
Combined BSA-Seq Based Mapping and RNA-Seq Profiling Reveal Candidate Genes Associated with Plant Architecture in Brassica napus

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Phenotypic Characteristics of 4942C-5
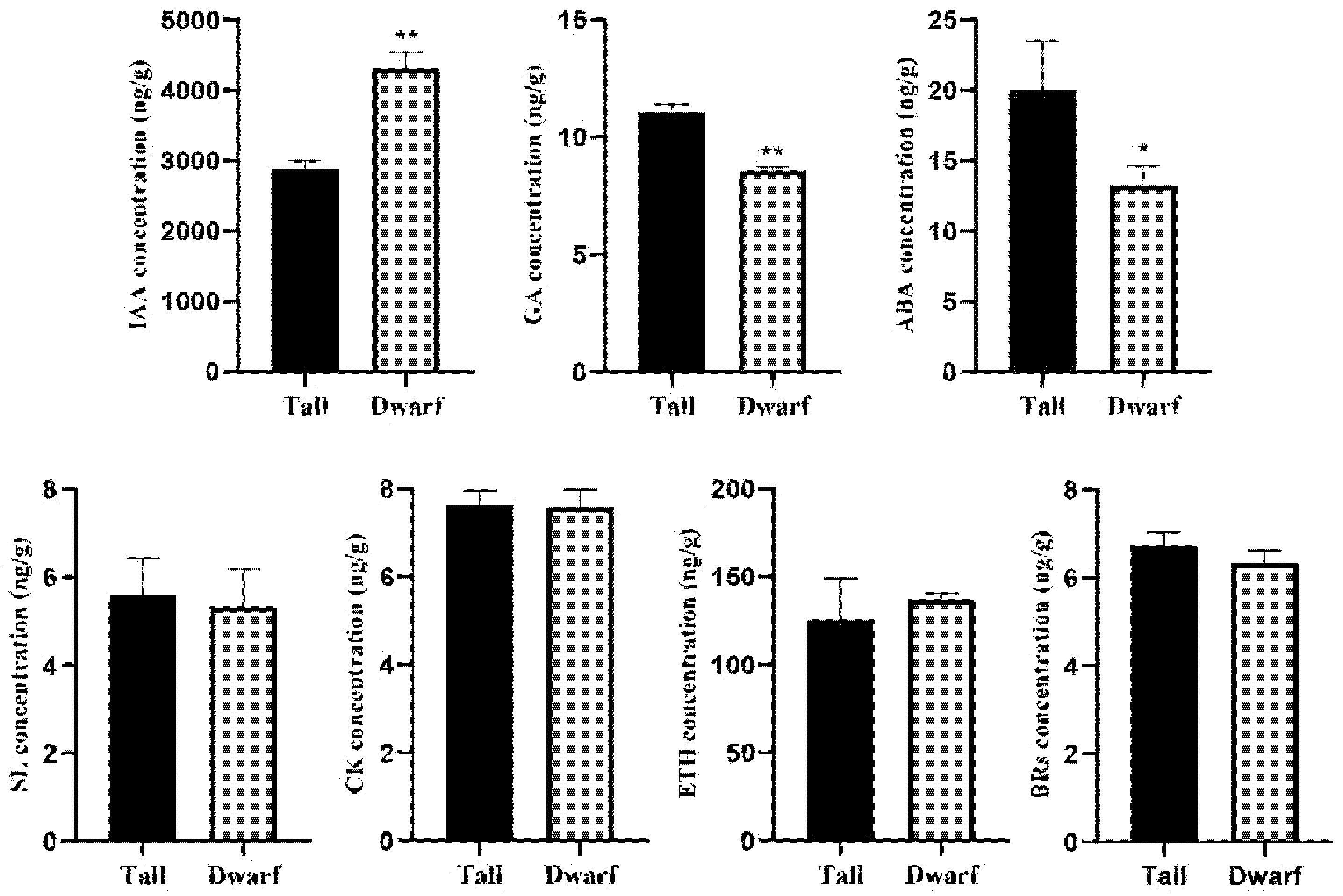
2.2. Identification of Phytohormones
2.3. BSA-Seq Analysis
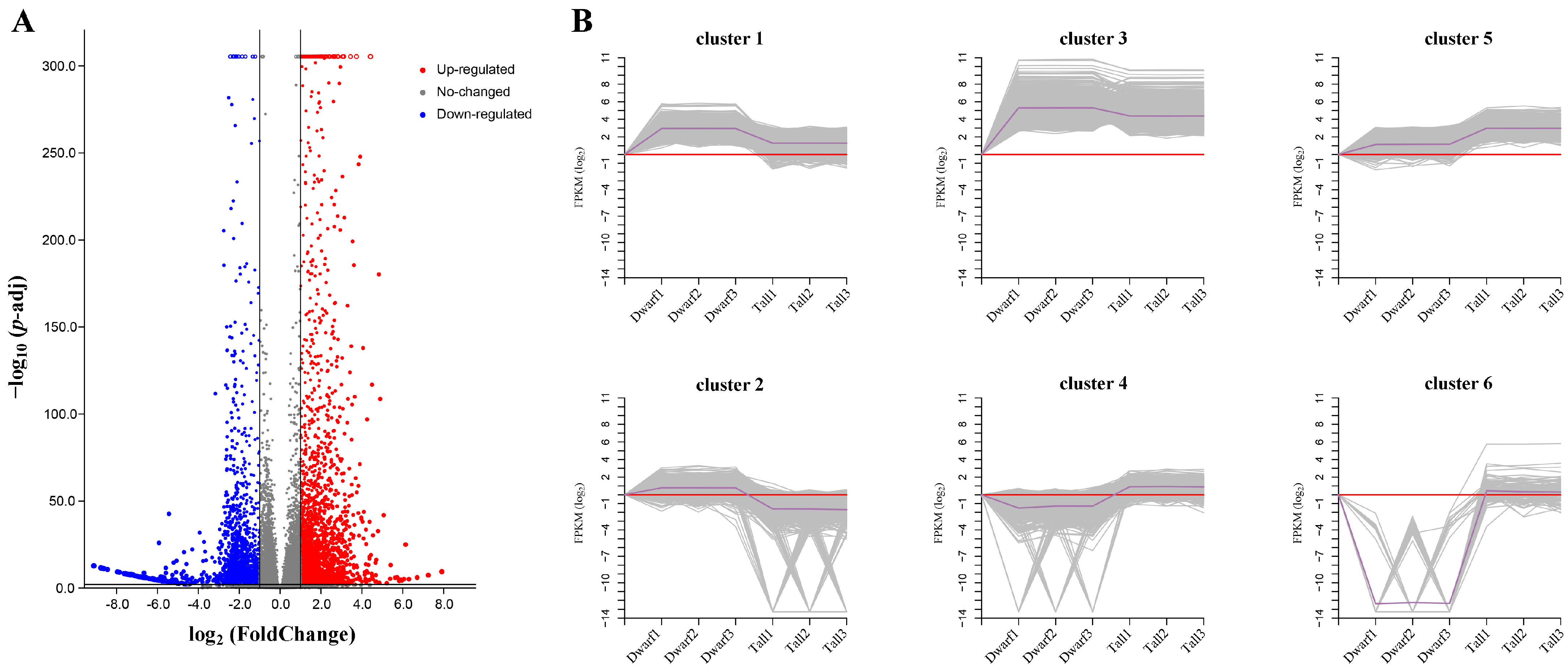
2.4. Transcriptome Sequencing Data Analysis
2.5. Expression Pattern Analysis of DEGs
2.6. Functional Analysis of DEGs
2.7. DEGs Involved in Plant Hormone Biosynthesis
2.8. DEGs Involved in Plant Hormone Signal Transduction
2.9. Analysis of DEGs Related to Cell Wall and Cell Expansion
2.10. Analysis of DEGs Related to the Phenylpropanoid Pathway
2.11. Association Analysis of BSA-Seq and Transcriptomic Data
3. Discussion
3.1. Phytohormones Might Play Major Roles in Determining Plant Architecture in B. napus
3.2. DEGs Involved in the Phenylpropanoid Pathway Are Associated with Phenotypic Differences between Tall and Dwarf Plants
3.3. WOX May Contribute to the Formation of the Dwarf Phenotype in B. napus
3.4. Chromosome Structural Variation Might Occur in 4942C-5 Plants
4. Materials and Methods
4.1. Plants Materials
4.2. Trait Evaluation
4.3. Phytohormone Profiling
4.4. DNA Library Construction and BSA-Seq Analysis
4.5. RNA Extraction, Library Construction, Sequencing, and Bioinformatics Analysis
4.6. Quantitative Real-Time PCR (qRT-PCR) Validation of DEGs
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Reinhardt, D.; Kuhlemeier, C. Plant architecture. EMBO Rep. 2002, 3, 846–851. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Smith, S.M.; Li, J. Genetic Regulation of Shoot Architecture. Annu. Rev. Plant Biol. 2018, 69, 437–468. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, A.; Ashikari, M.; Ueguchi-Tanaka, M.; Itoh, H.; Nishimura, A.; Swapan, D.; Ishiyama, K.; Saito, T.; Kobayashi, M.; Khush, G.; et al. Green revolution: A mutant gibberellin-synthesis gene in rice. Nature 2002, 416, 701–702. [Google Scholar] [CrossRef]
- Guo, Z.; Liu, X.; Zhang, B.; Yuan, X.; Xing, Y.; Liu, H.; Luo, L.; Chen, G.; Xiong, L. Genetic analyses of lodging resistance and yield provide insights into post-Green-Revolution breeding in rice. Plant Biotechnol. J. 2021, 19, 814–829. [Google Scholar] [CrossRef]
