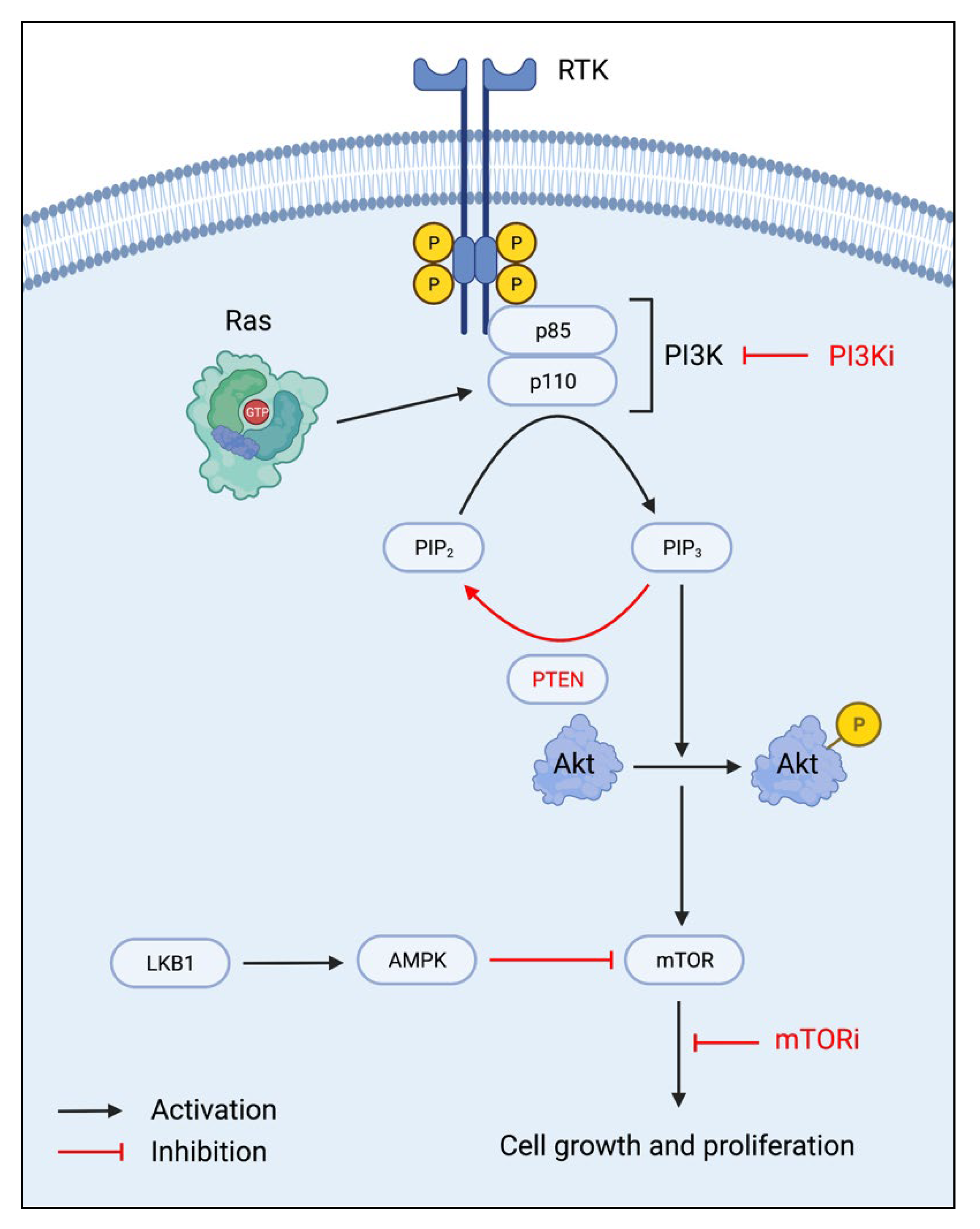
2.1. mTOR Inhibitors
2.2. PI3K Inhibitors
2.3. PTEN Upregulation
2.4. LKB1-AMPK Activation
3. HER2-Positive Targeted Therapies
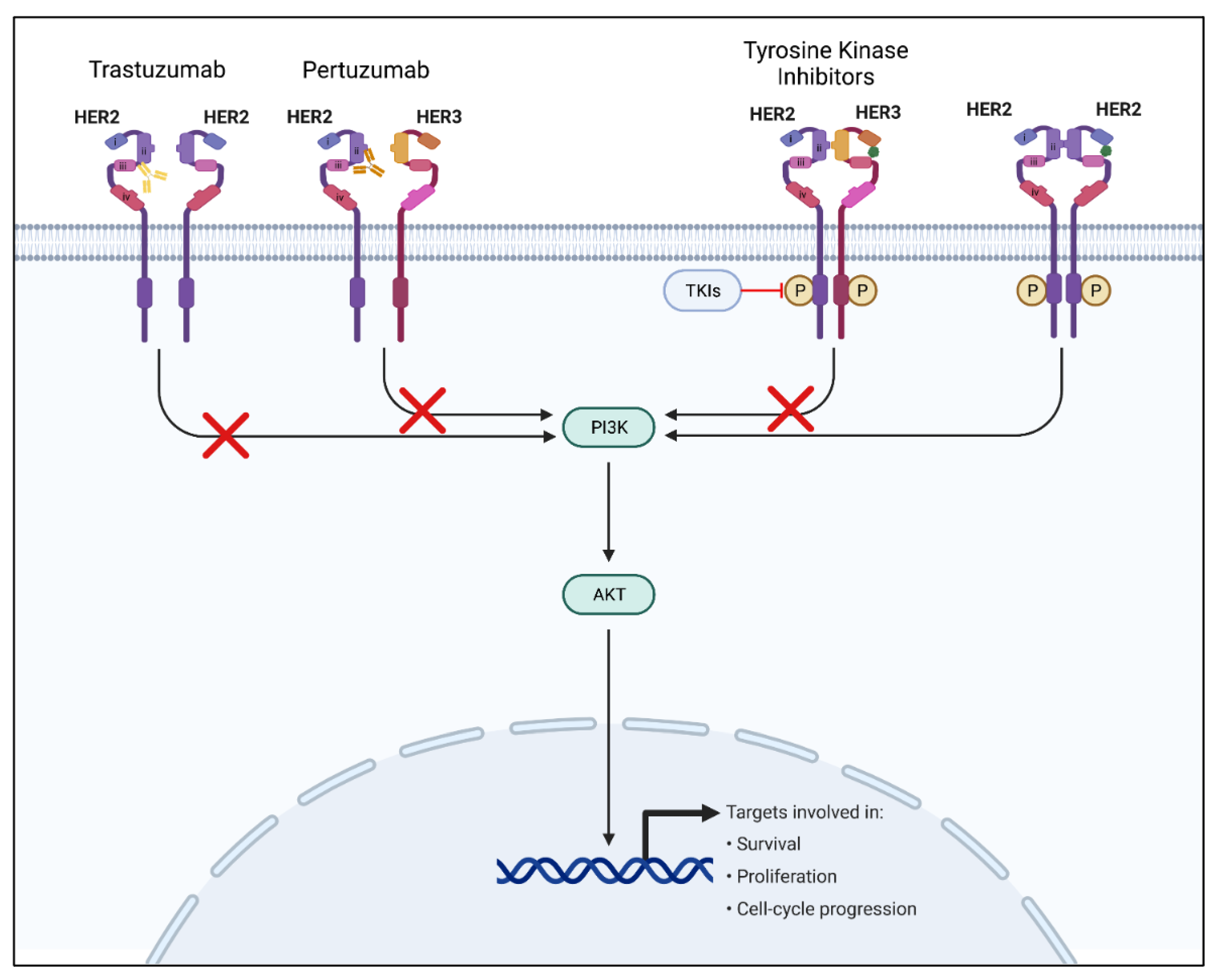
3.1. Tyrosine Kinase Inhibitors
3.2. Monoclonal Antibodies
3.3. Antibody-Drug Conjugates
4. HER2-Negative Targeted Therapies
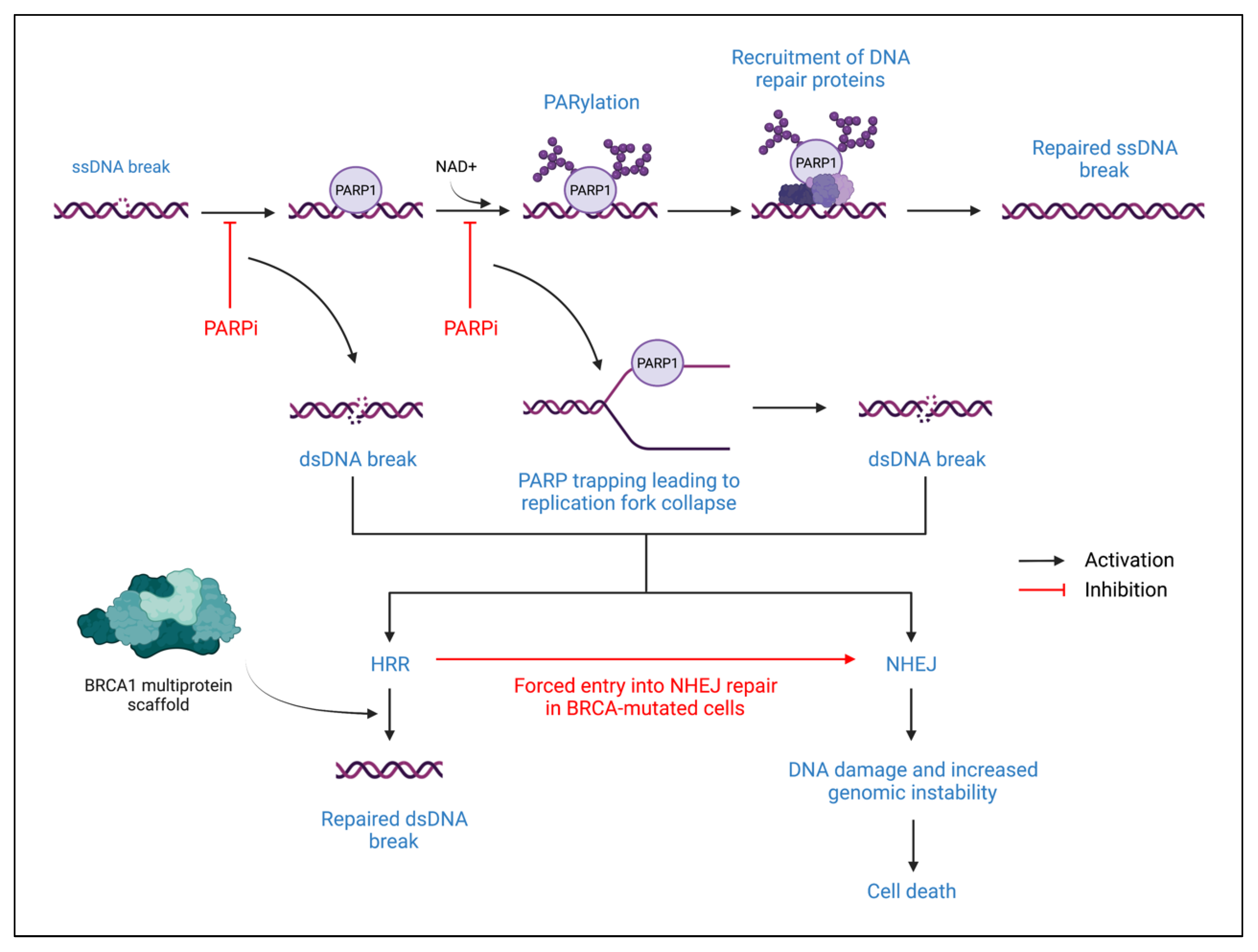
4.1. PARP Inhibitors
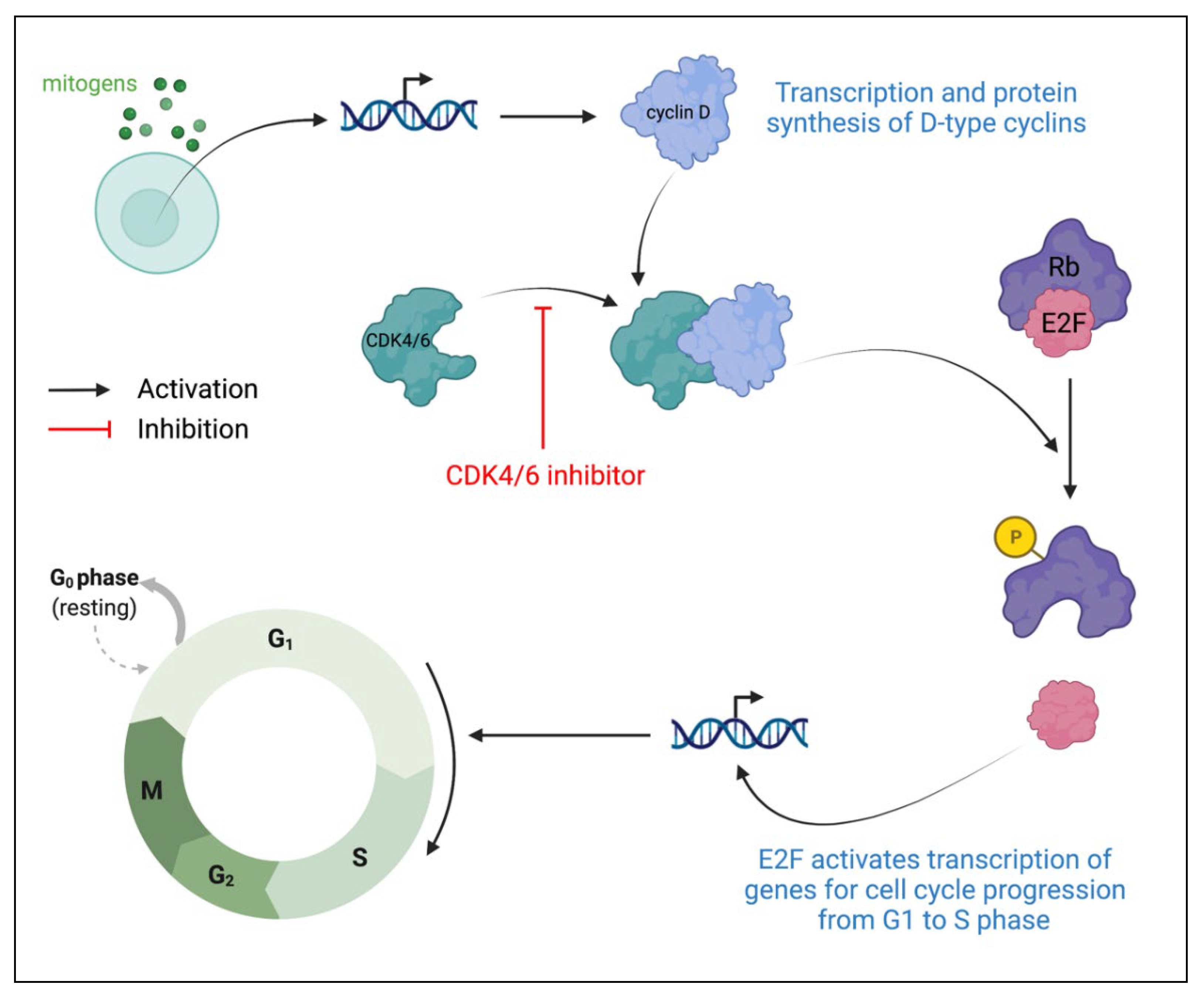
4.2. CDK4/6 Inhibitors
4.3. Antibody-Drug Conjugates
5. Immunotherapy
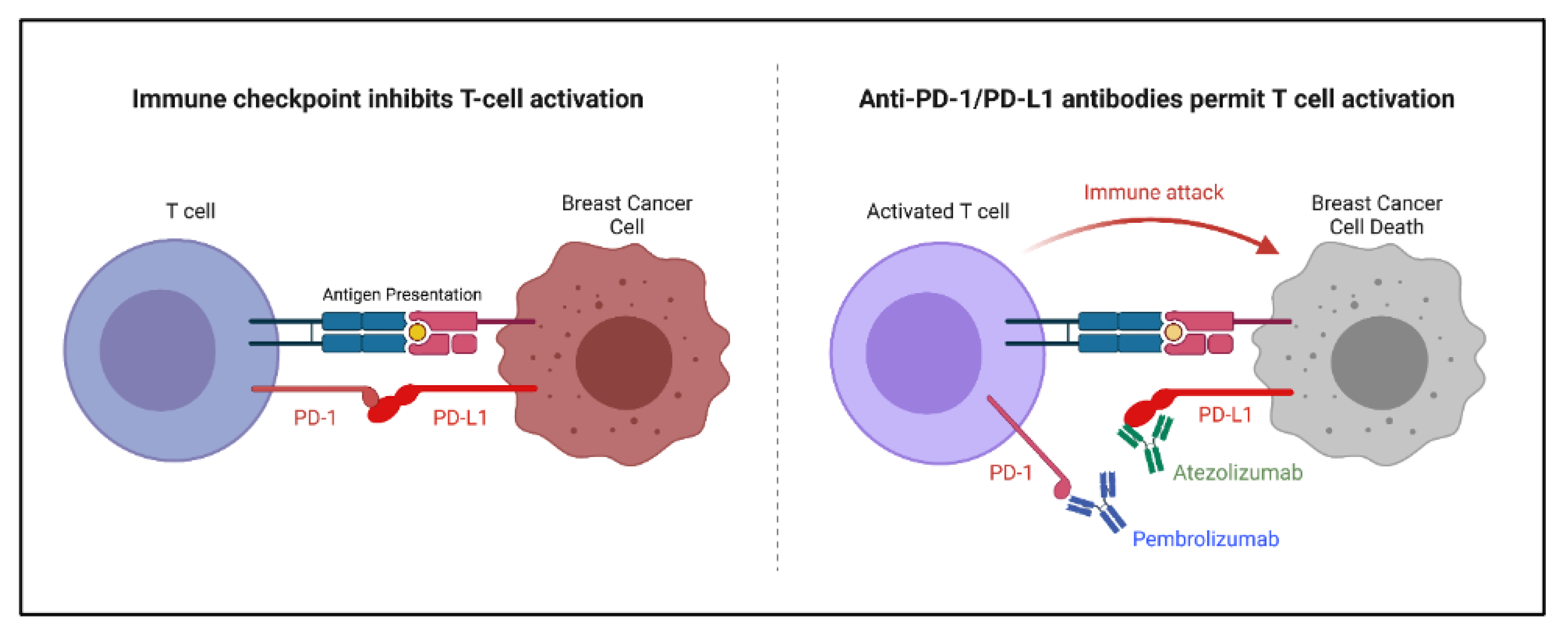
5.1. Immune Checkpoint Inhibitors
5.2. Cancer Vaccines
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ADC | Antibody-drug conjugate |
| ADCC | Antibody-dependent cell-mediated cytotoxicity |
| AI | Aromatase inhibitor |
| Akt | Protein kinase B |
| AMPK | Adenosine monophosphate-activated protein kinase |
| APC | Antigen-presenting cell |
| ATM | Ataxia telangiectasia-mutated kinase |
| ATP | Adenosine triphosphate |
| BC | Breast cancer |
| BRCA | Breast cancer-associated protein |
| CDK | Cyclin-dependent kinase |
| CK | Cytokeratins |
| CNS | Central nervous system |
| DNA | Deoxyribonucleic acid |
| dsDNA | Double-stranded deoxyribonucleic acid |
| EGFR/ERB | Epidermal growth factor receptor |
| ER | Estrogen receptor |
| FDA | Food and drug administration |
| gBRCA | Germline breast cancer associated protein gene |
| GM-CSF | Granulocyte-macrophage colony-stimulating factor |
| HER1 | Human epidermal growth factor receptor 1 |
| HER2 | Human epidermal growth factor receptor 2 |
| HER3 | Human epidermal growth factor receptor 3 |
| HER4 | Human epidermal growth factor receptor 4 |
| HLA-A2 | Human leukocyte antigen-A2 |
| HR | Hormone receptor |
| HRR | Homologous recombination repair |
| LKB1 | Liver kinase B1 |
| LumA | Luminal-A |
| LumB | Luminal-B |
| mTOR | Mammalian target of rapamycin |
| NAD+ | Nicotinamide adenine dinucleotide |
| NHEJ | Non-homologous end joining |
| PAM | PI3K/Akt/mTOR pathway |
| PARP | Poly-ADP-ribose polymerase |
| PARPi | Poly-ADP-ribose polymerase inhibitor |
| PARylation | Poly-ADP-ribosylation |
| PIK3CA | Phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha gene |
| PIP2 | Phosphatidylinositol-4,5-biphosphate |
| PIP3 | Phosphatidylinositol-3,4,5-triphosphate |
| PI3K | Phosphoinositide 3-kinase |
| PR | Progesterone receptor |
| PD-1 | Programmed cell death protein 1 |
| PD-L1 | Programmed cell death ligand 1 |
| PTEN | Phosphatase and tensin homolog |
| Rb | Retinoblastoma protein |
| RTK | Receptor tyrosine kinase |
| SERD | Selective estrogen receptor degrader |
| SERM | Selective estrogen receptor modulator |
| TAA | Tumor-associated antigen |
| T-DM1 | Trastuzumab emtansine |
| T-DXd | Trastuzumab deruxtecan |
| TGF-β | Transforming growth factor beta |
| TKI | Tyrosine kinase inhibitor |
| TNBC | Triple-negative breast cancer |
| Trop-2 | Trophoblast cell-surface antigen 2 |
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Breast Cancer Facts & Figures 2019–2020. Available online: https://www.cancer.org/research/cancer-facts-statistics/breast-cancer-facts-figures.html (accessed on 14 February 2022).
