
Deficiency in Retinal TGFβ Signaling Aggravates Neurodegeneration by Modulating Pro-Apoptotic and MAP Kinase Pathways

 ,
,  and
and
Abstract
:1. Introduction
2. Results
2.1. Deletion of TGFβ Signaling in Retinal Neurons and Müller Cells in Health and Disease
2.2. Deletion of TGFβ Signaling in Healthy Retinae Is Not Sufficient to Cause Morphological Changes
2.3. Deletion of TGFβ Signaling in Healthy Retinae Is Not Sufficient to Induce Major Transcriptional Changes
2.4. Deletion of TGFβ Signaling Increases the Susceptibility of Photoreceptors to Vpp-Induced Neurodegeneration
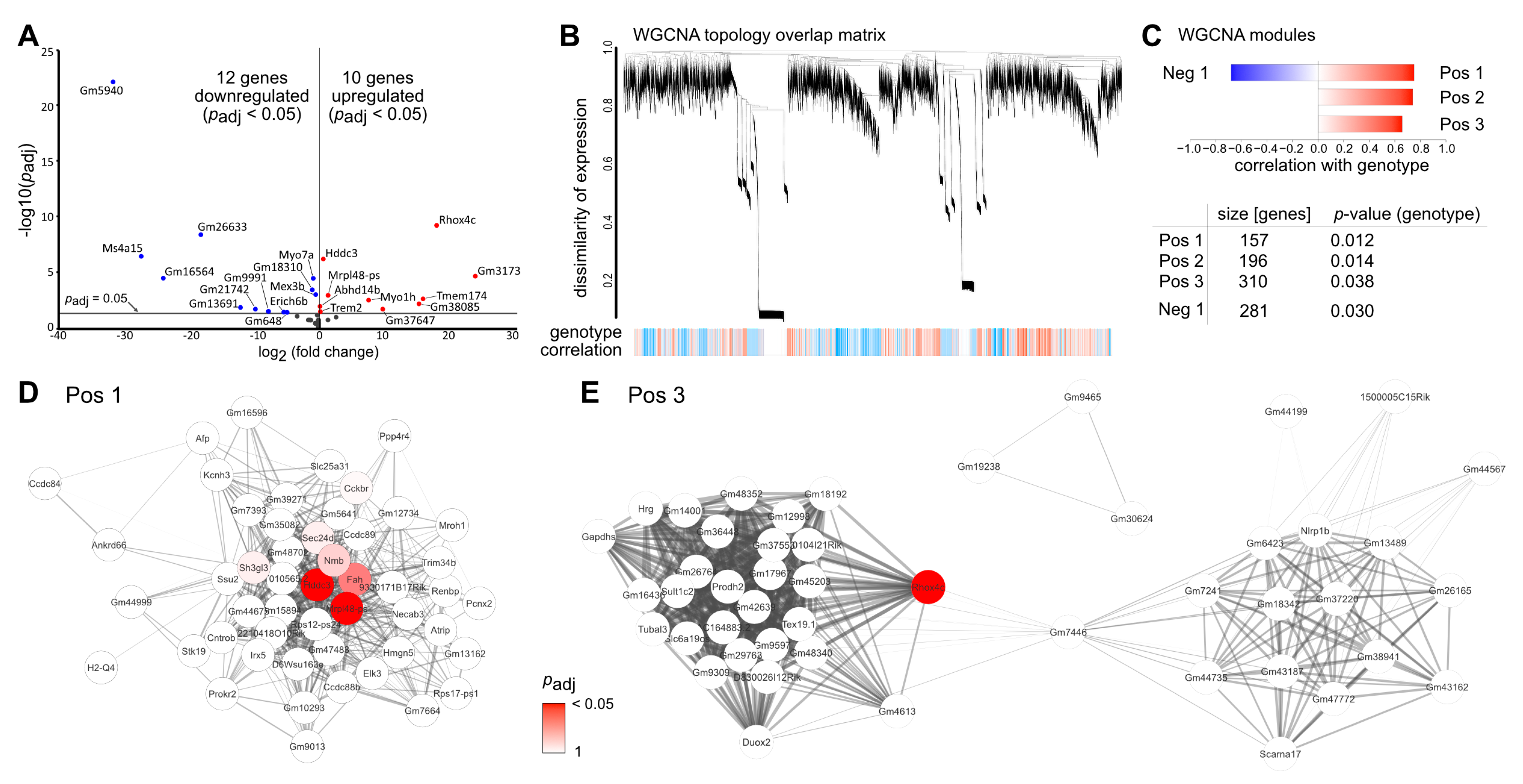
2.5. TGFβ-Mediated Effects on Vpp-Induced Transcriptomic Alterations
3. Discussion
3.1. TGFβ Signaling in Retinal Development and in the Healthy, Adult Retina
3.2. TGFβ Signaling Mediated Effects in Retinal Neurodegeneration
4. Conclusions
5. Material and Methods
5.1. Mice
5.2. Genotyping and Tgfbr2 Deletion
5.3. BaseScope®/In Situ Hybridization
5.4. RNA Sequencing
5.5. Bioinformatics
5.6. Cell Death Measurement by TdT-Mediated dUTP-Biotin Nick End Labeling (TUNEL)
5.7. Light Microscopy and Spider Diagram Analyses
5.8. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AAV | Adeno-associated virus |
| AMD | Age related macular degeneration |
| AP1 | Activator protein 1 |
| Bdnf | Brain derived neuroprotective factor |
| Cd29 | Integrin beta1 |
| CNS | Central nervous system |
| Cralbp | Cellular retinaldehyde-binding protein |
| Erk | Extracellular-signal regulated kinase |
| Fos | Fos proto-oncogene |
| GCL | Ganglion cell layer |
| Glul | Glutamine synthetase |
| Hddc | HD Domain containing 3 |
| Il6 | Interleukin-6 |
| INL | Inner nuclear layer |
| Itgb1 | Integrin beta-1 |
| Jnk3 | c-Jun N-terminal kinase3 |
| MAP | Mitogen-activated protein |
| MAPK | Mitogen-activated protein kinase |
| Mesh1 | HD Domain containing 3 |
| Mrpl48ps | Mitochondrial ribosomal protein L48 pseudogene |
| Myo7a | Myosin VIIA |
| Ngf | Neurotrophins nerve growth factor |
| OC | Optic nerve |
| ONH | Optic nerve head |
| ONL | Outer nuclear layer |
| OPL | Outer plexiform layer |
| OS | Ora serrata |
| PB | Phosphate buffer |
| PFA | Paraformaldehyde |
| pr. co. | Probe control |
| Rhox4c | Reproductive homeobox 4C |
| Rlbp1 | Retinaldehyde-binding protein 1 |
| RNAseq | Next generation RNA sequencing |
| RPE | Retinal pigment epithelium |
| Smad | Mothers against decapentaplegic homolog |
| Tgf | Transforming growth factor |
| Tgfbr2 | Transforming growth factor receptor |
| Tnf | Tumor necrosis factor |
| Trem2 | Triggering receptor expressed on Myeloid cells 2 |
| WGCNA | Weighted gene correlation network analysis |
References