- Zhao, S.Q.; Xiang, J.J.; Xue, H.W. Studies on the rice LEAF INCLINATION1 (LC1), an IAA-amido synthetase, reveal the effects of auxin in leaf inclination control. Mol. Plant 2013, 6, 174–187. [Google Scholar] [CrossRef] [Green Version]
- Ding, X.; Cao, Y.; Huang, L.; Zhao, J.; Xu, C.; Li, X.; Wang, S. Activation of the indole-3-acetic acid-amido synthetase GH3-8 suppresses expansin expression and promotes salicylate- and jasmonate-independent basal immunity in rice. Plant Cell 2008, 20, 228–240. [Google Scholar] [CrossRef] [Green Version]
- Fu, J.; Liu, H.; Li, Y.; Yu, H.; Li, X.; Xiao, J.; Wang, S. Manipulating broad-spectrum disease resistance by suppressing pathogen-induced auxin accumulation in rice. Plant Physiol. 2011, 155, 589–602. [Google Scholar] [CrossRef] [Green Version]
- Monna, L.; Kitazawa, N.; Yoshino, R.; Suzuki, J.; Masuda, H.; Maehara, Y.; Tanji, M.; Sato, M.; Nasu, S.; Minobe, Y. Positional Cloning of Rice Semidwarfing Gene, sd-1: Rice “Green Revolution Gene” Encodes a Mutant Enzyme Involved in Gibberellin Synthesis. DNA Res. 2002, 9, 11–17. [Google Scholar] [CrossRef] [Green Version]
- Heinrich, M.; Hettenhausen, C.; Lange, T.; Wunsche, H.; Fang, J.; Baldwin, I.T.; Wu, J. High levels of jasmonic acid antagonize the biosynthesis of gibberellins and inhibit the growth of Nicotiana attenuata stems. Plant J. 2013, 73, 591–606. [Google Scholar] [CrossRef]
- Depuydt, S.; Hardtke, C.S. Hormone signalling crosstalk in plant growth regulation. Curr. Biol. 2011, 21, R365–R373. [Google Scholar] [CrossRef]
- Ding, Z.; Lin, Z.; Li, Q.; Wu, H.; Xiang, C.; Wang, J. DNL1, encodes cellulose synthase-like D4, is a major QTL for plant height and leaf width in rice (Oryza sativa L.). Biochem. Biophys. Res. Commun. 2015, 457, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zheng, X.; Wang, C.; Hou, D.; Li, T.; Li, D.; Ma, C.; Sun, Z.; Tian, Y. Pear xyloglucan endotransglucosylase/hydrolases PcBRU1 promotes stem growth through regulating cell wall elongation. Plant Sci. 2021, 312, 111026. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.H.; Kang, K.; Lee, S.H.; Lee, I.J.; Paek, N.C. OsWOX3A is involved in negative feedback regulation of the gibberellic acid biosynthetic pathway in rice (Oryza sativa). J. Exp. Bot. 2016, 67, 1677–1687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, J.; Shi, Y.; Li, W.; Chen, S.; Wang, Y.; He, X.; Yin, X. RcPAL, a key gene in lignin biosynthesis in Ricinus communis L. BMC Plant Biol. 2019, 19, 181. [Google Scholar] [CrossRef]