- Li, C.; Fan, Z.; Lin, X.; Cao, M.; Song, F.; Song, F. Parity and risk of developing breast cancer according to tumor subtype: A systematic review and meta-analysis. Cancer Epidemiol. 2021, 75, 102050. [Google Scholar] [CrossRef] [PubMed]
- Carey, L.A.; Perou, C.M.; Livasy, C.A.; Dressler, L.G.; Cowan, D.; Conway, K.; Karaca, G.; Troester, M.A.; Tse, C.K.; Edmiston, S.; et al. Race, breast cancer subtypes, and survival in the Carolina Breast Cancer Study. JAMA 2006, 295, 2492–2502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haque, R.; Ahmed, S.A.; Inzhakova, G.; Shi, J.; Avila, C.; Polikoff, J.; Bernstein, L.; Enger, S.M.; Press, M.F. Impact of breast cancer subtypes and treatment on survival: An analysis spanning two decades. Cancer Epidemiol. Biomark. Prev. 2012, 21, 1848–1855. [Google Scholar] [CrossRef] [Green Version]
- Incidence and Relative Survival by Stage at Diagnosis for Common Cancers. Available online: https://www.cdc.gov/cancer/uscs/about/data-briefs/no25-incidence-relative-survival-stage-diagnosis.htm (accessed on 29 November 2021).
- Elston, C.W.; Ellis, I.O. Pathological prognostic factors in breast cancer. I. The value of histological grade in breast cancer: Experience from a large study with long-term follow-up. Histopathology 1991, 19, 403–410. [Google Scholar] [CrossRef]
- Harbeck, N.; Gnant, M. Breast cancer. Lancet 2017, 389, 1134–1150. [Google Scholar] [CrossRef]
- Koh, S.B.; Ellisen, L.W. Immune activation and evolution through chemotherapy plus checkpoint blockade in triple-negative breast cancer. Cancer Cell 2021, 39, 1562–1564. [Google Scholar] [CrossRef]
- Shanmugam, G.; Rakshit, S.; Sarkar, K. HDAC inhibitors: Targets for tumor therapy, immune modulation and lung diseases. Transl. Oncol. 2022, 16, 101312. [Google Scholar] [CrossRef]
- Manso, L.; Salvador, F.; Villagrasa, P.; Chic, N.; Bermejo, B.; Cejalvo, J.M.; Izarzugaza, Y.; Cantos, B.; Blanch, S.; Margeli, M.; et al. Abstract CT191: A window-of-opportunity study with atezolizumab and the oncolytic virus pelareorep in early breast cancer (AWARE-1). Cancer Res. 2021, 81, CT191. [Google Scholar] [CrossRef]
- Paplomata, E.; O’Regan, R. The PI3K/AKT/mTOR pathway in breast cancer: Targets, trials and biomarkers. Ther. Adv. Med. Oncol. 2014, 6, 154–166. [Google Scholar] [CrossRef] [Green Version]
- Chang, F.; Lee, J.T.; Navolanic, P.M.; Steelman, L.S.; Shelton, J.G.; Blalock, W.L.; Franklin, R.A.; McCubrey, J.A. Involvement of PI3K/Akt pathway in cell cycle progression, apoptosis, and neoplastic transformation: A target for cancer chemotherapy. Leukemia 2003, 17, 590–603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arena, F. Clinical implications of recent studies using mTOR inhibitors to treat advanced hormone receptor-positive breast cancer. Cancer Manag. Res. 2014, 6, 389–395. [Google Scholar] [CrossRef] [Green Version]
- Carbognin, L.; Miglietta, F.; Paris, I.; Dieci, M.V. Prognostic and Predictive Implications of PTEN in Breast Cancer: Unfulfilled Promises but Intriguing Perspectives. Cancers 2019, 11, 1401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steelman, L.S.; Martelli, A.M.; Cocco, L.; Libra, M.; Nicoletti, F.; Abrams, S.L.; McCubrey, J.A. The therapeutic potential of mTOR inhibitors in breast cancer. Br. J. Clin. Pharmacol. 2016, 82, 1189–1212. [Google Scholar] [CrossRef] [PubMed]
