
Role of Ionic Strength in the Formation of Stable Supramolecular Nanoparticle–Protein Conjugates for Biosensing




Abstract
:
1. Introduction
2. Results and Discussion
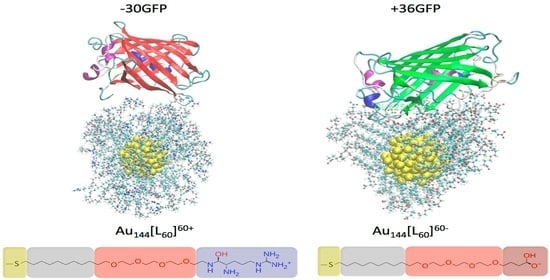
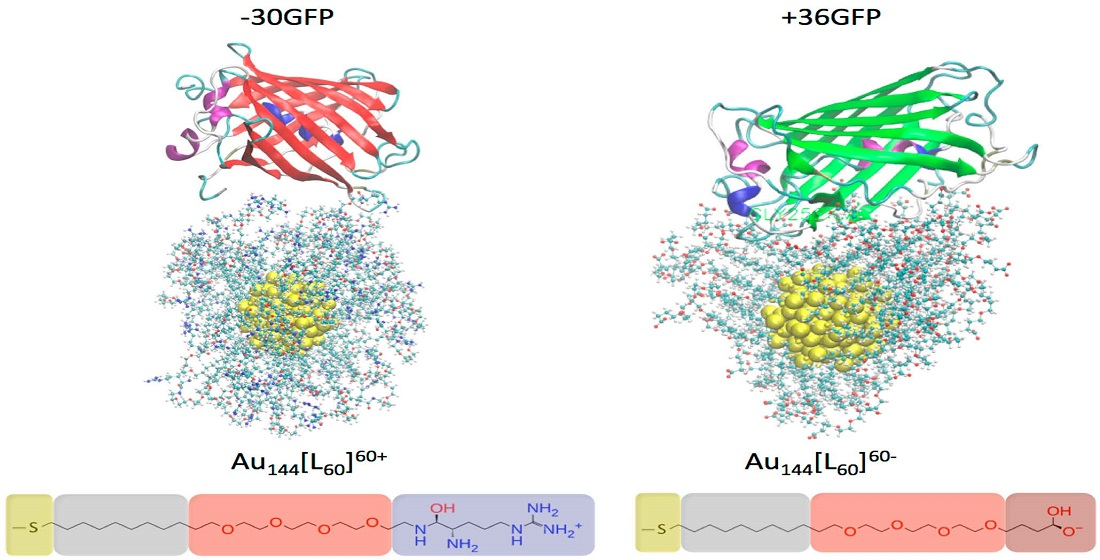
2.1. Investigated Systems
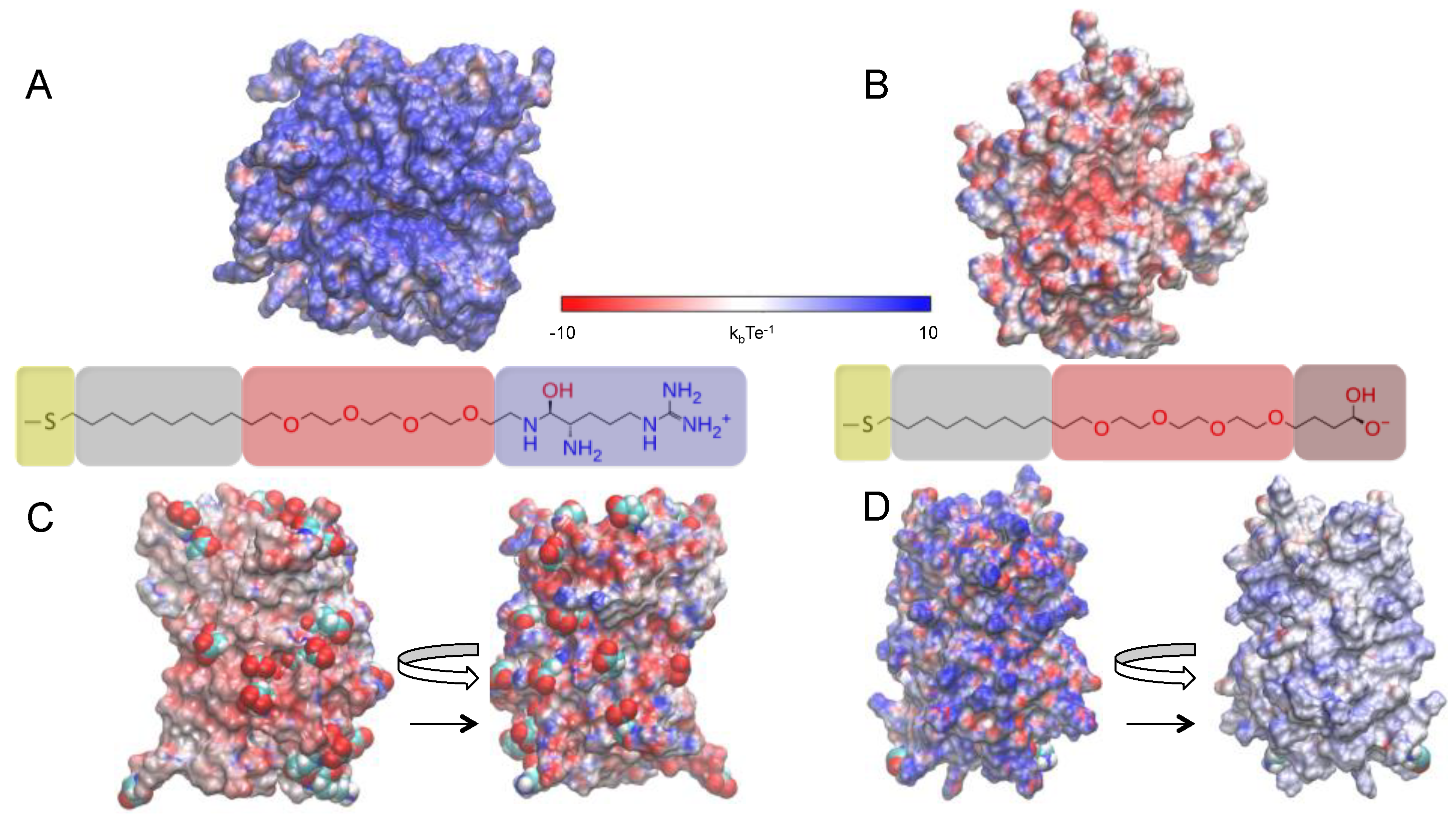
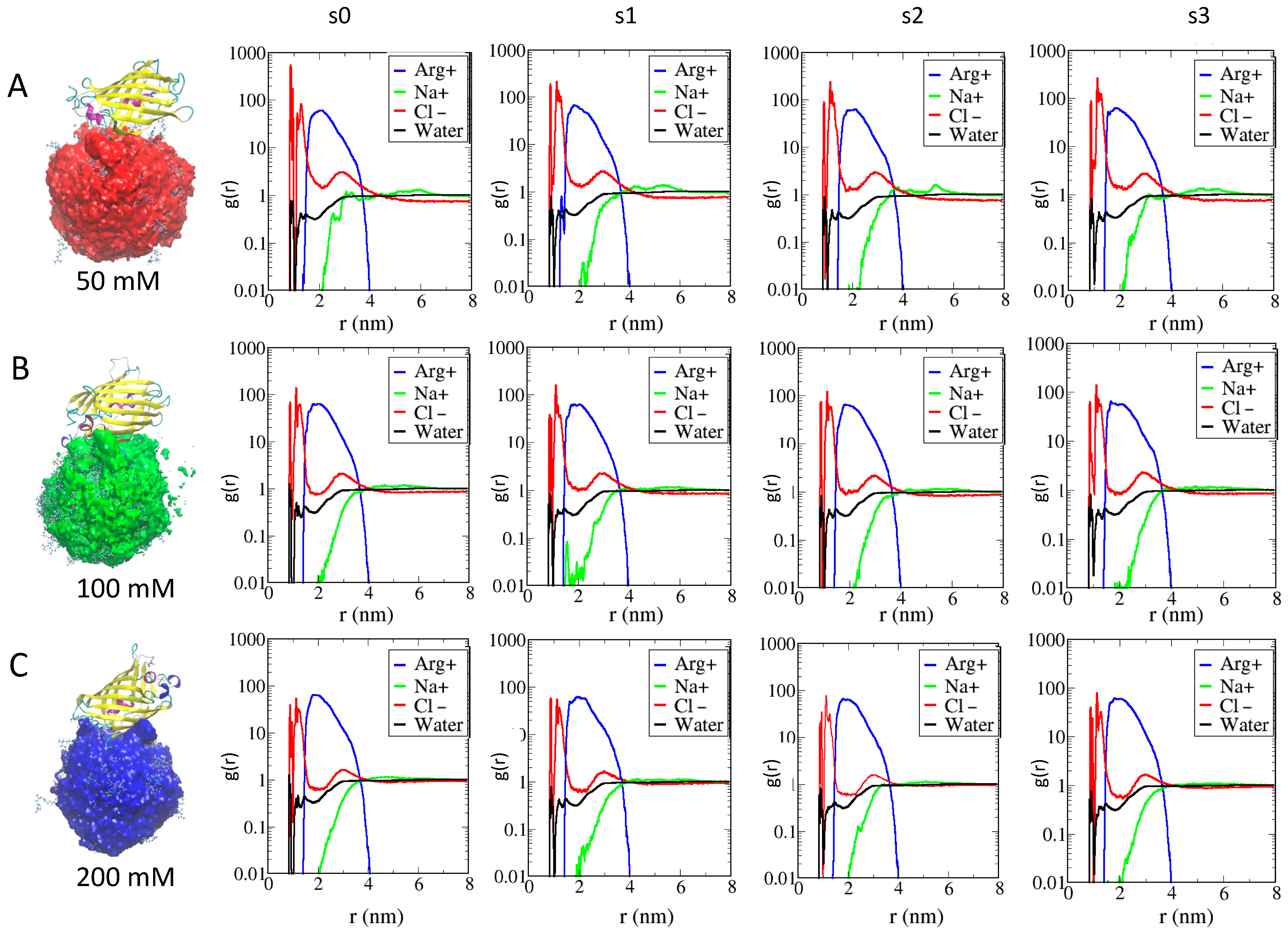
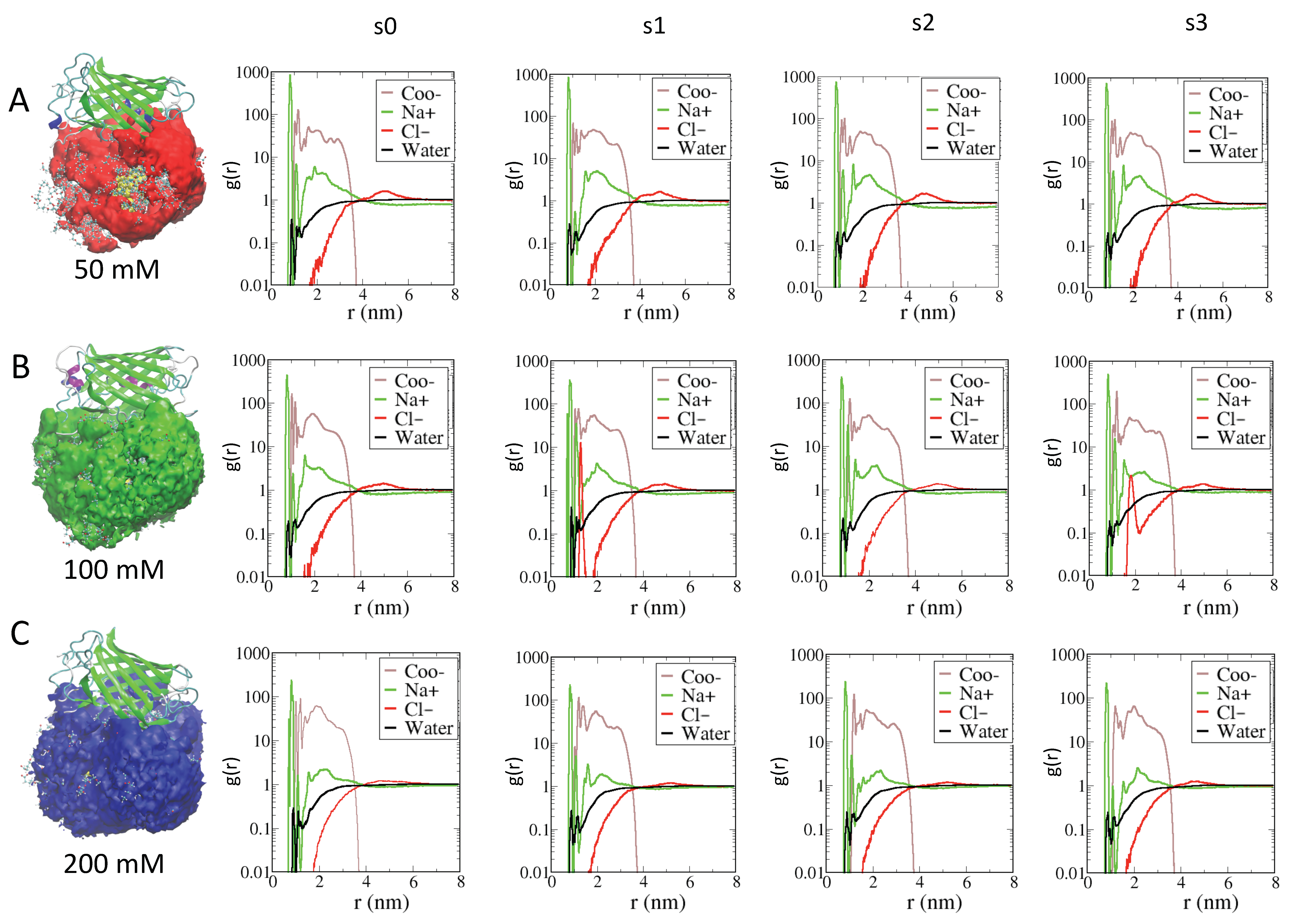
2.2. Effects of Ions on Single AuNP Nanoparticles
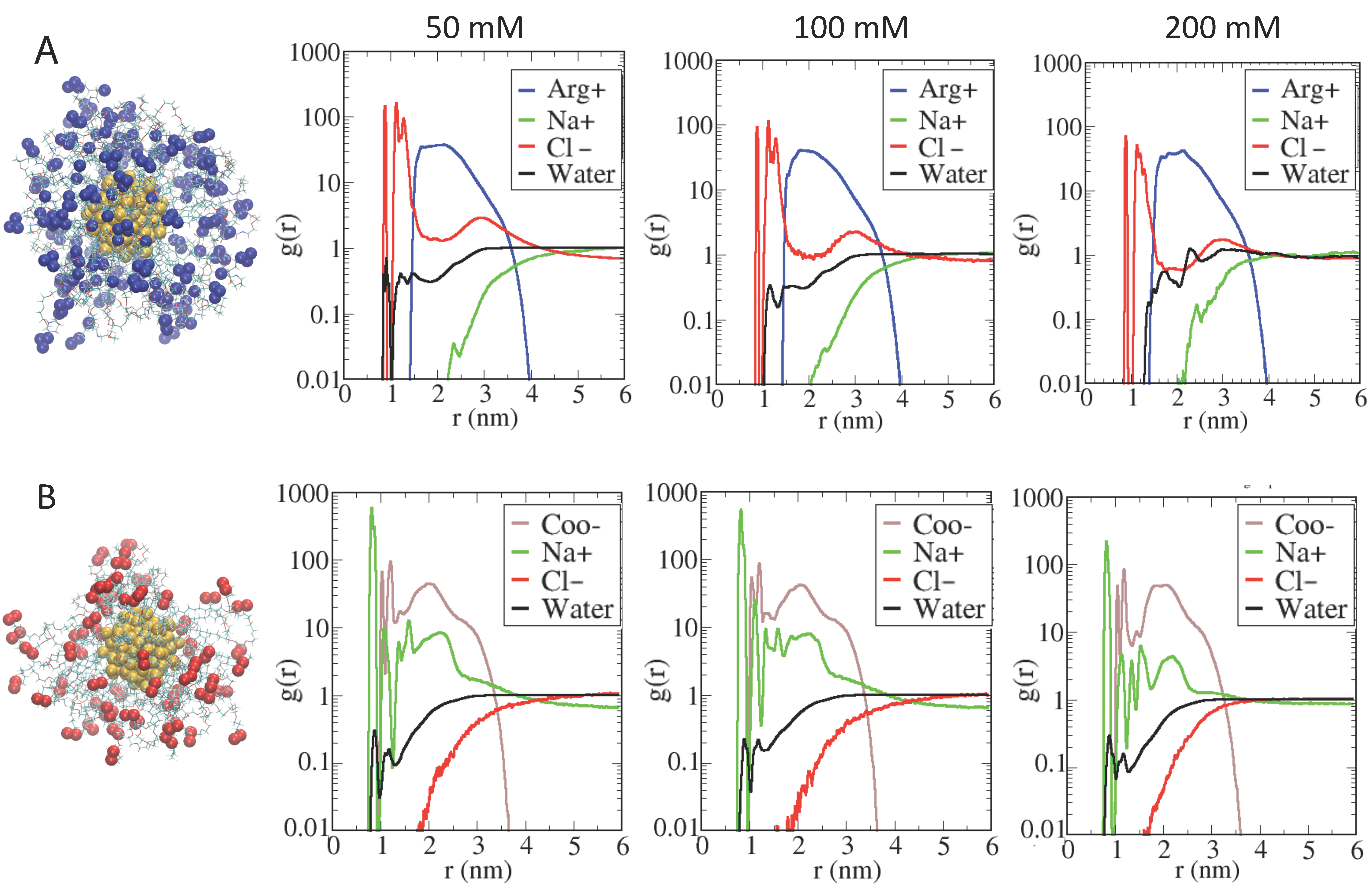
2.3. Role of Ionic Strength on Supramolecular Assemblies of Nanoparticles and Proteins
3. Materials and Methods
3.1. Development of Force Field Parameters for AuArg and AuCOO
3.2. Simulations Methodology and Setup
3.3. Setup of the Atomistic Simulations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Badia, A.; Singh, S.; Demers, L.; Cuccia, L.; Brown, G.R.; Lennox, R.B. Self-assembled monolayers on gold nanoparticles. Chem. Eur. J. 1996, 2, 359–363. [Google Scholar] [CrossRef]
- Saha, K.; Agasti, S.S.; Kim, C.; Li, X.; Rotello, V.M. Gold Nanoparticles in Chemical and Biological Sensing. Chem. Rev. 2012, 112, 2739–2779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dreaden, E.C.; Alkilany, A.M.; Huang, X.; Murphy, C.J.; El-Sayed, M.A. The golden age: Gold nanoparticles for biomedicine. Chem. Soc. Rev. 2012, 41, 2740–2779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mikolajczak, D.J.; Berger, A.A.; Koksch, B. Catalytically Active Peptide-Gold Nanoparticle Conjugates: Prospecting for Artificial Enzymes. Angew. Chem. Int. Ed. 2020, 59, 8776–8785. [Google Scholar] [CrossRef] [Green Version]
- Kotov, N.A. Inorganic nanoparticles as protein mimics. Science 2010, 330, 188–189. [Google Scholar] [CrossRef]
- De Biasi, F.; Mancin, F.; Rastrelli, F. Nanoparticle-assisted NMR spectroscopy: Achemosensing perspective. Prog. Nucl. Magn. Reson. Spectrosc. 2020, 117, 70–88. [Google Scholar] [CrossRef]
- De Biasi, F.; Rosa-Gastaldo, D.; Mancin, F.; Rastrelli, F. Hybrid nanoreceptors for high sensitivity detection of small molecules by NMR chemosensing. Chem. Commun. 2021, 57, 3002–3005. [Google Scholar] [CrossRef]
- Retout, M.; Blond, P.; Jabin, I.; Bruylants, G. Ultrastable PEGylated Calixarene-Coated Gold Nanoparticles with a Tunable Bioconjugation Density for Biosensing Applications. BioConjug. Chem. 2021, 32, 290–300. [Google Scholar] [CrossRef]