- Liu, C.; Wang, J.; Huang, T.; Wang, F.; Yuan, F.; Cheng, X.; Zhang, Y.; Shi, S.; Wu, J.; Liu, K. A missense mutation in the VHYNP motif of a DELLA protein causes a semi-dwarf mutant phenotype in Brassica napus. Theor. Appl. Genet. 2010, 121, 249–258. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Li, H.; Li, J.; Wang, B.; Dai, C.; Wang, J.; Liu, K. Brassica napus DS-3, encoding a DELLA protein, negatively regulates stem elongation through gibberellin signaling pathway. Theor. Appl. Genet. 2017, 130, 727–741. [Google Scholar] [CrossRef]
- Cheng, H.; Jin, F.; Zaman, Q.U.; Ding, B.; Hao, M.; Wang, Y.; Huang, Y.; Wells, R.; Dong, Y.; Hu, Q. Identification of Bna.IAA7.C05 as allelic gene for dwarf mutant generated from tissue culture in oilseed rape. BMC Plant Biol. 2019, 19, 500. [Google Scholar] [CrossRef]
- Zheng, M.; Hu, M.; Yang, H.; Tang, M.; Zhang, L.; Liu, H.; Li, X.; Liu, J.; Sun, X.; Fan, S.; et al. Three BnaIAA7 homologs are involved in auxin/brassinosteroid-mediated plant morphogenesis in rapeseed (Brassica napus L.). Plant Cell Rep. 2019, 38, 883–897. [Google Scholar] [CrossRef] [Green Version]
- Zhao, B.; Wang, B.; Li, Z.; Guo, T.; Zhao, J.; Guan, Z.; Liu, K. Identification and characterization of a new dwarf locus DS-4 encoding an Aux/IAA7 protein in Brassica napus. Theor. Appl. Genet. 2019, 132, 1435–1449. [Google Scholar] [CrossRef]
- Li, H.; Li, J.; Song, J.; Zhao, B.; Guo, C.; Wang, B.; Zhang, Q.; Wang, J.; King, G.J.; Liu, K. An auxin signaling gene BnaA3.IAA7 contributes to improved plant architecture and yield heterosis in rapeseed. New Phytol. 2019, 222, 837–851. [Google Scholar] [CrossRef]