- Royce, M.E.; Osman, D. Everolimus in the Treatment of Metastatic Breast Cancer. Breast Cancer 2015, 9, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Riccardi, F.; Colantuoni, G.; Diana, A.; Mocerino, C.; Carteni, G.; Lauria, R.; Febbraro, A.; Nuzzo, F.; Addeo, R.; Marano, O.; et al. Exemestane and Everolimus combination treatment of hormone receptor positive, HER2 negative metastatic breast cancer: A retrospective study of 9 cancer centers in the Campania Region (Southern Italy) focused on activity, efficacy and safety. Mol. Clin. Oncol. 2018, 9, 255–263. [Google Scholar] [CrossRef] [Green Version]
- Kwitkowski, V.E.; Prowell, T.M.; Ibrahim, A.; Farrell, A.T.; Justice, R.; Mitchell, S.S.; Sridhara, R.; Pazdur, R. FDA approval summary: Temsirolimus as treatment for advanced renal cell carcinoma. Oncologist 2010, 15, 428–435. [Google Scholar] [CrossRef] [Green Version]
- Chan, S.; Scheulen, M.E.; Johnston, S.; Mross, K.; Cardoso, F.; Dittrich, C.; Eiermann, W.; Hess, D.; Morant, R.; Semiglazov, V.; et al. Phase II study of temsirolimus (CCI-779), a novel inhibitor of mTOR, in heavily pretreated patients with locally advanced or metastatic breast cancer. J. Clin. Oncol. 2005, 23, 5314–5322. [Google Scholar] [CrossRef]
- Fleming, G.F.; Ma, C.X.; Huo, D.; Sattar, H.; Tretiakova, M.; Lin, L.; Hahn, O.M.; Olopade, F.O.; Nanda, R.; Hoffman, P.C.; et al. Phase II trial of temsirolimus in patients with metastatic breast cancer. Breast Cancer Res. Treat. 2012, 136, 355–363. [Google Scholar] [CrossRef] [Green Version]
- Andre, F.; Ciruelos, E.; Rubovszky, G.; Campone, M.; Loibl, S.; Rugo, H.S.; Iwata, H.; Conte, P.; Mayer, I.A.; Kaufman, B.; et al. Alpelisib for PIK3CA-Mutated, Hormone Receptor-Positive Advanced Breast Cancer. N. Engl. J. Med. 2019, 380, 1929–1940. [Google Scholar] [CrossRef]
- Rugo, H.S.; Lerebours, F.; Ciruelos, E.; Drullinsky, P.; Ruiz-Borrego, M.; Neven, P.; Park, Y.H.; Prat, A.; Bachelot, T.; Juric, D.; et al. Alpelisib plus fulvestrant in PIK3CA-mutated, hormone receptor-positive advanced breast cancer after a CDK4/6 inhibitor (BYLieve): One cohort of a phase 2, multicentre, open-label, non-comparative study. Lancet Oncol. 2021, 22, 489–498. [Google Scholar] [CrossRef]
- Turner, S.; Chia, S.; Kanakamedala, H.; Hsu, W.C.; Park, J.; Chandiwana, D.; Ridolfi, A.; Yu, C.L.; Zarate, J.P.; Rugo, H.S. Effectiveness of Alpelisib + Fulvestrant Compared with Real-World Standard Treatment Among Patients with HR+, HER2-, PIK3CA-Mutated Breast Cancer. Oncologist 2021, 26, e1133–e1142. [Google Scholar] [CrossRef] [PubMed]
- Janku, F. Phosphoinositide 3-kinase (PI3K) pathway inhibitors in solid tumors: From laboratory to patients. Cancer Treat. Rev. 2017, 59, 93–101. [Google Scholar] [CrossRef] [Green Version]