- Huang, R.; Luther, D.C.; Zhang, X.; Gupta, S.; Tufts, S.A.; Rotello, V.M. Engineering the Interface between Inorganic Nanoparticles and Biological Systems through Ligand Design. Nanomaterials 2021, 11, 1001. [Google Scholar] [CrossRef]
- Miranda, O.R.; Chen, H.-T.; You, C.-C.; Mortenson, D.E.; Yang, X.-C.; Bunz, U.H.F.; Rotello, V.M. Enzyme-amplified Array Sensing of Proteins in Solution and in Biofluids. J. Am. Chem. Soc. 2010, 132, 5285–5289. [Google Scholar] [CrossRef] [Green Version]
- Chen, R.; Neri, S.; Prins, L.J. Enhanced catalytic activity under non-equilibrium conditions. Nat. Nanotechnol. 2020, 15, 868–874. [Google Scholar] [CrossRef] [PubMed]
- Dutta, S.; Corni, S.; Brancolini, G. Molecular dynamics simulations of a Catalytic Multivalent Peptide-Nanoparticle Complex. Int. J. Mol. Sci. 2021, 22, 3624. [Google Scholar] [CrossRef] [PubMed]
- Lou-Franco, J.; Das, B.; Elliott, C.; Cao, C. Gold Nanozymes: From Concept to Biomedical Applications. Nano Micro Lett. 2021, 13, 10. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, P.; Han, G.; De, M.; Kim, C.; Rotello, V. Gold nanoparticles in delivery applications. Adv. Drug Deliv. Rev. 2008, 60, 1307–1315. [Google Scholar] [CrossRef]
- Mottas, I.; Bekdemir, A.; Cereghetti, A.; Spagnuolo, L.; Yang, Y.S.S.; Müller, M.; Irvine, D.J.; Stellacci, F.; Bourquin, C. Amphiphilic nanoparticle delivery enhances the anticancer efficacy of a TLR7 ligand via local immune activation. Biomaterials 2019, 190, 111–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kretzmann, J.A.; Luther, D.C.; Evans, C.W.; Jeon, T.; Jerome, W.; Gopalakrishnan, S.; Lee, Y.-W.; Norret, M.; Iyer, K.S.; Rotello, V.M. Regulation of Proteins to the Cytosol Using Delivery Systems with Engineered Polymer Architecture. JACS 2021, 143, 4758–4765. [Google Scholar] [CrossRef] [PubMed]
- Visalakshan, R.M.; García, L.E.G.; Benzigar, M.R.; Ghazaryan, A.; Simon, J.; Mierczynska-Vasilev, A.; Michl, T.D.; Vinu, A.; Mailänder, V.; Morsbach, S.; et al. The Influence of Nanoparticle Shape on Protein Corona Formation. Small 2020, 16, e2000285. [Google Scholar] [CrossRef]