- Buch, H.; Vinding, T.; La Cour, M.; Appleyard, M.; Jensen, G.B.; Nielsen, N.V. Prevalence and causes of visual impairment and blindness among 9980 Scandinavian adults: The Copenhagen City Eye Study. Ophthalmology 2004, 111, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Farrar, G.J.; Kenna, P.F.; Humphries, P. On the genetics of retinitis pigmentosa and on mutation-independent approaches to therapeutic intervention. EMBO J. 2002, 21, 857–864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartong, D.T.; Berson, E.L.; Dryja, T.P. Retinitis pigmentosa. Lancet 2006, 368, 1795–1809. [Google Scholar] [CrossRef]
- Ruzickova, S.; Stanek, D. Mutations in spliceosomal proteins and retina degeneration. RNA Biol. 2017, 14, 544–552. [Google Scholar] [CrossRef] [Green Version]
- de Jong, P.T. Age-related macular degeneration. N. Engl. J. Med. 2006, 355, 1474–1485. [Google Scholar] [CrossRef]
- Ambati, J.; Fowler, B.J. Mechanisms of age-related macular degeneration. Neuron 2012, 75, 26–39. [Google Scholar] [CrossRef] [Green Version]
- Antonetti, D.A.; Klein, R.; Gardner, T.W. Diabetic retinopathy. N. Engl. J. Med. 2012, 366, 1227–1239. [Google Scholar] [CrossRef] [Green Version]
- Kolb, H. Simple Anatomy of the Retina. In Webvision: The Organization of the Retina and Visual System; Kolb, H., Fernandez, E., Nelson, R., Eds.; University of Utah Health Sciences Center: Salt Lake City, UT, USA, 1995. [Google Scholar]
- Bielmeier, C.B.; Roth, S.; Schmitt, S.I.; Boneva, S.K.; Schlecht, A.; Vallon, M.; Tamm, E.R.; Ergun, S.; Neueder, A.; Braunger, B.M. Transcriptional Profiling Identifies Upregulation of Neuroprotective Pathways in Retinitis Pigmentosa. Int. J. Mol. Sci. 2021, 22, 6307. [Google Scholar] [CrossRef]
- Naash, M.I.; Hollyfield, J.G.; al-Ubaidi, M.R.; Baehr, W. Simulation of human autosomal dominant retinitis pigmentosa in transgenic mice expressing a mutated murine opsin gene. Proc. Natl. Acad. Sci. USA 1993, 90, 5499–5503. [Google Scholar] [CrossRef] [Green Version]
- Massague, J. TGFbeta signalling in context. Nat. Rev. Mol. Cell Biol. 2012, 13, 616–630. [Google Scholar] [CrossRef]
- Braunger, B.M.; Pielmeier, S.; Demmer, C.; Landstorfer, V.; Kawall, D.; Abramov, N.; Leibinger, M.; Kleiter, I.; Fischer, D.; Jägle, H.; et al. TGF-β Signaling Protects Retinal Neurons from Programmed Cell Death during the Development of the Mammalian Eye. J. Neurosci. 