- Dent, S.; Cortes, J.; Im, Y.H.; Dieras, V.; Harbeck, N.; Krop, I.E.; Wilson, T.R.; Cui, N.; Schimmoller, F.; Hsu, J.Y.; et al. Phase III randomized study of taselisib or placebo with fulvestrant in estrogen receptor-positive, PIK3CA-mutant, HER2-negative, advanced breast cancer: The SANDPIPER trial. Ann. Oncol. 2021, 32, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Jhaveri, K.; Chang, M.T.; Juric, D.; Saura, C.; Gambardella, V.; Melnyk, A.; Patel, M.R.; Ribrag, V.; Ma, C.X.; Aljumaily, R.; et al. Phase I Basket Study of Taselisib, an Isoform-Selective PI3K Inhibitor, in Patients with PIK3CA-Mutant Cancers. Clin. Cancer Res. 2021, 27, 447–459. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Jang, H.; Nussinov, R. PI3K inhibitors: Review and new strategies. Chem. Sci. 2020, 11, 5855–5865. [Google Scholar] [CrossRef]
- Schoffski, P.; Cresta, S.; Mayer, I.A.; Wildiers, H.; Damian, S.; Gendreau, S.; Rooney, I.; Morrissey, K.M.; Spoerke, J.M.; Ng, V.W.; et al. A phase Ib study of pictilisib (GDC-0941) in combination with paclitaxel, with and without bevacizumab or trastuzumab, and with letrozole in advanced breast cancer. Breast Cancer Res. 2018, 20, 109. [Google Scholar] [CrossRef]
- Garrido-Castro, A.C.; Saura, C.; Barroso-Sousa, R.; Guo, H.; Ciruelos, E.; Bermejo, B.; Gavila, J.; Serra, V.; Prat, A.; Pare, L.; et al. Phase 2 study of buparlisib (BKM120), a pan-class I PI3K inhibitor, in patients with metastatic triple-negative breast cancer. Breast Cancer Res. 2020, 22, 120. [Google Scholar] [CrossRef]
- Moses, C.; Nugent, F.; Waryah, C.B.; Garcia-Bloj, B.; Harvey, A.R.; Blancafort, P. Activating PTEN Tumor Suppressor Expression with the CRISPR/dCas9 System. Mol. Ther. Nucleic Acids 2019, 14, 287–300. [Google Scholar] [CrossRef] [Green Version]
- De Amicis, F.; Aquila, S.; Morelli, C.; Guido, C.; Santoro, M.; Perrotta, I.; Mauro, L.; Giordano, F.; Nigro, A.; Ando, S.; et al. Bergapten drives autophagy through the up-regulation of PTEN expression in breast cancer cells. Mol. Cancer 2015, 14, 130. [Google Scholar] [CrossRef] [Green Version]
- Wu, Q.X.; Yuan, S.X.; Ren, C.M.; Yu, Y.; Sun, W.J.; He, B.C.; Wu, K. Oridonin upregulates PTEN through activating p38 MAPK and inhibits proliferation in human colon cancer cells. Oncol. Rep. 2016, 35, 3341–3348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, N.; Huang, D.; Lu, N.; Luo, L. Role of the LKB1/AMPK pathway in tumor invasion and metastasis of cancer cells (Review). Oncol. Rep. 2015, 34, 2821–2826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, N.S.; Zou, J.R.; Lin, H.; Ke, R.; He, X.L.; Xiao, L.; Huang, D.; Luo, L.; Lv, N.; Luo, Z. LKB1/AMPK inhibits TGF-beta1 production and the TGF-beta signaling pathway in breast cancer cells. Tumor Biol. 2016, 37, 8249–8258. [Google Scholar] [CrossRef] [Green Version]
- Nagalingam, A.; Arbiser, J.L.; Bonner, M.Y.; Saxena, N.K.; Sharma, D. Honokiol activates AMP-activated protein kinase in breast cancer cells via an LKB1-dependent pathway and inhibits breast carcinogenesis. Breast Cancer Res. 2012, 14, R35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arora, S.; Singh, S.; Piazza, G.A.; Contreras, C.M.; Panyam, J.; Singh, A.P. Honokiol: A novel natural agent for cancer prevention and therapy. Curr. Mol. Med. 2012, 12, 1244–1252. [Google Scholar] [CrossRef] [PubMed]
- Schlam, I.; Swain, S.M. HER2-positive breast cancer and tyrosine kinase inhibitors: The time is now. NPJ Breast Cancer 2021, 7, 56. [Google Scholar] [CrossRef] [PubMed]
- Mitri, Z.; Constantine, T.; O’Regan, R. The HER2 Receptor in Breast Cancer: Pathophysiology, Clinical Use, and New Advances in Therapy. Chemother. Res. Pract. 2012, 2012, 743193. [Google Scholar] [CrossRef] [Green Version]