- Chen, W.; Zhang, Y.; Liu, X.; Chen, B.; Tu, J.; Tingdong, F. Detection of QTL for six yield-related traits in oilseed rape (Brassica napus) using DH and immortalized F(2) populations. Theor. Appl. Genet. 2007, 115, 849–858. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Song, X.; Cheng, Y.; Zou, X.; Zeng, L.; Qiao, X.; Lu, G.; Fu, G.; Qu, Z.; Zhang, X. Identification of QTLs for branch number in oilseed rape (Brassica napus L.). J. Genet. Genomics 2014, 41, 557–559. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Cheng, H.; Wang, W.; Liu, J.; Hao, M.; Mei, D.; Zhou, R.; Fu, L.; Hu, Q. Identification of BnaYUCCA6 as a candidate gene for branch angle in Brassica napus by QTL-seq. Sci. Rep. 2016, 6, 38493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Gao, J.; Chen, J.; Wang, Z.; Shen, W.; Yi, B.; Wen, J.; Ma, C.; Shen, J.; Fu, T.; et al. Identification and fine mapping of a major locus controlling branching in Brassica napus. Theor. Appl. Genet. 2020, 133, 771–783. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wang, W.; Mei, D.; Wang, H.; Fu, L.; Liu, D.; Li, Y.; Hu, Q. Characterizing Variation of Branch Angle and Genome-Wide Association Mapping in Rapeseed (Brassica napus L.). Front. Plant Sci. 2016, 7, 21. [Google Scholar] [CrossRef] [Green Version]
- Yan, J.; Liao, X.; He, R.; Zhong, M.; Feng, P.; Li, X.; Tang, D.; Liu, X.; Zhao, X. Ectopic expression of GA 2-oxidase 6 from rapeseed (Brassica napus L.) causes dwarfism, late flowering and enhanced chlorophyll accumulation in Arabidopsis thaliana. Plant Physiol. Biochem. 2017, 111, 10–19. [Google Scholar] [CrossRef]
- El-Sharkawy, I.; Ismail, A.; Darwish, A.; El Kayal, W.; Subramanian, J.; Sherif, S.M. Functional characterization of a gibberellin F-box protein, PslSLY1, during plum fruit development. J. Exp. Bot. 2021, 72, 371–384. [Google Scholar] [CrossRef]
- Benjamins, R.; Scheres, B. Auxin: The Looping Star in Plant Development. Annu. Rev. Plant Biol. 2008, 59, 443–465. [Google Scholar] [CrossRef]
- Zheng, Z.; Guo, Y.; Novak, O.; Chen, W.; Ljung, K.; Noel, J.P.; Chory, J. Local auxin metabolism regulates environment-induced hypocotyl elongation. Nat. Plants 2016, 2, 16025. [Google Scholar] [CrossRef]
- Ding, C.; Lin, X.; Zuo, Y.; Yu, Z.; Baerson, S.R.; Pan, Z.; Zeng, R.; Song, Y. Transcription factor OsbZIP49 controls tiller angle and plant architecture through the induction of indole-3-acetic acid-amido synthetases in rice. Plant J. 2021, 108, 1346–1364. [Google Scholar] [CrossRef]