- Engstrom, A.M.; Faase, R.A.; Marquart, G.W.; Baio, J.E.; Mackiewicz, M.R.; Harper, S.L. Size-Dependent Interactions of Lipid-Coated Gold Nanoparticles: Developing a Better Mechanistic Understanding Through Model Cell Membranes and in vivo Toxicity. Int. J. Nanomed. 2020, 15, 4091–4104. [Google Scholar] [CrossRef]
- Riccardi, L.; Gabrielli, L.; Sun, X.; De Biasi, F.; Rastrelli, F.; Mancin, F.; De Vivo, M. Nanoparticle-Based Receptors Mimic Protein-Ligand Recognition. Chem 2017, 3, 92–109. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.; Ganesh, S.; Wang, W.; Amiji, M. The role of surface chemistry in serum protein corona-mediated cellular delivery and gene silencing with lipid nanoparticles. Nanoscale 2019, 11, 8760–8775. [Google Scholar] [CrossRef]
- Srijampa, S.; Buddhisa, S.; Ngernpimai, S.; Leelayuwat, C.; Proungvitaya, S.; Chompoosor, A.; Tippayawat, P. Influence of Gold Nanoparticles with Different Surface Charges on Localization and Monocyte Behavior. BioConjug. Chem. 2020, 31, 1133–1143. [Google Scholar] [CrossRef] [PubMed]
- Auría-Soro, C.; Nesma, T.; Juanes-Velasco, P.; Landeira-Viñuela, A.; Fidalgo-Gomez, H.; Acebes-Fernandez, V.; Gongora, R.; Parra, M.J.A.; Manzano-Roman, R.; Fuentes, M. Interactions of Nanoparticles and Biosystems: Microenvironment of Nanoparticles and Biomolecules in Nanomedicine. Nanomaterials 2019, 9, 1365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, B.; Shin, J.; Wu, J.; Omstead, D.T.; Kiziltepe, T.; Littlepage, L.E.; Bilgicer, B. Engineering peptide-targeted liposomal nanoparticles optimized for improved selectivity for HER2-positive breast cancer cells to achieve enhanced in vivo efficacy. J. Control. Release 2020, 322, 530–541. [Google Scholar] [CrossRef]
- Pinals, R.L.; Chio, L.; Ledesma, F.; Landry, M.P. Engineering at the nano-bio interface: Harnessing the protein corona towards nanoparticle design and function. Analyst 2020, 145, 5090–5112. [Google Scholar] [CrossRef]
- Sun, X.; Riccardi, L.; De Biasi, F.; Rastrelli, F.; De Vivo, M.; Mancin, F. Molecular-Dynamics-Simulation-Directed Rational Design of Nanoreceptors with Targeted Affinity. Angew. Chem. Int. Ed. 2019, 58, 7702–7707. [Google Scholar] [CrossRef] [PubMed]
- Chew, A.K.; Van Lehn, R.C. Effect of Core Morphology on the Structural Asymmetry of Alkanethiol Monolayer Protected Gold Nanoparticles. J. Phys. Chem. C 2018, 122, 26288–26297. [Google Scholar] [CrossRef]