- Yu, S.; Liu, Q.; Han, X.; Qin, S.; Zhao, W.; Li, A.; Wu, K. Development and clinical application of anti-HER2 monoclonal and bispecific antibodies for cancer treatment. Exp. Hematol. Oncol. 2017, 6, 31. [Google Scholar] [CrossRef] [Green Version]
- Xuhong, J.C.; Qi, X.W.; Zhang, Y.; Jiang, J. Mechanism, safety and efficacy of three tyrosine kinase inhibitors lapatinib, neratinib and pyrotinib in HER2-positive breast cancer. Am. J. Cancer Res. 2019, 9, 2103–2119. [Google Scholar]
- Ryan, Q.; Ibrahim, A.; Cohen, M.H.; Johnson, J.; Ko, C.W.; Sridhara, R.; Justice, R.; Pazdur, R. FDA drug approval summary: Lapatinib in combination with capecitabine for previously treated metastatic breast cancer that overexpresses HER-2. Oncologist 2008, 13, 1114–1119. [Google Scholar] [CrossRef]
- Voigtlaender, M.; Schneider-Merck, T.; Trepel, M. Lapatinib. Recent Results Cancer Res. 2018, 211, 19–44. [Google Scholar] [CrossRef] [PubMed]
- Incorvati, J.A.; Shah, S.; Mu, Y.; Lu, J. Targeted therapy for HER2 positive breast cancer. J. Hematol. Oncol. 2013, 6, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomez, H.L.; Doval, D.C.; Chavez, M.A.; Ang, P.C.; Aziz, Z.; Nag, S.; Ng, C.; Franco, S.X.; Chow, L.W.; Arbushites, M.C.; et al. Efficacy and safety of lapatinib as first-line therapy for ErbB2-amplified locally advanced or metastatic breast cancer. J. Clin. Oncol. 2008, 26, 2999–3005. [Google Scholar] [CrossRef]
- Oliveira, M.; Garrigos, L.; Assaf, J.D.; Escriva-de-Romani, S.; Saura, C. Neratinib plus capecitabine for the treatment of advanced HER2-positive breast cancer. Expert Rev. Anticancer Ther. 2020, 20, 731–741. [Google Scholar] [CrossRef]
- Neratinib for breast cancer. Aust. Prescr. 2019, 42, 209–210. [CrossRef] [PubMed] [Green Version]
- Blair, H.A. Pyrotinib: First Global Approval. Drugs 2018, 78, 1751–1755. [Google Scholar] [CrossRef]
- Ulrich, L.; Okines, A.F.C. Treating Advanced Unresectable or Metastatic HER2-Positive Breast Cancer: A Spotlight on Tucatinib. Breast Cancer 2021, 13, 361–381. [Google Scholar] [CrossRef]
- Wuerstlein, R.; Harbeck, N. Neoadjuvant Therapy for HER2-positive Breast Cancer. Rev. Recent Clin. Trials 2017, 12, 81–92. [Google Scholar] [CrossRef]
- Richard, S.; Selle, F.; Lotz, J.P.; Khalil, A.; Gligorov, J.; Soares, D.G. Pertuzumab and trastuzumab: The rationale way to synergy. An. Acad. Bras. Cienc. 2016, 88 (Suppl. 1), 565–577. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Miao, W.; He, D.; Wang, S.; Lou, J.; Jiang, Y.; Wang, S. Recent Progress on Immunotherapy for Breast Cancer: Tumor Microenvironment, Nanotechnology and More. Front. Bioeng. Biotechnol. 2021, 9, 680315. [Google Scholar] [CrossRef]
- Rugo, H.S.; Im, S.A.; Cardoso, F.; Cortes, J.; Curigliano, G.; Musolino, A.; Pegram, M.D.; Wright, G.S.; Saura, C.; Escriva-de-Romani, S.; et al. Efficacy of Margetuximab vs Trastuzumab in Patients with Pretreated ERBB2-Positive Advanced Breast Cancer: A Phase 3 Randomized Clinical Trial. JAMA Oncol. 2021, 7, 573–584. [Google Scholar] [CrossRef] [PubMed]