2013, 33, 14246–14258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walshe, T.E.; Leach, L.L.; D’Amore, P.A. TGF-beta signaling is required for maintenance of retinal ganglion cell differentiation and survival. Neuroscience 2011, 189, 123–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walshe, T.E.; Saint-Geniez, M.; Maharaj, A.S.R.; Sekiyama, E.; Maldonado, A.E.; D’Amore, P.A. TGF-beta is required for vascular barrier function, endothelial survival and homeostasis of the adult microvasculature. PLoS ONE 2009, 4, e5149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, M.; Ma, Y.; Yi, X.; Guo, R.; Zhu, W.; Fan, X.; Xu, G.; Frey, W.H., 2nd; Liu, X. Intranasal delivery of transforming growth factor-beta1 in mice after stroke reduces infarct volume and increases neurogenesis in the subventricular zone. BMC Neurosci. 2008, 9, 117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.M.; Jung, J.S.; Jang, M.S.; Kang, K.S.; Kang, S.K. Transforming growth factor-beta1 regulates the fate of cultured spinal cord-derived neural progenitor cells. Cell Prolif. 2008, 41, 248–264. [Google Scholar] [CrossRef]
- Gabriel, C.; Ali, C.; Lesne, S.; Fernandez-Monreal, M.; Docagne, F.; Plawinski, L.; MacKenzie, E.T.; Buisson, A.; Vivien, D. Transforming growth factor alpha-induced expression of type 1 plasminogen activator inhibitor in astrocytes rescues neurons from excitotoxicity. FASEB J. 2003, 17, 277–279. [Google Scholar] [CrossRef]
- Shi, Y.; Massague, J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell 2003, 113, 685–700. [Google Scholar] [CrossRef] [Green Version]
- Massague, J. How cells read TGF-beta signals. Nat. Rev. Mol. Cell Biol. 2000, 1, 169–178. [Google Scholar] [CrossRef]
- Clayton, S.W.; Ban, G.I.; Liu, C.; Serra, R. Canonical and noncanonical TGF-beta signaling regulate fibrous tissue differentiation in the axial skeleton. Sci. Rep. 2020, 10, 21364. [Google Scholar] [CrossRef]
- Zhang, Y.E. Non-Smad pathways in TGF-beta signaling. Cell Res. 2009, 19, 128–139. [Google Scholar] [CrossRef] [Green Version]
- Levy, G.; Levi-Acobas, F.; Blanchard, S.; Gerber, S.; Larget-Piet, D.; Chenal, V.; Liu, X.Z.; Newton, V.; Steel, K.P.; Brown, S.D.; et al. Myosin VIIA gene: Heterogeneity of the mutations responsible for Usher syndrome type IB. Hum. Mol. Genet. 1997, 6, 111–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, D.; Lee, G.; Lee, J.H.; Kim, H.Y.; Rhee, H.W.; Park, S.Y.; Kim, K.J.; Kim, Y.; Kim, B.Y.; Hong, J.I.; et al. A metazoan ortholog of SpoT hydrolyzes ppGpp and functions in starvation responses. Nat. Struct. Mol. Biol. 2010, 17, 1188–1194. [Google Scholar] [CrossRef] [PubMed]
- Ding, C.C.; Rose, J.; Sun, T.; Wu, J.; Chen, P.H.; Lin, C.C.; Yang, W.H.; Chen, K.Y.; Lee, H.; Xu, E.; et al. MESH1 is a cytosolic NADPH phosphatase that regulates ferroptosis. Nat. Metab. 2020, 2, 270–277. [Google Scholar] [CrossRef] [Green Version]