- Leyser, O. Auxin Signaling. Plant Physiol. 2018, 176, 465–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lerouxel, O.; Cavalier, D.M.; Liepman, A.H.; Keegstra, K. Biosynthesis of plant cell wall polysaccharides—A complex process. Curr. Opin. Plant Biol. 2006, 9, 621–630. [Google Scholar] [CrossRef]
- Wolf, S.; van der Does, D.; Ladwig, F.; Sticht, C.; Kolbeck, A.; Schürholz, A.K.; Augustin, S.; Keinath, N.; Rausch, T.; Greiner, S.; et al. A receptor-like protein mediates the response to pectin modification by activating brassinosteroid signaling. Proc. Natl. Acad. Sci. USA 2014, 111, 15261–15266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergonci, T.; Silva-Filho, M.C.; Moura, D.S. Antagonistic relationship between AtRALF1 and brassinosteroid regulates cell expansion-related genes. Plant Signal. Behav. 2014, 9, e976146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castorina, G.; Consonni, G. The Role of Brassinosteroids in Controlling Plant Height in Poaceae: A Genetic Perspective. Int. J. Mol. Sci. 2020, 21, 1191. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, T.; Mogami, J.; Yamaguchi-Shinozaki, K. ABA-dependent and ABA-independent signaling in response to osmotic stress in plants. Curr. Opin. Plant Biol. 2014, 21, 133–139. [Google Scholar] [CrossRef]
- Haecker, A.; Groß-Hardt, R.; Geiges, B.; Sarkar, A.; Breuninger, H.; Herrmann, M.; Laux, T. Expression dynamics of WOX genes mark cell fate decisions during early embryonic patterning in Arabidopsis thaliana. Development 2004, 131, 657–668. [Google Scholar] [CrossRef] [Green Version]
- Si, X.; Wang, W.; Wang, K.; Liu, Y.; Bai, J.; Meng, Y.; Zhang, X.; Liu, H. A Sheathed Spike Gene, TaWUS-like Inhibits Stem Elongation in Common Wheat by Regulating Hormone Levels. Int. J. Mol. Sci. 2021, 22, 11210. [Google Scholar] [CrossRef]
- Leibfried, A.; To, J.P.; Busch, W.; Stehling, S.; Kehle, A.; Demar, M.; Kieber, J.J.; Lohmann, J.U. WUSCHEL controls meristem function by direct regulation of cytokinin-inducible response regulators. Nature 2005, 438, 1172–1175. [Google Scholar] [CrossRef]
- Spielmann, M.; Lupianez, D.G.; Mundlos, S. Structural variation in the 3D genome. Nat. Rev. Genet. 2018, 19, 453–467. [Google Scholar] [CrossRef]
