
Adenosine Kinase Isoforms in the Developing Rat Hippocampus after LiCl/Pilocarpine Status Epilepticus


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
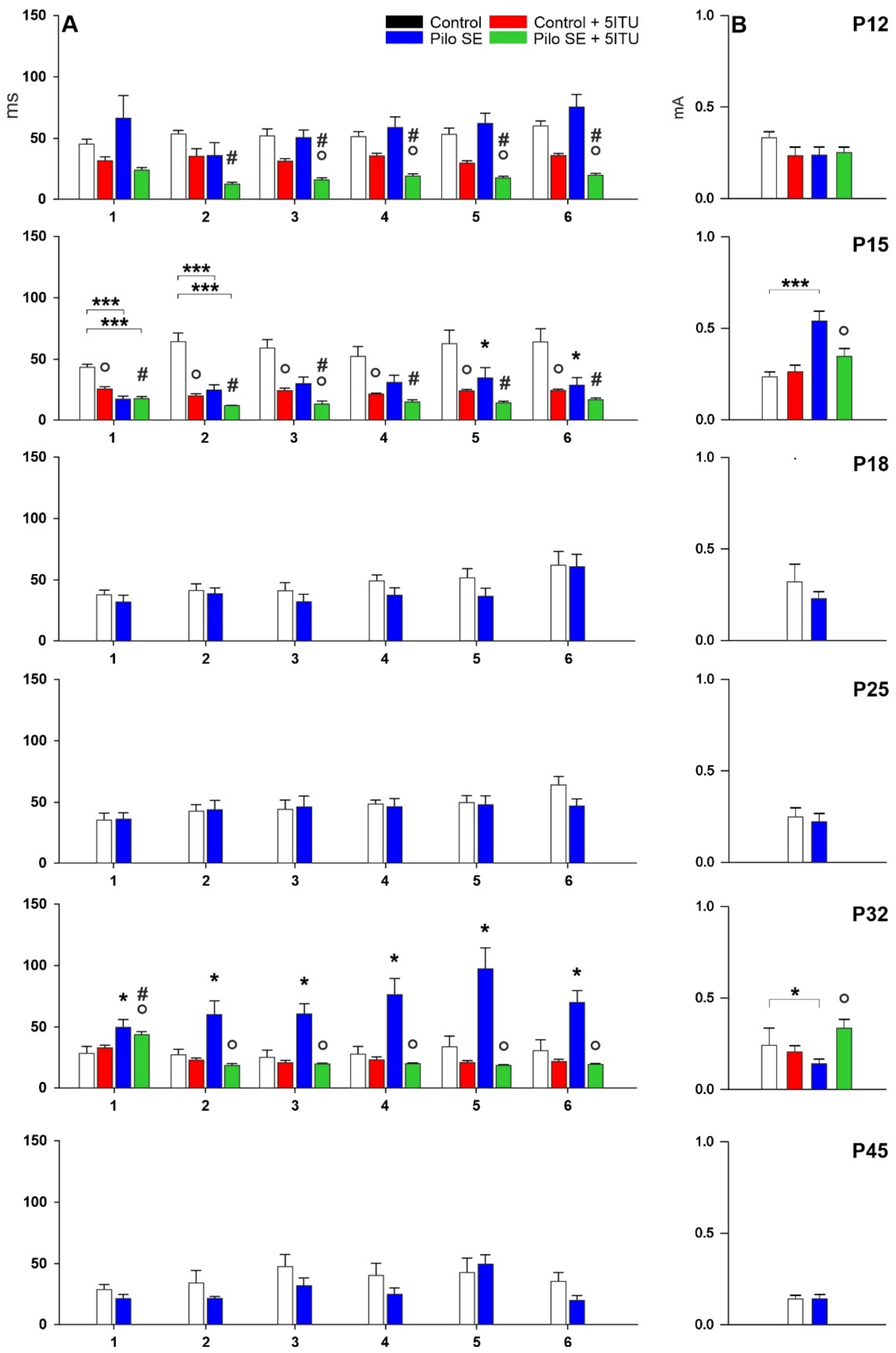
2.1. Threshold Intensity
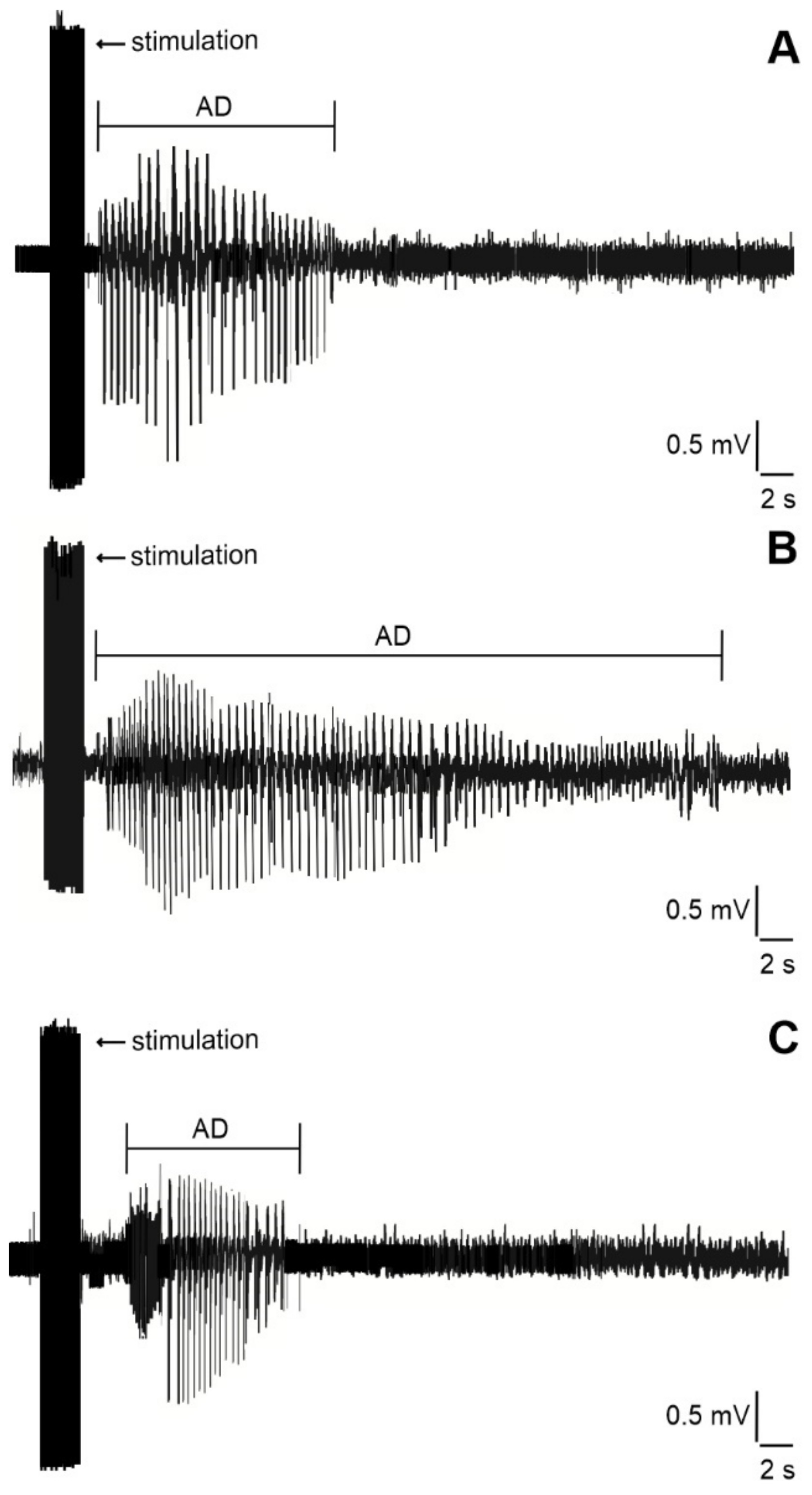
2.2. Duration of Hippocampal Afterdischarges
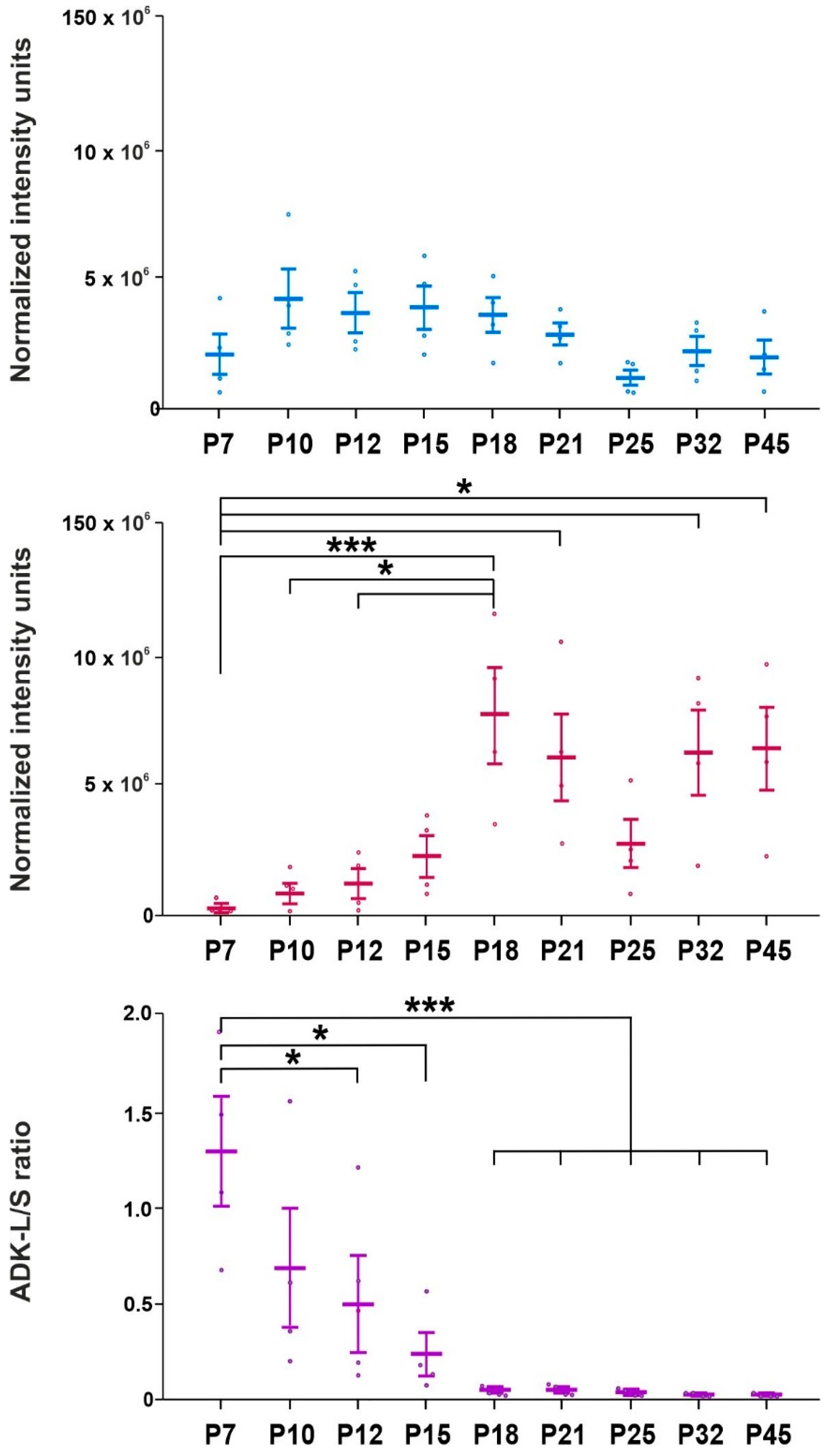
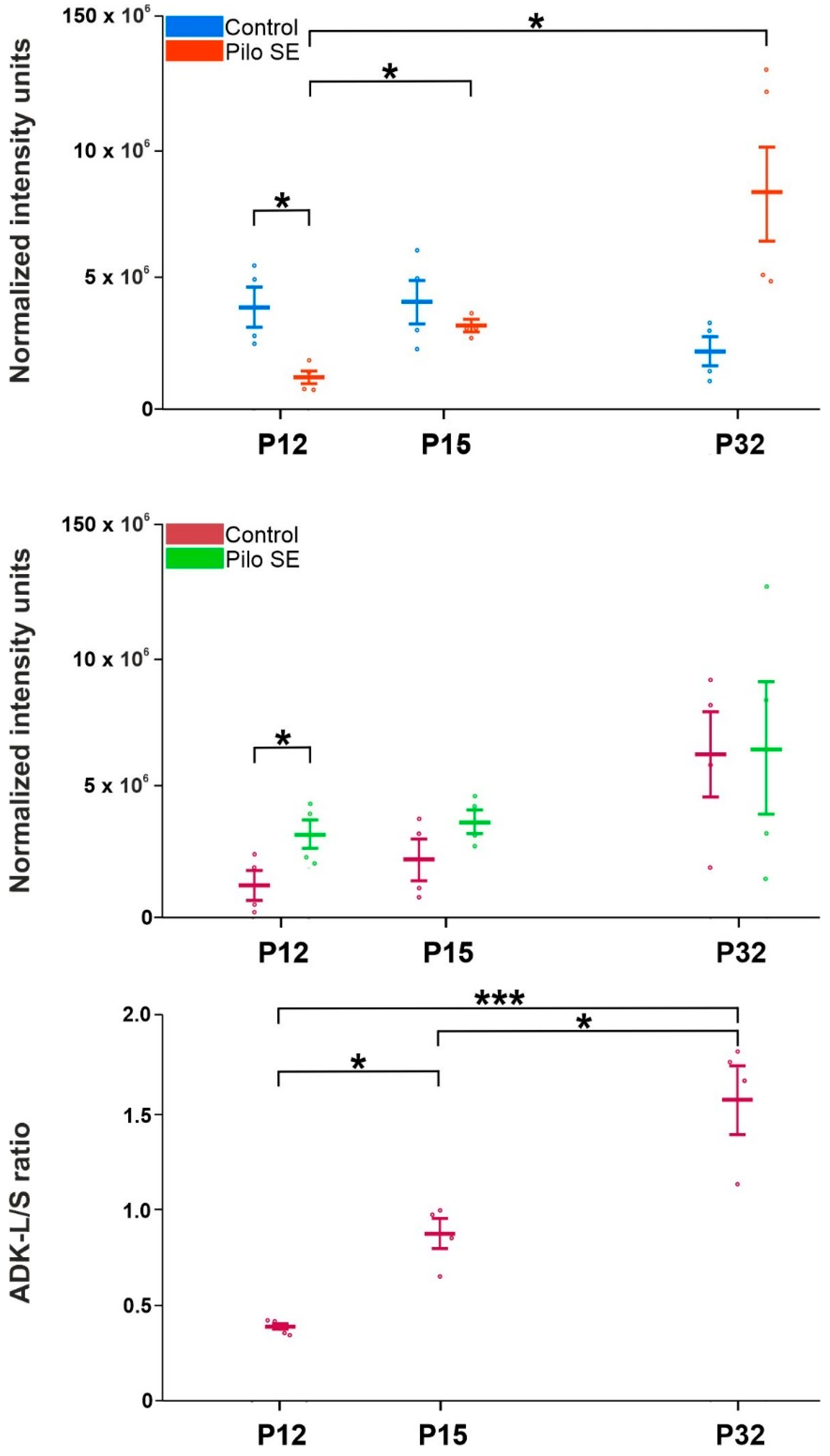
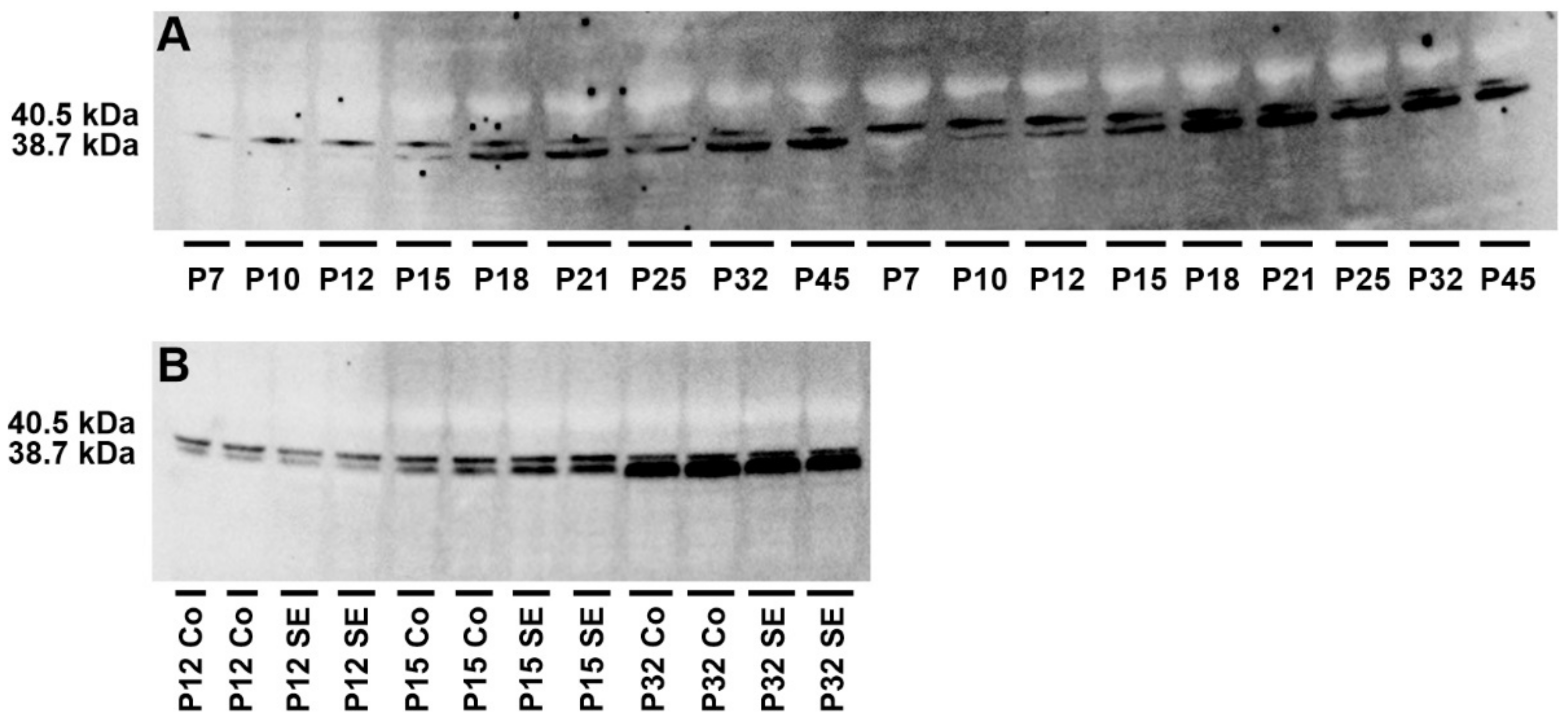
2.3. Analysis of Adenosine Kinase Isoforms
3. Discussion
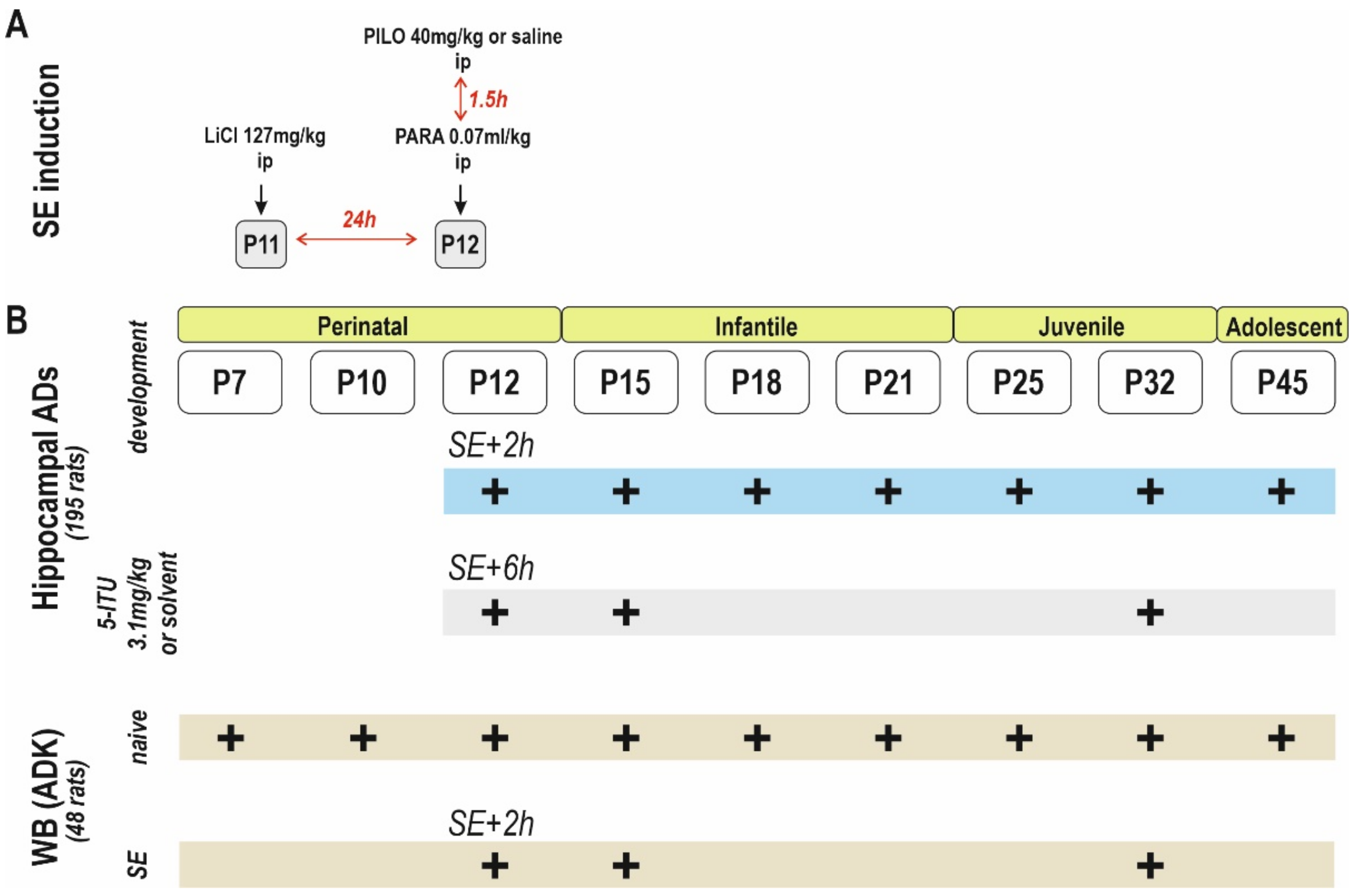
4. Materials and Methods
4.1. Animals
4.2. Status Epilepticus
4.3. Surgery
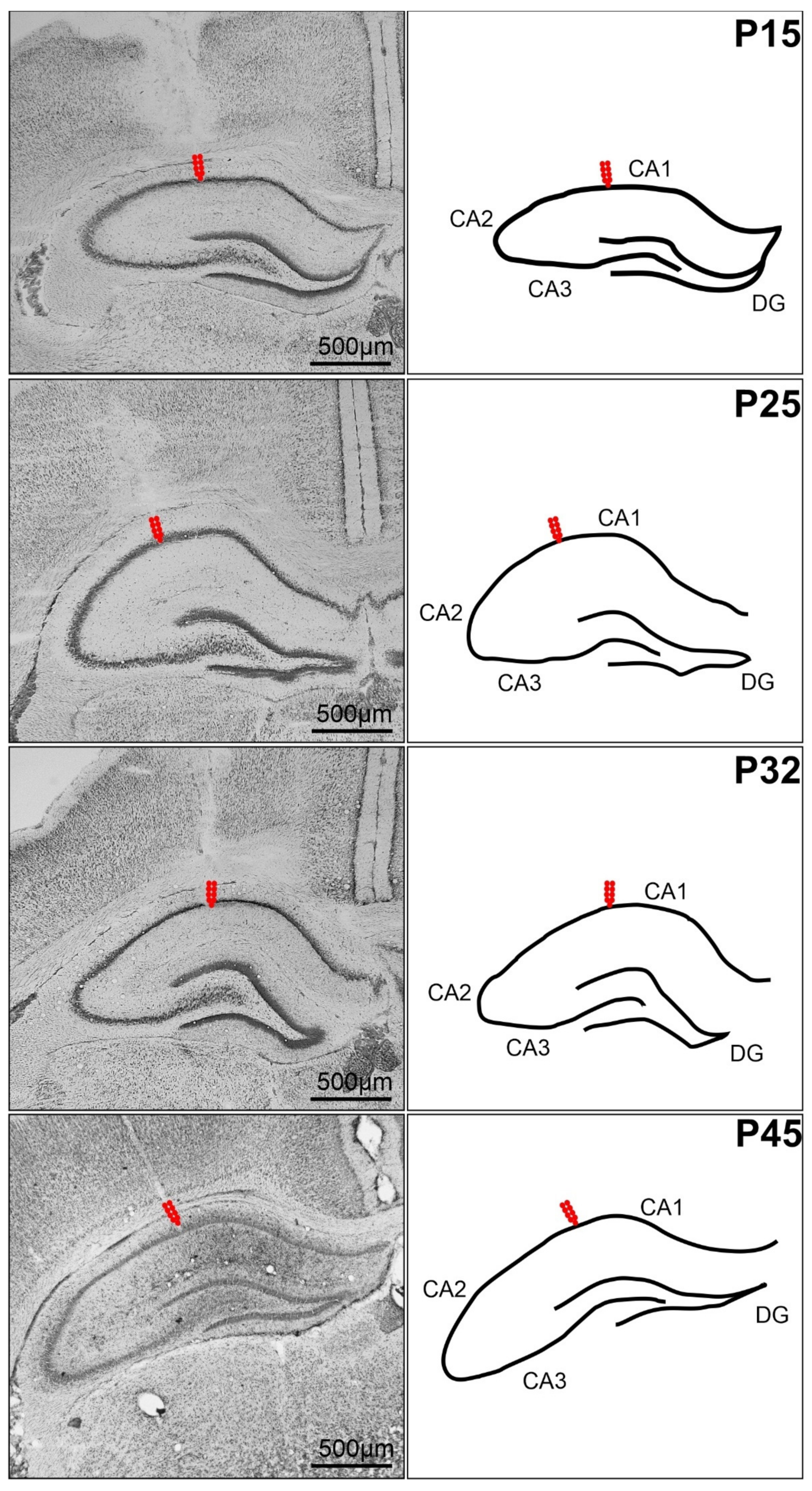
4.4. Stimulation Procedure, Afterdischarges and Threshold Intensity
4.5. Recording
4.6. Drugs
4.7. Western Blot Analysis
4.8. Statistics
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wiesner, J.B.; Ugarkar, B.G.; Castellino, A.J.; Barankiewicz, J.; Dumas, D.P.; Gruber, H.E.; Foster, A.C.; Erion, M.D. Adenosine kinase inhibitors as a novel approach to anticonvulsant therapy. J. Pharmacol. Exp. Ther. 1999, 289, 1669–1677. [Google Scholar]
- Gorter, J.A.; Van Vliet, E.A.; Aronica, E.; Breit, T.; Rauwerda, H.; Da Silva, F.H.L.; Wadman, W.J. Potential New Antiepileptogenic Targets Indicated by Microarray Analysis in a Rat Model for Temporal Lobe Epilepsy. J. Neurosci. 2006, 26, 11083–11110. [Google Scholar] [CrossRef] [PubMed]
- Lowry, O.H.; Rosebrough, N.J.; Farr, A.L.; Randall, R.J. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 1951, 193, 265–275. [Google Scholar] [CrossRef]
- Gaudin, A.; Lepetre-Mouelhi, S.; Mougin, J.; Parrod, M.; Pieters, G.; Garcia-Argote, S.; Loreau, O.; Goncalves, J.; Chacun, H.; Courbebaisse, Y.; et al. Pharmacokinetics, biodistribution and metabolism of squalenoyl adenosine nanoparticles in mice using dual radio-labeling and radio-HPLC analysis. J. Control. Release 2015, 212, 50–58. [Google Scholar] [CrossRef] [Green Version]
- Fritz, H.; Hess, R. Ossification of the rat and mouse skeleton in the perinatal period. Teratology 1970, 3, 331–337. [Google Scholar] [CrossRef] [PubMed]
- Dunwiddie, T.V.; Masino, S.A. The role and regulation of adenosine in the central nervous system. Annu. Rev. Neurosci. 2001, 24, 31–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fredholm, B.B.; Chen, J.F.; Cunha, R.A.; Svenningsson, P.; Vaugeois, J.M. Adenosine and brain function. Int. Rev. Neurobiol. 2005, 63, 191–270. [Google Scholar] [CrossRef]
- Fredholm, B.B.; Chen, J.F.; Masino, S.A.; Vaugeois, J.M. Actions of adenosine at its receptors in the CNS: Insights from knockouts and drugs. Annu. Rev. Pharmacol. Toxicol. 2005, 45, 385–412. [Google Scholar] [CrossRef]
- Boison, D. Adenosine and epilepsy: From therapeutic rationale to new therapeutic strategies. Neuroscientist 2005, 11, 25–36. [Google Scholar] [CrossRef]