- Kleinberger, G.; Yamanishi, Y.; Suarez-Calvet, M.; Czirr, E.; Lohmann, E.; Cuyvers, E.; Struyfs, H.; Pettkus, N.; Wenninger-Weinzierl, A.; Mazaheri, F.; et al. TREM2 mutations implicated in neurodegeneration impair cell surface transport and phagocytosis. Sci. Transl. Med. 2014, 6, 243ra286. [Google Scholar] [CrossRef] [PubMed]
- Erta, M.; Quintana, A.; Hidalgo, J. Interleukin-6, a major cytokine in the central nervous system. Int. J. Biol. Sci. 2012, 8, 1254–1266. [Google Scholar] [CrossRef]
- Cuadrado, A.; Nebreda, A.R. Mechanisms and functions of p38 MAPK signalling. Biochem. J. 2010, 429, 403–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drew, B.A.; Burow, M.E.; Beckman, B.S. MEK5/ERK5 pathway: The first fifteen years. Biochim. Biophys. Acta 2012, 1825, 37–48. [Google Scholar] [CrossRef] [Green Version]
- Gibbs, D.; Azarian, S.M.; Lillo, C.; Kitamoto, J.; Klomp, A.E.; Steel, K.P.; Libby, R.T.; Williams, D.S. Role of myosin VIIa and Rab27a in the motility and localization of RPE melanosomes. J. Cell Sci. 2004, 117, 6473–6483. [Google Scholar] [CrossRef] [Green Version]
- Kremer, H.; van Wijk, E.; Marker, T.; Wolfrum, U.; Roepman, R. Usher syndrome: Molecular links of pathogenesis, proteins and pathways. Hum. Mol. Genet. 2006, 15 (Suppl. S2), R262–R270. [Google Scholar] [CrossRef] [Green Version]
- Goumans, M.J.; Liu, Z.; ten Dijke, P. TGF-beta signaling in vascular biology and dysfunction. Cell Res. 2009, 19, 116–127. [Google Scholar] [CrossRef] [Green Version]
- Goumans, M.J.; Mummery, C. Functional analysis of the TGFbeta receptor/Smad pathway through gene ablation in mice. Int. J. Dev. Biol. 2000, 44, 253–265. [Google Scholar]
- Massague, J. G1 cell-cycle control and cancer. Nature 2004, 432, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Schlecht, A.; Vallon, M.; Wagner, N.; Ergun, S.; Braunger, B.M. TGFbeta-Neurotrophin Interactions in Heart, Retina, and Brain. Biomolecules 2021, 11, 1360. [Google Scholar] [CrossRef]
- Marquardt, T.; Ashery-Padan, R.; Andrejewski, N.; Scardigli, R.; Guillemot, F.; Gruss, P. Pax6 is required for the multipotent state of retinal progenitor cells. Cell 2001, 105, 43–55. [Google Scholar] [CrossRef] [Green Version]
- Cepko, C.L.; Austin, C.P.; Yang, X.; Alexiades, M.; Ezzeddine, D. Cell fate determination in the vertebrate retina. Proc. Natl. Acad. Sci. USA 1996, 93, 589–595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ameyar, M.; Wisniewska, M.; Weitzman, J.B. A role for AP-1 in apoptosis: The case for and against. Biochimie 2003, 85, 747–752. [Google Scholar] [CrossRef]
- Mahlknecht, U.; Will, J.; Varin, A.; Hoelzer, D.; Herbein, G. Histone deacetylase 3, a class I histone deacetylase, suppresses MAPK11-mediated activating transcription factor-2 activation and represses TNF gene expression. J. Immunol. 2004, 173, 3979–3990. [Google Scholar] [CrossRef] [Green Version]