- Costa, R.L.B.; Czerniecki, B.J. Clinical development of immunotherapies for HER2+ breast cancer: A review of HER2-directed monoclonal antibodies and beyond. NPJ Breast Cancer 2020, 6, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakada, T.; Sugihara, K.; Jikoh, T.; Abe, Y.; Agatsuma, T. The Latest Research and Development into the Antibody-Drug Conjugate, [fam-] Trastuzumab Deruxtecan (DS-8201a), for HER2 Cancer Therapy. Chem. Pharm. Bull. 2019, 67, 173–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferraro, E.; Drago, J.Z.; Modi, S. Implementing antibody-drug conjugates (ADCs) in HER2-positive breast cancer: State of the art and future directions. Breast Cancer Res. 2021, 23, 84. [Google Scholar] [CrossRef]
- Herceg, Z.; Wang, Z.Q. Functions of poly(ADP-ribose) polymerase (PARP) in DNA repair, genomic integrity and cell death. Mutat. Res. 2001, 477, 97–110. [Google Scholar] [CrossRef]
- Cortesi, L.; Rugo, H.S.; Jackisch, C. An Overview of PARP Inhibitors for the Treatment of Breast Cancer. Target. Oncol. 2021, 16, 255–282. [Google Scholar] [CrossRef]
- Goncalves, A.; Bertucci, A.; Bertucci, F. PARP Inhibitors in the Treatment of Early Breast Cancer: The Step Beyond? Cancers 2020, 12, 1378. [Google Scholar] [CrossRef]
- Goulooze, S.C.; Cohen, A.F.; Rissmann, R. Olaparib. Br. J. Clin. Pharmacol. 2016, 81, 171–173. [Google Scholar] [CrossRef] [Green Version]
- Rose, M.; Burgess, J.T.; O’Byrne, K.; Richard, D.J.; Bolderson, E. PARP Inhibitors: Clinical Relevance, Mechanisms of Action and Tumor Resistance. Front. Cell Dev. Biol. 2020, 8, 564601. [Google Scholar] [CrossRef]
- Sun, T.; Shi, Y.; Cui, J.; Yin, Y.; Ouyang, Q.; Liu, Q.; Zhang, Q.; Chen, Y.; Zhimin, S.; Wang, S.; et al. A phase 2 study of pamiparib in the treatment of patients with locally advanced or metastatic HER2-negative breast cancer with germline BRCA mutation. J. Clin. Oncol. 2021, 39, 1087. [Google Scholar] [CrossRef]
- Tigan, A.S.; Bellutti, F.; Kollmann, K.; Tebb, G.; Sexl, V. CDK6-a review of the past and a glimpse into the future: From cell-cycle control to transcriptional regulation. Oncogene 2016, 35, 3083–3091. [Google Scholar] [CrossRef] [PubMed]
- Du, Q.; Guo, X.; Wang, M.; Li, Y.; Sun, X.; Li, Q. The application and prospect of CDK4/6 inhibitors in malignant solid tumors. J. Hematol. Oncol. 2020, 13, 41. [Google Scholar] [CrossRef] [PubMed]
- Corona, S.P.; Generali, D. Abemaciclib: A CDK4/6 inhibitor for the treatment of HR+/HER2- advanced breast cancer. Drug Des. Dev. Ther. 2018, 12, 321–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, M.; Nunes, M.R.; Stearns, V. CDK4/6 Inhibitors: Game Changers in the Management of Hormone Receptor-Positive Advanced Breast Cancer? Oncology 2018, 32, 216–222. [Google Scholar] [PubMed]
- Abraham, J.; Coleman, R.; Elias, A.; Holmes, F.A.; Kalinsky, K.; Kittaneh, M.; Lower, E.; Mahtani, R.; Terry Mamounas, E.; Pegram, M.; et al. Use of cyclin-dependent kinase (CDK) 4/6 inhibitors for hormone receptor-positive, human epidermal growth factor receptor 2-negative, metastatic breast cancer: A roundtable discussion by The Breast Cancer Therapy Expert Group (BCTEG). Breast Cancer Res. Treat. 2018, 171, 11–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Leary, B.; Finn, R.S.; Turner, N.C. Treating cancer with selective CDK4/6 inhibitors. Nat. Rev. Clin. Oncol. 2016, 13, 417–430. [Google Scholar] [CrossRef]