- Cunha, R.A. Neuroprotection by adenosine in the brain: From A(1) receptor activation to A (2A) receptor blockade. Purinergic Signal. 2005, 1, 111–134. [Google Scholar] [CrossRef] [Green Version]
- Lado, F.A.; Moshe, S. How do seizures stop? Epilepsia 2008, 49, 1651–1664. [Google Scholar] [CrossRef]
- Williams, M. Purines: From premise to promise. J. Auton. Nerv. Syst. 2000, 81, 285–288. [Google Scholar] [CrossRef]
- Boison, D. Adenosine Kinase: Exploitation for Therapeutic Gain. Pharmacol. Rev. 2013, 65, 906–943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.; Gupta, R.S. Adenosine kinase and ribokinase—The RK family of proteins. Cell. Mol. Life Sci. 2008, 65, 2875–2896. [Google Scholar] [CrossRef]
- Fedele, D.E.; Koch, P.; Scheurer, L.; Simpson, E.; Möhler, H.; Brüstle, O.; Boison, D. Engineering embryonic stem cell derived glia for adenosine delivery. Neurosci. Lett. 2004, 370, 160–165. [Google Scholar] [CrossRef]
- Güttinger, M.; Fedele, D.; Koch, P.; Padrun, V.; Pralong, W.F.; Brüstle, O.; Boison, D. Suppression of Kindled Seizures by Paracrine Adenosine Release from Stem Cell-Derived Brain Implants. Epilepsia 2005, 46, 1162–1169. [Google Scholar] [CrossRef] [Green Version]
- Huber, A.; Güttinger, M.; Möhler, H.; Boison, D. Seizure suppression by adenosine A2A receptor activation in a rat model of audiogenic brainstem epilepsy. Neurosci. Lett. 2002, 329, 289–292. [Google Scholar] [CrossRef]
- Pak, M.; Haas, H.; Decking, U.; Schrader, J. Inhibition of adenosine kinase increases endogenous adenosine and depresses neuronal activity in hippocampal slices. Neuropharmacology 1994, 33, 1049–1053. [Google Scholar] [CrossRef]
- Kowaluk, E.A.; Bhagwat, S.S.; Jarvis, M.F. Adenosine kinase inhibitors. Curr. Pharm. Des. 1998, 4, 403–416. [Google Scholar]
- Ugarkar, B.G.; DaRe, J.M.; Kopcho, J.J.; Browne, C.E.; Schanzer, J.M.; Wiesner, J.B.; Erion, M.D. Adenosine Kinase Inhibitors. 1. Synthesis, Enzyme Inhibition, and Antiseizure Activity of 5-Iodotubercidin Analogues. J. Med. Chem. 2000, 43, 2883–2893. [Google Scholar] [CrossRef]
- Boison, D. The adenosine kinase hypothesis of epileptogenesis. Prog. Neurobiol. 2008, 84, 249–262. [Google Scholar] [CrossRef] [Green Version]
- Gouder, N.; Scheurer, L.; Fritschy, J.-M.; Boison, D. Overexpression of Adenosine Kinase in Epileptic Hippocampus Contributes to Epileptogenesis. J. Neurosci. 2004, 24, 692–701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, X.A.; Singh, B.; Park, J.; Gupta, R.S. Subcellular localization of adenosine kinase in mammalian cells: The long isoform of AdK is localized in the nucleus. Biochem. Biophys. Res. Commun. 2009, 388, 46–50. [Google Scholar] [CrossRef]
- Williams-Karnesky, R.L.; Sandau, U.S.; Lusardi, T.; Lytle, N.K.; Farrell, J.M.; Pritchard, E.M.; Kaplan, D.L.; Boison, D. Epigenetic changes induced by adenosine augmentation therapy prevent epileptogenesis. J. Clin. Investig. 2013, 123, 3552–3563. [Google Scholar] [CrossRef] [Green Version]