- Hassan, S.A. Strong Dependence of the Nano-Bio Interactions on Core Morphology and Layer Composition of Ultrasmall Nanostructures. J. Chem. Phys. 2019, 151, 105102. [Google Scholar] [CrossRef]
- Sousa, A.A.; Hassan, S.A.; Knittel, L.L.; Balbo, A.; Aronova, M.A.; Brown, P.H.; Schuckc, P.; Leapman, R.D. Biointeractions of ultrasmall glutathione-coated gold nanoparticles: Effect of small size variations. Nanoscale 2016, 8, 6577–6588. [Google Scholar] [CrossRef] [Green Version]
- Kyrychenko, A.; Pasko, D.A.; Kalugin, O.N. Poly(vinyl alcohol) as a water protecting agent for silver nanoparticles: The role of polymer size and structure. Phys. Chem. Chem. Phys. 2017, 19, 8742–8756. [Google Scholar] [CrossRef]
- Villareal, O.D.; Chen, K.Y.; Whetten, R.L.; Yacaman, M.J. Ligand-modulated interactions between charged monolayer-protected Au144(SR)60 gold nanoparticles in physiological saline. Phys. Chem. Chem. Phys. 2015, 17, 3680–3688. [Google Scholar] [CrossRef] [Green Version]
- Lira, A.L.; Ferreira, R.S.; Torquato, R.J.S.; Zhao, H.; Oliva, M.L.V.; Hassan, S.A.; Schuck, P.; Sousa, A.A. Binding kinetics of ultrasmall gold nanoparticles with proteins. Nanoscale 2018, 10, 3235–3244. [Google Scholar] [CrossRef] [PubMed]
- Hoek, E.M.V.; Agarwal, G.K. Extended DLVO interactions between spherical particles and rough surfaces. J. Colloid Interface Sci. 2006, 298, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Mout, R.; Moyano, D.F.; Rana, S.; Rotello, V.M. Surface Functionalization of Nanoparticles for Nanomedicine. Chem. Soc. Rev. 2012, 41, 2539–2544. [Google Scholar] [CrossRef] [PubMed]
- Lynch, I.; Salvati, A.; Dawson, K.A. Protein-Nanoparticle Interactions: What Does the Cell See? Nat. Nanotech. 2009, 4, 546–547. [Google Scholar] [CrossRef]
- Nel, A.E.; Mädler, L.; Velegol, D.; Xia, T.; Hoek, E.M.V.; Somasundaran, P.; Klaessig, F.; Castranova, V.; Thompson, M. Understanding Biophysiocochemical Interactions at the Nano-Bio Interface. Nat. Mater. 2009, 8, 543–557. [Google Scholar] [CrossRef]
- Franco-Ulloa, S.; Tatulli, G.; Bore, S.L.; Moglianetti, M.; Pompa, P.P.; Cascella, M.; De Vivo, M. Dispersion state phase diagram of citrate-coated metallic nanoparticles in saline solutions. Nat. Commun. 2020, 11, 5422. [Google Scholar] [CrossRef]