- Bayraktar, S.; Batoo, S.; Al-Hattab, E.; Basu, S.; Okuno, S.; Gluck, S. Future perspectives and challenges with CDK4/6 inhibitors in hormone receptor-positive metastatic breast cancer. Future Oncol. 2020, 16, 2661–2672. [Google Scholar] [CrossRef]
- Shah, A.N.; Cristofanilli, M. The Growing Role of CDK4/6 Inhibitors in Treating Hormone Receptor-Positive Advanced Breast Cancer. Curr. Treat. Options Oncol. 2017, 18, 6. [Google Scholar] [CrossRef]
- Li, J.; Fu, F.; Yu, L.; Huang, M.; Lin, Y.; Mei, Q.; Lv, J.; Wang, C. Cyclin-dependent kinase 4 and 6 inhibitors in hormone receptor-positive, human epidermal growth factor receptor-2 negative advanced breast cancer: A meta-analysis of randomized clinical trials. Breast Cancer Res. Treat. 2020, 180, 21–32. [Google Scholar] [CrossRef]
- Cersosimo, R.J. Cyclin-dependent kinase 4/6 inhibitors for the management of advanced or metastatic breast cancer in women. Am. J. Health Syst. Pharm. 2019, 76, 1183–1202. [Google Scholar] [CrossRef]
- Bardia, A.; Mayer, I.A.; Vahdat, L.T.; Tolaney, S.M.; Isakoff, S.J.; Diamond, J.R.; O’Shaughnessy, J.; Moroose, R.L.; Santin, A.D.; Abramson, V.G.; et al. Sacituzumab Govitecan-hziy in Refractory Metastatic Triple-Negative Breast Cancer. N. Engl. J. Med. 2019, 380, 741–751. [Google Scholar] [CrossRef] [PubMed]
- Syed, Y.Y. Sacituzumab Govitecan: First Approval. Drugs 2020, 80, 1019–1025. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.; Heimes, A.S. Immunomodulating Therapies in Breast Cancer-From Prognosis to Clinical Practice. Cancers 2021, 13, 4883. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.T.; Lee, S.H.; Heo, Y.S. Molecular Interactions of Antibody Drugs Targeting PD-1, PD-L1, and CTLA-4 in Immuno-Oncology. Molecules 2019, 24, 1190. [Google Scholar] [CrossRef] [Green Version]
- Behravan, J.; Razazan, A.; Behravan, G. Towards Breast Cancer Vaccines, Progress and Challenges. Curr. Drug Discov. Technol. 2019, 16, 251–258. [Google Scholar] [CrossRef]
- Solinas, C.; Aiello, M.; Migliori, E.; Willard-Gallo, K.; Emens, L.A. Breast cancer vaccines: Heeding the lessons of the past to guide a path forward. Cancer Treat. Rev. 2020, 84, 101947. [Google Scholar] [CrossRef]
- Pallerla, S.; Abdul, A.; Comeau, J.; Jois, S. Cancer Vaccines, Treatment of the Future: With Emphasis on HER2-Positive Breast Cancer. Int. J. Mol. Sci. 2021, 22, 779. [Google Scholar] [CrossRef]
- Corti, C.; Giachetti, P.; Eggermont, A.M.M.; Delaloge, S.; Curigliano, G. Therapeutic vaccines for breast cancer: Has the time finally come? Eur. J. Cancer 2022, 160, 150–174. [Google Scholar] [CrossRef]
- Criscitiello, C.; Viale, G.; Curigliano, G. Peptide vaccines in early breast cancer. Breast 2019, 44, 128–134. [Google Scholar] [CrossRef]
- Clifton, G.T.; Peoples, G.E.; Mittendorf, E.A. The development and use of the E75 (HER2 369-377) peptide vaccine. Future Oncol. 2016, 12, 1321–1329. [Google Scholar] [CrossRef]
- Anixa Biosciences and Cleveland Clinic File IND Application for Breast Cancer Vaccine. Available online: https://www.biospace.com/article/releases/anixa-biosciences-and-cleveland-clinic-file-ind-application-for-breast-cancer-vaccine/ (accessed on 25 December 2021).