- Kobow, K.; Blümcke, I. The methylation hypothesis: Do epigenetic chromatin modifications play a role in epileptogenesis? Epilepsia 2011, 52, 15–19. [Google Scholar] [CrossRef] [PubMed]
- Qureshi, I.A.; Mehler, M.F. Epigenetic mechanisms underlying human epileptic disorders and the process of epileptogenesis. Neurobiol. Dis. 2010, 39, 53–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fedele, D.E.; Gouder, N.; Güttinger, M.; Gabernet, L.; Scheurer, L.; Rülicke, T.; Crestani, F.; Boison, D. Astrogliosis in epilepsy leads to overexpression of adenosine kinase, resulting in seizure aggravation. Brain 2005, 128, 2383–2395. [Google Scholar] [CrossRef] [Green Version]
- Studer, F.; Fedele, D.; Marowsky, A.; Schwerdel, C.; Wernli, K.; Vogt, K.; Fritschy, J.-M.; Boison, D. Shift of adenosine kinase expression from neurons to astrocytes during postnatal development suggests dual functionality of the enzyme. Neuroscience 2006, 142, 125–137. [Google Scholar] [CrossRef]
- Kiese, K.; Jablonski, J.; Boison, D.; Kobow, K. Dynamic Regulation of the Adenosine Kinase Gene during Early Postnatal Brain Development and Maturation. Front. Mol. Neurosci. 2016, 9, 99. [Google Scholar] [CrossRef] [Green Version]
- Gebril, H.M.; Rose, R.M.; Gesese, R.; Emond, M.P.; Huo, Y.; Aronica, E.; Boison, D. Adenosine kinase inhibition promotes proliferation of neural stem cells after traumatic brain injury. Brain Commun. 2020, 2, fcaa017. [Google Scholar] [CrossRef] [Green Version]
- Semple, B.D.; Blomgren, K.; Gimlin, K.; Ferriero, D.M.; Noble-Haeusslein, L.J. Brain development in rodents and humans: Identifying benchmarks of maturation and vulnerability to injury across species. Prog. Neurobiol. 2013, 106–107, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Chauvel, P.; Landre, E.; Trottier, S.; Vignel, J.P.; Biraben, A.; Devaux, B.; Bancaud, J. Electrical stimulation with intracerebral electrodes to evoke seizures. Adv. Neurol. 1993, 63, 115–121. [Google Scholar]
- Valentin, A.; Anderson, M.; Alarcón, G.; Seoane, J.J.G.; Selway, R.; Binnie, C.D.; Polkey, C.E. Responses to single pulse electrical stimulation identify epileptogenesis in the human brain in vivo. Brain 2002, 125, 1709–1718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kandratavicius, L.; Balista, P.A.; Lopes-Aguiar, C.; Ruggiero, R.N.; Umeoka, E.H.; Garcia-Cairasco, N.; Bueno-Junior, L.S.; Leite, J.P. Animal models of epilepsy: Use and limitations. Neuropsychiatr. Dis. Treat. 2014, 10, 1693–1705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maresova, D.; Mares, P. Hippocampo-cortical after-discharges during ontogenesis in rats. In Ontogenesis of the Brain; Trojan, S., Stastny, F., Eds.; University Carolina Press: Prague, Czech Republic, 1987; pp. 279–283. [Google Scholar]