- Tavanti, F.; Pedone, A.; Menziani, M.C.; Alexander-Katz, A. Computational Insights into the Binding of Monolayer-Capped Gold Nanoparticles onto Amyloid-beta Fibrils. ACS Chem. Neurosci. 2020, 11, 3153–3160. [Google Scholar] [CrossRef]
- Sen, S.; Vukovic, L.; Kral, P. Computational screening of nanoparticles coupling to Abeta40 peptides and fibrils. Sci. Rep. 2019, 9, 17804. [Google Scholar] [CrossRef] [Green Version]
- Sousa, A.A.; Schuck, P.; Hassan, S.A. Biomolecular interactions of ultrasmall metallic nanoparticles and nanoclusters. Nanoscale Adv. 2021, 3, 2995. [Google Scholar] [CrossRef]
- Brancolini, G.; Tozzini, V. Multiscale modeling of protein interactions with functionalized nanoparticles. Curr. Opin. Colloid Interface Sci. 2019, 41, 66–73. [Google Scholar] [CrossRef]
- Pfeiffer, C.; Rehbock, C.; Hühn, D.; Carrillo-Carrion, C.; de Aberasturi, D.J.; Merk, V.; Barcikowski, S.; Parak, W.J. Interaction of colloidal nanoparticles with their local environment: The (ionic) nanoenvironment around nanoparticles is different from bulk and determines the physico-chemical properties of the nanoparticles. J. R. Soc. Interface 2014, 11, 20130931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ray, M.; Brancolini, G.; Luther, D.; Jiang, Z.; Cao-Milán, R.; Cuadros, A.; Burden, A.; Clark, V.; Rana, S.; Mout, R.; et al. High affinity protein surface binding through co-engineering of nanoparticles and proteins. Nanoscale 2022, 14, 2411–2418. [Google Scholar] [CrossRef] [PubMed]
- Mout, R.; Ray, M.; Tay, T.; Sasaki, K.; Yesilbag Tonga, G.; Rotello, V.M. General Strategy for Direct Cytosolic Protein Delivery via Protein-Nanoparticle Co-engineering. ACS Nano 2017, 11, 6416–6421. [Google Scholar] [CrossRef] [PubMed]
- Miranda, O.R.; Creran, B.; Rotello, V.M. Array-based Sensing with Nanoparticles: ‘Chemical Noses’ for Sensing Biomolecules and Cell Surfaces. Curr. Opin. Chem. Biol. 2010, 14, 728–736. [Google Scholar] [CrossRef] [Green Version]
- Lawrence, M.S.; Phillips, K.J.; Liu, D.R. Supercharging Proteins Can Impart Unusual Resilience. JACS 2007, 129, 10110–10112. [Google Scholar] [CrossRef] [Green Version]