- Ying, J.; Li, H.; Cui, Y.; Wong, A.H.; Langford, C.; Tao, Q. Epigenetic disruption of two proapoptotic genes MAPK10/JNK3 and PTPN13/FAP-1 in multiple lymphomas and carcinomas through hypermethylation of a common bidirectional promoter. Leukemia 2006, 20, 1173–1175. [Google Scholar] [CrossRef] [Green Version]
- Curran, T.; Morgan, J.I. Fos: An immediate-early transcription factor in neurons. J. Neurobiol. 1995, 26, 403–412. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, D.; McQuade, J.S.; Behbehani, M.; Tsien, J.Z.; Xu, M. c-fos regulates neuronal excitability and survival. Nat. Genet. 2002, 30, 416–420. [Google Scholar] [CrossRef]
- Garcia, T.B.; Hollborn, M.; Bringmann, A. Expression and signaling of NGF in the healthy and injured retina. Cytokine Growth Factor Rev. 2017, 34, 43–57. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Silverman, S.M.; Zhao, L.; Villasmil, R.; Campos, M.M.; Amaral, J.; Wong, W.T. Absence of TGFbeta signaling in retinal microglia induces retinal degeneration and exacerbates choroidal neovascularization. eLife 2019, 8, e42049. [Google Scholar] [CrossRef] [PubMed]
- Sometani, A.; Kataoka, H.; Nitta, A.; Fukumitsu, H.; Nomoto, H.; Furukawa, S. Transforming growth factor-beta1 enhances expression of brain-derived neurotrophic factor and its receptor, TrkB, in neurons cultured from rat cerebral cortex. J. Neurosci. Res. 2001, 66, 369–376. [Google Scholar] [CrossRef] [PubMed]
- Krieglstein, K.; Unsicker, K. Distinct modulatory actions of TGF-beta and LIF on neurotrophin-mediated survival of developing sensory neurons. Neurochem. Res. 1996, 21, 843–850. [Google Scholar] [CrossRef]
- Qin, D.; Wang, J.; Le, A.; Wang, T.J.; Chen, X.; Wang, J. Traumatic Brain Injury: Ultrastructural Features in Neuronal Ferroptosis, Glial Cell Activation and Polarization, and Blood-Brain Barrier Breakdown. Cells 2021, 10, 1009. [Google Scholar] [CrossRef]
- Wang, S.K.; Xue, Y.; Cepko, C.L. Microglia modulation by TGF-beta1 protects cones in mouse models of retinal degeneration. J. Clin. Investig. 2020, 130, 4360–4369. [Google Scholar] [CrossRef]
- Chytil, A.; Magnuson, M.A.; Wright, C.V.; Moses, H.L. Conditional inactivation of the TGF-beta type II receptor using Cre:Lox. Genesis 2002, 32, 73–75. [Google Scholar] [CrossRef]
- Kugler, M.; Schlecht, A.; Fuchshofer, R.; Kleiter, I.; Aigner, L.; Tamm, E.R.; Braunger, B.M. Heterozygous modulation of TGF-beta signaling does not influence Muller glia cell reactivity or proliferation following NMDA-induced damage. Histochem. Cell Biol. 2015, 144, 443–455. [Google Scholar] [CrossRef]