- Velisek, L.; Mares, P. Hippocampal afterdischarges in rats. I. Effects of antiepileptics. Physiol. Res. 2004, 53, 453–461. [Google Scholar] [PubMed]
- Kotloski, R.; Lynch, M.; Lauersdorf, S.; Sutula, T. Repeated brief seizures induce progressive hippocampal neuron loss and memory deficits. Prog. Brain Res. 2002, 135, 95–110. [Google Scholar] [CrossRef]
- Norwood, B.A.; Bumanglag, A.V.; Osculati, F.; Sbarbati, A.; Marzola, P.; Nicolato, E.; Fabene, P.F.; Sloviter, R.S. Classic hippocampal sclerosis and hippocampal-onset epilepsy produced by a single “cryptic” episode of focal hippocampal excitation in awake rats. J. Comp. Neurol. 2010, 518, 3381–3407. [Google Scholar] [CrossRef] [Green Version]
- Gollwitzer, S.; Hopfengärtner, R.; Rössler, K.; Müller, T.; Olmes, D.G.; Lang, J.; Köhn, J.; Onugoren, M.D.; Heyne, J.; Schwab, S.; et al. Afterdischarges elicited by cortical electric stimulation in humans: When do they occur and what do they mean? Epilepsy Behav. 2018, 87, 173–179. [Google Scholar] [CrossRef]
- Curia, G.; Longo, D.; Biagini, G.; Jones, R.S.; Avoli, M. The pilocarpine model of temporal lobe epilepsy. J. Neurosci. Methods 2008, 172, 143–157. [Google Scholar] [CrossRef]
- Kubová, H.; Mareš, P.; Suchomelová, L.; Brožek, G.; Druga, R.; Pitkänen, A. Status epilepticus in immature rats leads to behavioural and cognitive impairment and epileptogenesis. Eur. J. Neurosci. 2004, 19, 3255–3265. [Google Scholar] [CrossRef]
- DeLorenzo, R.; Hauser, W.A.; Towne, A.R.; Boggs, J.G.; Pellock, J.M.; Penberthy, L.; Garnett, L.; Fortner, C.A.; Ko, D. A prospective, population-based epidemiologic study of status epilepticus in Richmond, Virginia. Neurology 1996, 46, 1029–1035. [Google Scholar] [CrossRef]
- Chin, R.F.; Neville, B.G.; Peckham, C.; Bedford, H.; Wade, A.; Scott, R.C. Incidence, cause, and short-term outcome of convulsive status epilepticus in childhood: Prospective population-based study. Lancet 2006, 368, 222–229. [Google Scholar] [CrossRef]
- Sengupta, P. The Laboratory Rat: Relating Its Age with Human’s. Int. J. Prev. Med. 2013, 4, 624–630. [Google Scholar]
- Etherington, L.-A.V.; Patterson, G.E.; Meechan, L.; Boison, D.; Irving, A.J.; Dale, N.; Frenguelli, B.G. Astrocytic adenosine kinase regulates basal synaptic adenosine levels and seizure activity but not activity-dependent adenosine release in the hippocampus. Neuropharmacology 2009, 56, 429–437. [Google Scholar] [CrossRef] [PubMed]
- Peterson, G.L. A simplification of the protein assay method of Lowry et al. which is more generally applicable. Anal. Biochem. 1977, 83, 346–356. [Google Scholar] [CrossRef]
- Colella, A.D.; Chegenii, N.; Tea, M.N.; Gibbins, I.L.; Williams, K.A.; Chataway, T.K. Comparison of Stain-Free gels with traditional immunoblot loading control methodology. Anal. Biochem. 2012, 430, 108–110. [Google Scholar] [CrossRef]