- McNaughton, B.R.; Cronican, J.J.; Thompson, D.B.; Liu, D.R. Mammalian Cell Penetration, siRNA Transfection, and DNA Transfection by Supercharged Proteins. Proc. Natl. Acad. Sci. USA 2009, 106, 6111–6116. [Google Scholar] [CrossRef] [Green Version]
- Brancolini, G.; Toroz, D.; Corni, S. Can small hydrophobic gold nanoparticles inhibit beta(2)-microglobulin fibrillation? Nanoscale 2014, 6, 7903–7911. [Google Scholar] [CrossRef]
- Verma, A.; Simard, J.M.; Rotello, V.M. Effect of Ionic Strength on the Binding of α-Chymotrypsin to Nanoparticle Receptors. Langmuir 2004, 20, 4178–4181. [Google Scholar] [CrossRef]
- Franco-Ulloa, S.; Riccardi, L.; Rimembrana, F.; Pini, M.; De Vivo, M. NanoModeler: A Webserver for Molecular Simulations and Engineering of Nanoparticles. J. Chem. Theory Comput. 2019, 15, 2022–2032. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Smith, J.C.; Hess, B.; Lindahl, E. Gromacs: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Bayly, C.I.; Cieplak, P.; Cornell, W.D.; Kollman, P.A. A Well-Behaved Electrostatic Potential Based Method Using Charge Restraints for Deriving Atomic Charges: The RESP Model. J. Phys. Chem. 1993, 97, 10269–10280. [Google Scholar] [CrossRef]
- Vanquelef, E.; Simon, S.; Marquant, G.; Garcia, E.; Klimerak, G.; Delepine, J.C.; Cieplak, P.; Dupradeau, F.Y. RED Server: A web service for deriving RESP and ESP charges and building force field libraries for new molecules and molecular fragments. Nucleic Acids Res. 2011, 39, W511–W517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An Nlog(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Frenkel, D.; Smit, B. Understanding Molecular Simulation from Algorithms to Applications; Academic Press: Cambridge, MA, USA, 2002. [Google Scholar]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parrinello, M.; Rahman, A. Polymorphic Transitions in Single-Crystals—A New Molecular-Dynamics Method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Martinez, M.; Bruce, N.J.; Romanowska, J.; Kokh, D.B.; Ozboyaci, M.; Yu, X.; Öztürk, M.A.; Richter, S.; Wade, R.C.J. SDA 7: A modular and parallel implementation of the simulation of diffusional association software. Comput. Chem. 2015, 36, 1631–1645. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