- Kugler, M.; Schlecht, A.; Fuchshofer, R.; Schmitt, S.I.; Kleiter, I.; Aigner, L.; Tamm, E.R.; Braunger, B.M. SMAD7 deficiency stimulates Muller progenitor cell proliferation during the development of the mammalian retina. Histochem. Cell Biol. 2017, 148, 21–32. [Google Scholar] [CrossRef]
- Braunger, B.M.; Ohlmann, A.; Koch, M.; Tanimoto, N.; Volz, C.; Yang, Y.; Bösl, M.R.; Cvekl, A.; Jägle, H.; Seeliger, M.W.; et al. Constitutive overexpression of Norrin activates Wnt/β-catenin and endothelin-2 signaling to protect photoreceptors from light damage. Neurobiol. Dis. 2013, 50, 1–12. [Google Scholar] [CrossRef]
- Braunger, B.M.; Leimbeck, S.V.; Schlecht, A.; Volz, C.; Jägle, H.; Tamm, E.R. Deletion of ocular transforming growth factor β signaling mimics essential characteristics of diabetic retinopathy. Am. J. Pathol. 2015, 185, 1749–1768. [Google Scholar] [CrossRef] [PubMed]
- Karnovsky, M.J. A formaldehyde-glutaraldehyde fixative of high osmolarity for use in electron microscopy. J. Cell Biol. 1965, 27, 137–138. [Google Scholar]
- Richardson, K.C.; Jarett, L.; Finke, E.H. Embedding in epoxy resins for ultrathin sectioning in electron microscopy. Stain Technol. 1960, 35, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Boneva, S.K.; Gross, T.R.; Schlecht, A.; Schmitt, S.I.; Sippl, C.; Jagle, H.; Volz, C.; Neueder, A.; Tamm, E.R.; Braunger, B.M. Cre recombinase expression or topical tamoxifen treatment do not affect retinal structure and function, neuronal vulnerability or glial reactivity in the mouse eye. Neuroscience 2016, 325, 188–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dysregulation Analysis | Enriched Pathways 1: WikiPathways 2019 Mouse; 2: KEGG 2019 Mouse; 3: BioPlanet 2019 | Gene Ontology Enrichment (Biological Process 2021) | Potential Regulators 1: ChEA; 2: Encode TF ChIP-Seq 2015 |
|---|---|---|---|
| 310 genes significantly downregulated in double mutant, not regulated in VPP | 1: Electron Transport Chain 20.83, Oxidative phosphorylation 18.40, Translation Factors 12.91, Proteasome Degradation 12.00, Cytoplasmic Ribosomal Proteins 8.45 2: Oxidative phosphorylation 18.90, Ribosome 16.84, RNA polymerase 13.09, Ubiquitin mediated proteolysis 11.34, Thermogenesis 8.26 3: Chemiosmotic coupling formation of ATP 134.04, Valine, leucine and isoleucine biosynthesis 62.79, Cytoplasmic ribosomal proteins 29.21, Cap-dependent translation initiation 24.60, Activation of mRNA upon binding of the cap-binding complex and eIFs, and subsequent binding to 43S 21.53 | tRNA pseudouridine synthesis 181.21, regulation of stem cell division 107.93, sarcomere organization 75.56, phosphorylated carbohydrate dephosphorylation 62.79, inositol phosphate dephosphorylation 62.79 | 1: HCFC1 19.91, JARID1A 18.65, YY1 10.50, BCL3 7.26 2: KAT2A 43.14, EP300 19.63, SIN3A 19.63, POLR2AphosphoS5 17.07, MYC 16.31 |