- Sloviter, R.S. The functional organization of the hippocampal dentate gyrus and its relevance to the pathogenesis of temporal lobe epilepsy. Ann. Neurol. 1994, 35, 640–654. [Google Scholar] [CrossRef]
- Dudek, F.E.; Staley, K.J. Seizure probability in animal models of acquired epilepsy: A perspective on the concept of the preictal state. Epilepsy Res. 2011, 97, 324–331. [Google Scholar] [CrossRef]
- Löscher, W.; Hirsch, L.; Schmidt, D. The enigma of the latent period in the development of symptomatic acquired epilepsy—Traditional view versus new concepts. Epilepsy Behav. 2015, 52, 78–92. [Google Scholar] [CrossRef] [PubMed]
- Tsenov, G.; Mares, P. Depression and/or potentiation of cortical responses after status epilepticus in immature rats. Physiol. Res. 2007, 56, 485–491. [Google Scholar] [CrossRef]
- Sankar, R.; Shin, D.H.; Liu, H.; Mazarati, A.; De Vasconcelos, A.P.; Wasterlain, C.G. Patterns of Status Epilepticus-Induced Neuronal Injury during Development and Long-Term Consequences. J. Neurosci. 1998, 18, 8382–8393. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Baram, T.Z.; Soltesz, I. Febrile seizures in the developing brain result in persistent modification of neuronal excitability in limbic circuits. Nat. Med. 1999, 5, 888–894. [Google Scholar] [CrossRef]
- Dube, C.; Chen, K.; Eghbal-Ahmadi, M.; Brunson, K.; Soltesz, I.; Baram, T.Z. Prolonged febrile seizures in the immature rat model enhance hippocampal excitability long term. Ann. Neurol. 2000, 47, 336–344. [Google Scholar] [CrossRef]
- Dinocourt, C.; Petanjek, Z.; Freund, T.F.; Ben-Ari, Y.; Esclapez, M. Loss of interneurons innervating pyramidal cell dendrites and axon initial segments in the CA1 region of the hippocampus following pilocarpine-induced seizures. J. Comp. Neurol. 2003, 459, 407–425. [Google Scholar] [CrossRef] [PubMed]
- Nairismagi, J.; Pitkanen, A.; Kettunen, M.; Kauppinen, R.A.; Kubova, H. Status Epilepticus in 12-day-old Rats Leads to Temporal Lobe Neurodegeneration and Volume Reduction: A Histologic and MRI Study. Epilepsia 2006, 47, 479–488. [Google Scholar] [CrossRef]
- Scholl, E.; Dudek, F.; Ekstrand, J. Neuronal degeneration is observed in multiple regions outside the hippocampus after lithium pilocarpine-induced status epilepticus in the immature rat. Neuroscience 2013, 252, 45–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furtado, M.; Castro, O.; Del Vecchio, F.; de Oliveira, J.C.; Garcia-Cairasco, N. Study of spontaneous recurrent seizures and morphological alterations after status epilepticus induced by intrahippocampal injection of pilocarpine. Epilepsy Behav. 2011, 20, 257–266. [Google Scholar] [CrossRef]
- Müller, C.J.; Bankstahl, M.; Gröticke, I.; Löscher, W. Pilocarpine vs. lithium–pilocarpine for induction of status epilepticus in mice: Development of spontaneous seizures, behavioral alterations and neuronal damage. Eur. J. Pharmacol. 2009, 619, 15–24. [Google Scholar] [CrossRef]