- Carvalho, C.M.; Lupski, J.R. Mechanisms underlying structural variant formation in genomic disorders. Nat. Rev. Genet. 2016, 17, 224–238. [Google Scholar] [CrossRef] [PubMed]
- Polsoni, L. Large-scale microspore culture technique for mutation-selection studies in Brassica napus. Can. J. Bot. 1988, 66, 1681–1685. [Google Scholar] [CrossRef]
- Bednarek, P.T.; Pachota, K.A.; Dynkowska, W.M.; Machczynska, J.; Orłowska, R. Understandingin vitro tissue culture-induced variation phenomenon in microspore system. Int. J. Mol. Sci. 2021, 22, 7546. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, J. Genes controlling plant architecture. Curr. Opin. Biotechnol. 2006, 17, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhou, C.; Yan, X.; Zhang, J.; Xu, J. Simultaneous analysis of ten phytohormones in Sargassum horneri by high-performance liquid chromatography with electrospray ionization tandem mass spectrometry. J. Sep. Sci. 2016, 39, 1804–1813. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ye, S.; Yan, L.; Ma, X.; Chen, Y.; Wu, L.; Ma, T.; Zhao, L.; Yi, B.; Ma, C.; Tu, J.; et al. Combined BSA-Seq Based Mapping and RNA-Seq Profiling Reveal Candidate Genes Associated with Plant Architecture in Brassica napus. Int. J. Mol. Sci. 2022, 23, 2472. https://doi.org/10.3390/ijms23052472
Ye S, Yan L, Ma X, Chen Y, Wu L, Ma T, Zhao L, Yi B, Ma C, Tu J, et al. Combined BSA-Seq Based Mapping and RNA-Seq Profiling Reveal Candidate Genes Associated with Plant Architecture in Brassica napus. International Journal of Molecular Sciences. 2022; 23(5):2472. https://doi.org/10.3390/ijms23052472
Chicago/Turabian StyleYe, Shenhua, Lei Yan, Xiaowei Ma, Yanping Chen, Lumei Wu, Tiantian Ma, Lun Zhao, Bin Yi, Chaozhi Ma, Jinxing Tu, and et al. 2022. "Combined BSA-Seq Based Mapping and RNA-Seq Profiling Reveal Candidate Genes Associated with Plant Architecture in Brassica napus" International Journal of Molecular Sciences 23, no. 5: 2472. https://doi.org/10.3390/ijms23052472
APA StyleYe, S., Yan, L., Ma, X., Chen, Y., Wu, L., Ma, T., Zhao, L., Yi, B., Ma, C., Tu, J., Shen, J., Fu, T., & Wen, J. (2022). Combined BSA-Seq Based Mapping and RNA-Seq Profiling Reveal Candidate Genes Associated with Plant Architecture in Brassica napus. International Journal of Molecular Sciences, 23(5), 2472. https://doi.org/10.3390/ijms23052472