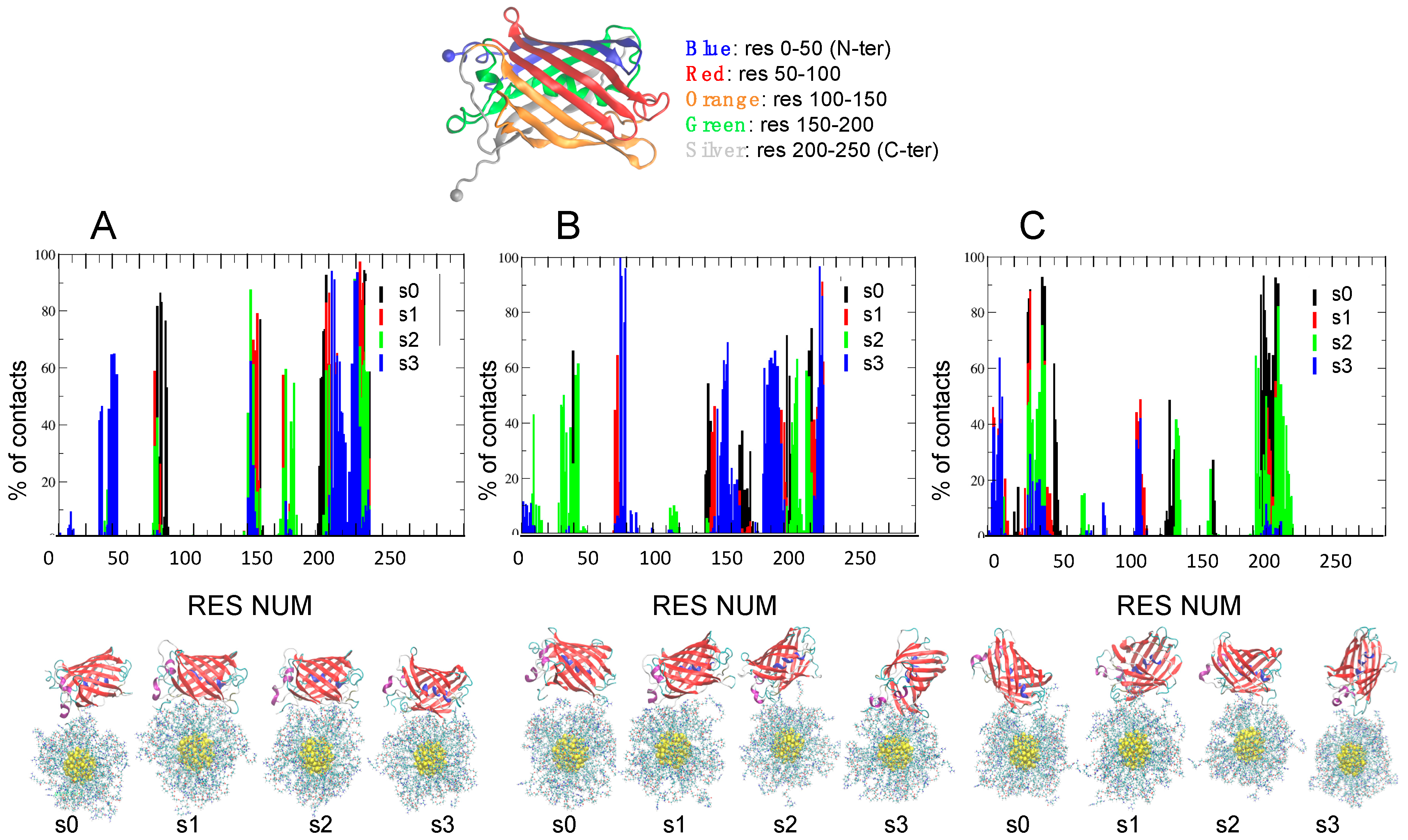
| IOS | Label | Contact Residues for AuArg (>70% of Frames) |
|---|---|---|
| 50 mM | s0 | TYR149 HIS79 HIS197 ALA225 MET76 PHE221 GLY230 PRO73 ILE227 ASP196 HIS75 TYR198 GLY226 THR223 ASP228 |
| s1 | ALA225 GLU202 PHE221 ASN142 ILE227 THR223 ALA224 HIS229 TYR198 ASP147 LEU219 ASP228 SER200 | |
| s2 | ASN142 ILE227 ALA204 LEU219 | |
| s3 | ALA204 LEU219 GLU202 PHE221 | |
| 100 mM | s0 | GLU202 PHE221 |
| s1 | ILE227 HIS229 | |
| s2 | - | |
| s3 | HIS79 HIS75 ILE227 GLN78 HIS229 | |
| 200 mM | s0 | ASN210 LEU219 GLY31 LYS43 LEU42 PRO209 GLU30 SER206 GLU32 ASP208 THR41 VAL217 |
| s1 | GLU32 THR41 | |
| s2 | LEU214 | |
| s3 | - |
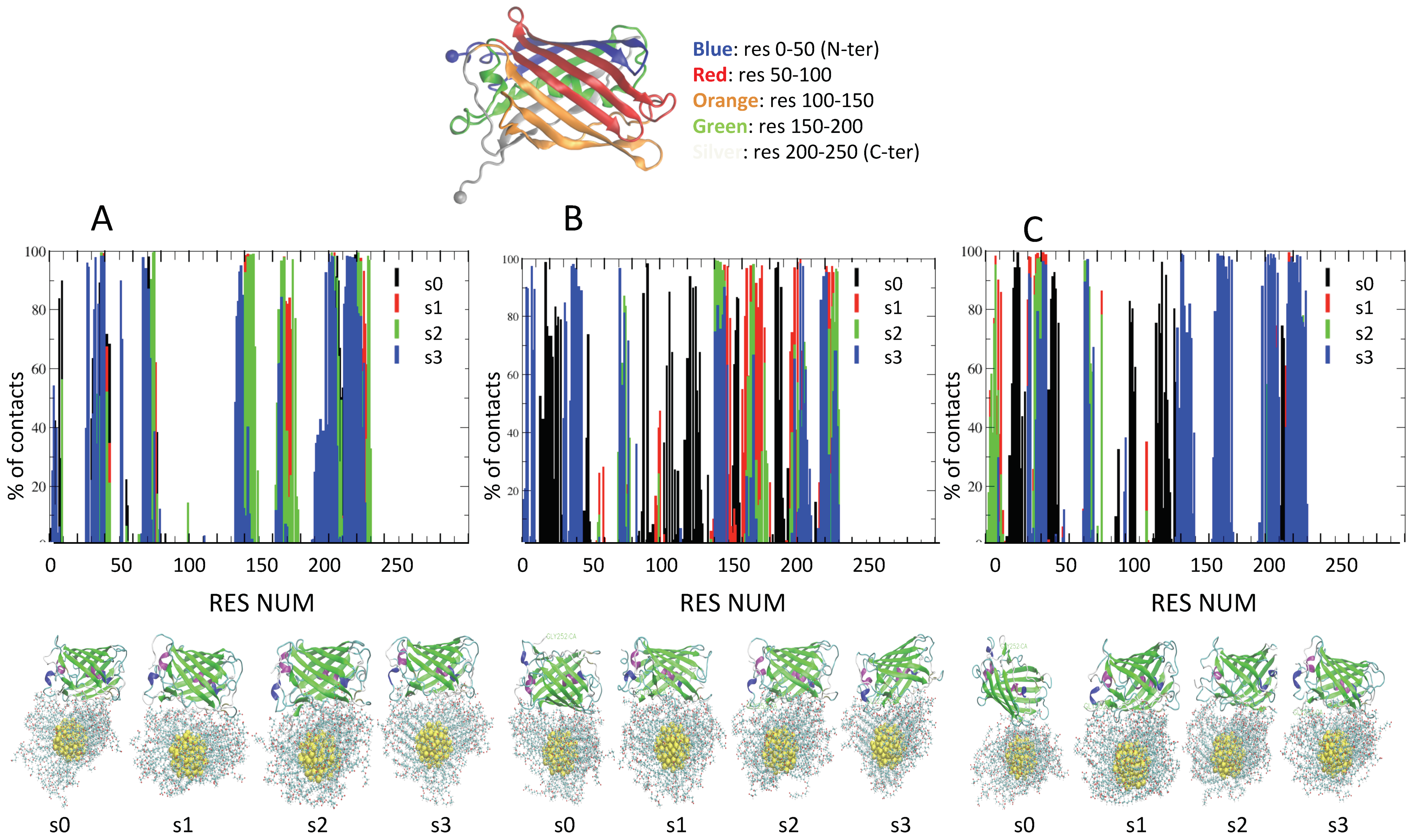
| IOS | Label | Contact Residues for AuCOO (>70% of Frames) |
|---|---|---|
| 50 mM | s0 | LYS204 HIS146 THR41 PHE221 LYS39 ASN168 PHE143 ASN144 LYS32 LEU219 ARG202 ARG140 SER200 ARG7 LEU218 ARG37 SER145 ASN142 VAL217 TYR141 LYS207 THR201 SER206 MET216 THR223 THR36 LEU205 ARG71 ASN142 LYS9 |
| s1 | PHE221 LYS204 SER203 HIS146 ASN168 LYS228 ASP171 ARG202 LYS39 LEU219 ARG140 ASN144 THR201 ASN142 GLY172 GLY226 VAL169 SER200 ARG37 ALA225 THR223 VAL174 SER173 THR223 LYS170 ARG71 | |
| s2 | HIS229 SER203 HIS146 LYS204 PHE221 LYS228 ARG202 SER145 ASN168 LYS39 LEU219 ARG140 LEU219 ARG166 ARG202 ASN144 PHE143 HIS75 GLY38 ASN142 LYS74 LYS164 TYR141 PRO73 LYS74 ARG37 SER206 VAL174 THR223 VAL222 | |
| s3 | LYS204 ARG202 ILE227 LEU219 GLY31 PRO209 LYS170 LYS39 HIS75 ASN142 SER206 GLY226 LEU218 LYS74 ARG37 LYS30 ALA225 VAL217 ARG37 PRO73 LYS207 VAL217 MET216 LYS43 LEU42 ARG140 TYR72 PRO73 THR36 ALA224 THR223 LEU205 HIS215 THR41 ARG71 PHE221 LYS204 ASP208 TRP55 HIS215 LYS32 LYS138 | |
| 100 mM | s0 | LYS124 ARG155 ASP19 ARG107 ASP19 THR48 VAL91 SER26 GLY18 ARG188 LYS122 PHE25 LYS24 LYS17 ASN157 ILE186 LYS88 LYS156 THR184 PRO185 ARG126 |
| s1 | VAL174 ASN196 SER203 ARG202 LYS39 ARG166 LEU219 ASN144 THR151 SER173 GLY172 ASN142 GLY226 SER200 TYR149 ALA225 TYR141 LYS170 TYR149 ALA224 ARG140 VAL169 VAL148 PHE163 LYS162 TYR198 LYS147 THR223 LEU176 ASN168 LYS147 PHE221 LYS204 ARG166 GLU220 LYS228 | |
| s2 | LYS204 PHE221 LYS228 ARG202 ILE227 ARG140 ARG166 HIS75 ASN142 ASN144 PHE143 GLY226 LEU219 LYS74 ALA225 TYR141 ARG37 THR223 LEU205 LYS147 ASN168 HIS146 HIS229 | |