- Cavalheiro, E.A.; Silva, D.F.; Turski, W.A.; Calderazzo-Filho, L.S.; Bortolotto, Z.A.; Turski, L. The susceptibility of rats to pilocarpine-induced seizures is age-dependent. Dev. Brain Res. 1987, 37, 43–58. [Google Scholar] [CrossRef]
- Racine, R.J. Modification of seizure activity by electrical stimulation: II. Motor seizure. Electroencephalogr. Clin. Neurophysiol. 1972, 32, 281–294. [Google Scholar] [CrossRef]
- Bragin, A.; Penttonen, M.; Buzsaki, G. Termination of Epileptic Afterdischarge in the Hippocampus. J. Neurosci. 1997, 17, 2567–2579. [Google Scholar] [CrossRef]
- Ekstrand, J.; Pouliot, W.; Scheerlinck, P.; Dudek, F. Lithium pilocarpine-induced status epilepticus in postnatal day 20 rats results in greater neuronal injury in ventral versus dorsal hippocampus. Neuroscience 2011, 192, 699–707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lynch, M.; Sayin, U.; Bownds, J.; Janumpalli, S.; Sutula, T. Long-term consequences of early postnatal seizures on hippocampal learning and plasticity. Eur. J. Neurosci. 2000, 12, 2252–2264. [Google Scholar] [CrossRef]
- Liu, C.; Yu, T.; Ren, Z.-W.; Xu, C.-P.; Wang, X.-Y.; Qiao, L.; Ni, D.-Y.; Zhang, G.-J.; Li, Y.-J. Properties of afterdischarges from electrical stimulation in patients with epilepsy. Epilepsy Res. 2017, 137, 39–44. [Google Scholar] [CrossRef]
- Dos Santos, N.F.; Arida, R.M.; Filho, E.M.T.; Priel, M.R.; Cavalheiro, E.A. Epileptogenesis in immature rats following recurrent status epilepticus. Brain Res. Rev. 2000, 32, 269–276. [Google Scholar] [CrossRef]
- Baram, T.Z.; Jensen, F.E.; Brooks-Kayal, A. Does Acquired Epileptogenesis in the Immature Brain Require Neuronal Death? Epilepsy Curr. 2011, 11, 21–26. [Google Scholar] [CrossRef]
- Brandt, C.; Potschka, H.; Löscher, W.; Ebert, U. N-methyl-d-aspartate receptor blockade after status epilepticus protects against limbic brain damage but not against epilepsy in the kainate model of temporal lobe epilepsy. Neuroscience 2003, 118, 727–740. [Google Scholar] [CrossRef]
- Zhang, G.; Raol, Y.S.H.; Hsu, F.-C.; Brooks-Kayal, A.R. Long-term alterations in glutamate receptor and transporter expression following early-life seizures are associated with increased seizure susceptibility. J. Neurochem. 2003, 88, 91–101. [Google Scholar] [CrossRef]
- Sankar, R.; Shin, N.; Liu, H.; Katsumori, H.; Wasterlain, C.G. Granule cell neurogenesis after status epilepticus in the immature rat brain. Epilepsia 2000, 41, S53–S56. [Google Scholar] [CrossRef] [Green Version]
- Folbergrova, J.; Jesina, P.; Kubova, H.; Druga, R.; Otahal, J. Status epilepticus in immature rats is associated with oxidative stress and mitochondrial dysfunction. Front. Cell. Neurosci. 2016, 10, 136. [Google Scholar] [CrossRef] [Green Version]
- Vizuete, A.F.K.; Hennemann, M.M.; Gonçalves, C.A.; De Oliveira, D.L. Phase-Dependent Astroglial Alterations in Li–Pilocarpine-Induced Status Epilepticus in Young Rats. Neurochem. Res. 2017, 42, 2730–2742. [Google Scholar] [CrossRef]