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Breast Cancer Subtype | Drug Category | Drug Name | Patient Population | Therapy Given |
|---|---|---|---|---|
| HR-positive | mTOR Inhibitors | Everolimus | HR+, HER2− Postmenopausal | Everolimus + Exemestane |
| HR+, HER2− Postmenopausal AI-resistant | Everolimus + Tamoxifen | |||
| Temsirolimus | ER+, HER2+ PTEN-deficient | Single-agent temsirolimus | ||
| ER+, HER2+ | Temsirolimus + Letrozole | |||
| Sirolimus | HR+, HER2− | Sirolimus + Tamoxifen | ||
| PI3K Inhibitors | Alpelisib | HR+, HER2− PIK3CA mutant Postmenopausal | Alpelisib + Fulvestrant | |
| Taselisib | ER+, HER2− PIK3CA mutant | Taselisib + Fulvestrant | ||
| Pictilisib | HER2+/− PIK3CA mutant | Pictilisib + Trastuzumab | ||
| Buparlisib | ER−, PR−, HER2− (TNBC) | Single-agent buparlisib | ||
| HER2-positive | TKIs | Lapatinib | ER−, PR−, HER2+ | Lapatinib + Capecitabine |
| HR+, HER2+ | Lapatinib + Letrozole | |||
| HR+, HER2+ | Lapatinib + Trastuzumab | |||
| Neratinib | HER2+ Early-stage | Single-agent neratinib | ||
| Pyrotinib | HER2+ | Pyrotinib + Capecitabine | ||
| Tucatinib | HER2+ | Single-agent tucatinib | ||
| Monoclonal Antibodies | Trastuzumab | HER2+ | Single-agent trastuzumab | |
| Pertuzumab | HER2+ | Pertuzumab + Trastuzumab | ||
| Margetuximab | HER2+ | Margetuximab + chemotherapy | ||
| Antibody-drug Conjugates | Trastuzumab Emtansine (T-DM1) | HER2+ Early-stage residual disease Post-neoadjuvant tx | Single-agent T-DM1 | |
| Trastuzumab Deruxtecan (T-DXd) | HER2+ Tx with at least two prior HER2-targeted therapies | Single-agent T-DXd | ||
| HER2-negative | PARP Inhibitors | Olaparib | HER2− Deleterious gBRCA mutant | Single-agent Olaparib |
| Talazoparib | HER2− Deleterious gBRCA mutant | Single-agent talazoparib | ||
| Veliparib | HER2− gBRCA mutant | Veliparib + platinum-based chemotherapy | ||
| Niraparib | HER2− gBRCA mutant | Niraparib as neoadjuvant chemotherapy | ||
| Rucaparib | gBRCA mutant | Single-agent rucaparib | ||
| gBRCA mutant or TNBC | Rucaparib + anticancer agent | |||
| Pamiparib | TNBC, gBRCA muant or HER2−, gBRCA mutant | Single-agent pamiparib | ||
| CDK4/CDK6 Inhibitors | Palbociclib | HER2− | Palbociclib + Letrozole | |
| HR+, HER2− Postmenopausal | Palbociclib + Fulvestrant | |||
| Ribociclib | HR+, HER2− Post/premenopausal | Ribociclib + Letrozole | ||
| HR+, HER2− Postmenopausal | Ribociclib + Fulvestrant | |||
| Abemaciclib | HR+, HER2− | Abemaciclib + AI | ||
| HR+, HER2− | Single-agent abemaciclib | |||
| Antibody-drug Conjugates | Sacituzumab Govitecan (IMMU-132) | TNBC Hx of two prior metastatic tx | Single-agent IMMU-132 | |
| TNBC | Immune Checkpoint Inhibitors | Atezolizumab | TNBC | Atezolizumab + chemotherapy |
| Pembrolizumab | TNBC | Pembrolizumab + chemotherapy | ||
| Cancer Vaccines | E75 | HER2+ | E75 + GM-CSF | |
| GP2 | HER2+ | Single-agent GP2 | ||
| Other | LKB1-AMPK Pathway Activator | Honokiol | Endocrine-resistant BC | Honokiol + rapamycin |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lau, K.H.; Tan, A.M.; Shi, Y. New and Emerging Targeted Therapies for Advanced Breast Cancer. Int. J. Mol. Sci. 2022, 23, 2288. https://doi.org/10.3390/ijms23042288
Lau KH, Tan AM, Shi Y. New and Emerging Targeted Therapies for Advanced Breast Cancer. International Journal of Molecular Sciences. 2022; 23(4):2288. https://doi.org/10.3390/ijms23042288
Chicago/Turabian StyleLau, Kristie H., Alexandra M. Tan, and Yihui Shi. 2022. "New and Emerging Targeted Therapies for Advanced Breast Cancer" International Journal of Molecular Sciences 23, no. 4: 2288. https://doi.org/10.3390/ijms23042288
APA StyleLau, K. H., Tan, A. M., & Shi, Y. (2022). New and Emerging Targeted Therapies for Advanced Breast Cancer. International Journal of Molecular Sciences, 23(4), 2288. https://doi.org/10.3390/ijms23042288