| s3 | LYS204 LYS39 LEU219 GLY31 ARG202 ARG7 ASN142 ARG37 VAL217 LYS74 ARG37 ASN144 TYR141 THR36 LEU42 LYS43 ARG140 ARG4 ARG71 LYS147 THR41 GLU3 PHE221 LYS9 HIS146 LYS77 LYS32 | |
| 200 mM | s0 | THR48 LYS124 THR47 LYS129 LYS122 LYS105 LYS24 HIS23 LYS103 TYR104 PRO52 GLY22 LEU135 ASN21 ARG126 LYS50 VAL20 LYS212 GLY49 GLY125 |
| s1 | ARG7 LEU219 PRO209 LYS83 GLY31 LYS32 ASP208 ARG37 TYR141 LEU205 HIS215 PHE6 LEU218 LYS30 LYS207 VAL217 ARG37 PRO11 THR36 ARG71 SER206 THR41 LYS43 THR223 ARG71 LYS9 ALA35 PHE221 LYS204 LEU42 THR41 PHE221 LYS204 LYS207 LYS39 | |
| s2 | ARG7 LEU219 PRO209 LYS83 GLY31 LYS32 ASP208 ARG37 TYR141 LEU205 HIS215 PHE6 LEU218 LYS30 LYS207 VAL217 ARG37 PRO11 THR36 ARG71 SER206 THR41 LYS43 THR223 ARG71 LYS9 ALA35 PHE221 LYS204 LEU42 THR41 PHE221 LYS204 LYS207 LYS39 | |
| s3 | LYS204 ASN196 HIS137 LYS228 LYS39 ARG202 VAL174 LEU219 GLY31 PRO209 LYS170 ARG166 SER203 ASN144 ILE227 PRO209 ASN142 LYS164 LEU218 GLY226 VAL217 TYR141 LYS207 ASP171 SER200 ARG37 ASN168 THR36 ALA225 SER206 ARG140 MET216 LYS43 LEU42 TYR72 PRO73 ALA224 LEU205 LEU176 LYS147 THR41 GLY172 SER173 ARG71 TYR198 THR223 HIS229 VAL169 PHE221 HIS229 HIS146 LEU205 HIS146 LYS32 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brancolini, G.; Rotello, V.M.; Corni, S. Role of Ionic Strength in the Formation of Stable Supramolecular Nanoparticle–Protein Conjugates for Biosensing. Int. J. Mol. Sci. 2022, 23, 2368. https://doi.org/10.3390/ijms23042368
Brancolini G, Rotello VM, Corni S. Role of Ionic Strength in the Formation of Stable Supramolecular Nanoparticle–Protein Conjugates for Biosensing. International Journal of Molecular Sciences. 2022; 23(4):2368. https://doi.org/10.3390/ijms23042368
Chicago/Turabian StyleBrancolini, Giorgia, Vincent M. Rotello, and Stefano Corni. 2022. "Role of Ionic Strength in the Formation of Stable Supramolecular Nanoparticle–Protein Conjugates for Biosensing" International Journal of Molecular Sciences 23, no. 4: 2368. https://doi.org/10.3390/ijms23042368
APA StyleBrancolini, G., Rotello, V. M., & Corni, S. (2022). Role of Ionic Strength in the Formation of Stable Supramolecular Nanoparticle–Protein Conjugates for Biosensing. International Journal of Molecular Sciences, 23(4), 2368. https://doi.org/10.3390/ijms23042368