- Klein, P.; Dingledine, R.; Aronica, E.; Bernard, C.; Blümcke, I.; Boison, D.; Brodie, M.J.; Brooks-Kayal, A.R.; Engel, J., Jr.; Forcelli, P.A.; et al. Commonalities in epileptogenic processes from different acute brain insults: Do they translate? Epilepsia 2018, 59, 37–66. [Google Scholar] [CrossRef]
- Rizzi, M.; Perego, C.; Aliprandi, M.; Richichi, C.; Ravizza, T.; Colella, D.; Velískǒvá, J.; Moshe, S.; de Simoni, M.G.; Vezzani, A. Glia activation and cytokine increase in rat hippocampus by kainic acid-induced status epilepticus during postnatal development. Neurobiol. Dis. 2003, 14, 494–503. [Google Scholar] [CrossRef]
- Sofroniew, M.V.; Vinters, H.V. Astrocytes: Biology and pathology. Acta Neuropathol. 2010, 119, 7–35. [Google Scholar] [CrossRef] [Green Version]
- Vezzani, A. Epileptogenic Role of Astrocyte Dysfunction. Epilepsy Curr. 2008, 8, 46–47. [Google Scholar] [CrossRef] [Green Version]
- Boison, D.; Steinhäuser, C. Epilepsy and astrocyte energy metabolism. Glia 2018, 66, 1235–1243. [Google Scholar] [CrossRef] [PubMed]
- George, B.; Kulkarni, S.K. Modulation of lithium-pilocarpine-induced status epilepticus by adenosinergic agents. Methods Find. Exp. Clin. Pharmacol. 1997, 19, 329–333. [Google Scholar] [PubMed]
- Khan, G.M.; Smolders, I.; Ebinger, G.; Michotte, Y. Anticonvulsant effect and neurotransmitter modulation of focal and systemic 2-chloroadenosine against the development of pilocarpine-induced seizures. Neuropharmacology 2000, 39, 2418–2432. [Google Scholar] [CrossRef]
- Boison, D. Adenosine kinase, epilepsy and stroke: Mechanisms and therapies. Trends Pharmacol. Sci. 2006, 27, 652–658. [Google Scholar] [CrossRef]
- Li, T.; Lan, J.Q.; Fredholm, B.B.; Simon, R.P.; Boison, D. Adenosine dysfunction in astrogliosis: Cause for seizure generation? Neuron Glia Biol. 2007, 3, 353–366. [Google Scholar] [CrossRef] [Green Version]
- Theofilas, P.; Brar, S.; Stewart, K.-A.; Shen, H.-Y.; Sandau, U.S.; Poulsen, D.; Boison, D. Adenosine kinase as a target for therapeutic antisense strategies in epilepsy. Epilepsia 2011, 52, 589–601. [Google Scholar] [CrossRef] [Green Version]
- Veliskova, J.; Velisek, L.; Mares, P. Epileptic phenomena produced by kainic acid in laboratory rats during ontogenesis. Physiol. Bohemoslov. 1988, 37, 395–405. [Google Scholar]
- Aronica, E.; Zurolo, E.; Iyer, A.; de Groot, M.; Anink, J.; Carbonell, C.; van Vliet, E.A.; Baayen, J.C.; Boison, D.; Gorter, J.A. Upregulation of adenosine kinase in astrocytes in experimental and human temporal lobe epilepsy. Epilepsia 2011, 52, 1645–1655. [Google Scholar] [CrossRef] [Green Version]
- Boison, D.; Chen, J.-F.; Fredholm, B.B. Adenosine signaling and function in glial cells. Cell Death Differ. 2009, 17, 1071–1082. [Google Scholar] [CrossRef] [Green Version]
- Frush, D.; McNamara, J. Evidence implicating dentate granule cells in wet dog shakes produced by kindling stimulations of entorhinal cortex. Exp. Neurol. 1986, 92, 102–113. [Google Scholar] [CrossRef]
- Pignataro, G.; Maysami, S.; Studer, F.E.; Wilz, A.; Simon, R.P.; Boison, D. Downregulation of Hippocampal Adenosine Kinase after Focal Ischemia as Potential Endogenous Neuroprotective Mechanism. J. Cereb. Blood Flow Metab. 2007, 28, 17–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller-Delaney, S.F.C.; Das, S.; Sano, T.; Jimenez-Mateos, E.M.; Bryan, K.; Buckley, P.G.; Stallings, R.L.; Henshall, D.C. Differential DNA Methylation Patterns Define Status Epilepticus and Epileptic Tolerance. J. Neurosci. 2012, 32, 1577–1588. [Google Scholar] [CrossRef] [Green Version]
- Zang, G.; Franklin, P.H.; Murray, T.F. Manipulation of endogenous adenosine in the rat prepiriform cortex modulates seizure susceptibility. J. Pharmacol. Exp. Ther. 1993, 264, 1415–1424. [Google Scholar]
- Jarvis, M.F. Therapeutic potential of adenosine kinase inhibition—Revisited. Pharmacol. Res. Perspect. 2019, 7, e00506. [Google Scholar] [CrossRef] [Green Version]
- Lietsche, J.; Imran, I.; Klein, J. Extracellular levels of ATP and acetylcholine during lithium-pilocarpine induced status epilepticus in rats. Neurosci. Lett. 2016, 611, 69–73. [Google Scholar] [CrossRef] [PubMed]
- Pazzagli, M.; Corsi, C.; Fratti, S.; Pedata, F.; Pepeu, G. Regulation of extracellular adenosine levels in the striatum of aging rats. Brain Res. 1995, 684, 103–106. [Google Scholar] [CrossRef]
- Sandau, U.S.; Yahya, M.; Bigej, R.; Friedman, J.L.; Saleumvong, B.; Boison, D. Transient use of a systemic adenosine kinase inhibitor attenuates epilepsy development in mice. Epilepsia 2019, 60, 615–625. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fábera, P.; Uttl, L.; Kubová, H.; Tsenov, G.; Mareš, P. Adenosine Kinase Isoforms in the Developing Rat Hippocampus after LiCl/Pilocarpine Status Epilepticus. Int. J. Mol. Sci. 2022, 23, 2510. https://doi.org/10.3390/ijms23052510
Fábera P, Uttl L, Kubová H, Tsenov G, Mareš P. Adenosine Kinase Isoforms in the Developing Rat Hippocampus after LiCl/Pilocarpine Status Epilepticus. International Journal of Molecular Sciences. 2022; 23(5):2510. https://doi.org/10.3390/ijms23052510
Chicago/Turabian StyleFábera, Petr, Libor Uttl, Hana Kubová, Grygoriy Tsenov, and Pavel Mareš. 2022. "Adenosine Kinase Isoforms in the Developing Rat Hippocampus after LiCl/Pilocarpine Status Epilepticus" International Journal of Molecular Sciences 23, no. 5: 2510. https://doi.org/10.3390/ijms23052510
APA StyleFábera, P., Uttl, L., Kubová, H., Tsenov, G., & Mareš, P. (2022). Adenosine Kinase Isoforms in the Developing Rat Hippocampus after LiCl/Pilocarpine Status Epilepticus. International Journal of Molecular Sciences, 23(5), 2510. https://doi.org/10.3390/ijms23052510