| 337 genes significantly upregulated in double mutant, not regulated in VPP | 1: Serotonin and anxiety 24.32, Dysregulated miRNA Targeting in Insulin/PI3K-AKT Signaling 12.95, IL-6 signaling Pathway 11.43, Oxidative Stress 11.37, Matrix Metalloproteinases 10.69, 2: Dopaminergic synapse 32.52, cAMP signaling pathway 17.02, IL-17 signaling pathway 13.61, Circadian entrainment 11.43, Aldosterone synthesis and secretion 10.74, 3: Activation of the AP-1 family of transcription factors 65.31, Erythropoietin-mediated neuroprotection through NF-kB 55.58, Signaling by Robo receptor 52.51, Inactivation of APC/C via direct inhibition of the APC/C complex 51.87, Kinesins 33.65 | sinoatrial node cell differentiation 230.87, microtubule nucleation by microtubule organizing center 230.87, negative regulation of synapse organization 230.87, aromatic amino acid transport 161.57, snoRNA localization 161.57 | 1: SUZ12 30.34, JARID2 20.32, MTF2 17.81, EZH2 15.66, RING1B 15.25, 2: POLR2A 9.20 |
| Dysregulation Analysis | Enriched Pathways 1: WikiPathways 2019 Mouse; 2: KEGG 2019 Mouse; 3: BioPlanet 2019 | Gene Ontology Enrichment (Biological Process 2021) | Potential Regulators 1: ChEA; 2: Encode TF ChIP-Seq 2015 |
|---|---|---|---|
| 1127 genes significantly downregulated in VPP, not regulated in double mutant | 1: mRNA processing 15.87, Mismatch repair 11.66, Fatty Acid Biosynthesis 5.53, Eukaryotic Transcription Initiation 5.25 2: Basal transcription factors 23.11, RNA transport 18.65, Nucleotide excision repair 15.19, Mismatch repair 12.78, Lysine degradation 10.73 3: Small interfering RNA (siRNA) biogenesis 96.77, Cytoskeletal remodeling regulation and cell spreading by IPP complex components 48.54, RNA polymerase II C-terminal domain phosphorylation and interaction with capping enzyme 48.54, ATM-mediated phosphorylation of repair proteins 39.86, NOSTRIN-mediated endothelial NOS trafficking 39.86 | mRNA cleavage involved in gene silencing by miRNA 161.54, cellular lipid biosynthetic process 96.77, snRNA modification 96.77, transcription-dependent tethering of RNA polymerase II gene DNA at nuclear periphery 96.77, mRNA splice site selection 76.01 | 1: KDM5B 36.25, CREM 24.91, FOXO3 20.56, BCL3 18.68 ERG 17.39 2: GABPA 59.59, KAT2A 49.90, MAX 44.05, FLI1 32.08, HCFC1 27.13 |
| 979 genes significantly upregulated in VPP, not regulated in double mutants | 1: Glutathione metabolism 20.55, Fatty Acid Biosynthesis 16.81, Prostaglandin Synthesis and Regulation 15.42, ACE Inhibitor Pathway 14.90, Heme Biosynthesis 14.90 2: Folate biosynthesis 38.75, Propanoate metabolism 15.42, Oxidative phosphorylation 14.95, beta-Alanine metabolism 14.38, Nitrogen metabolism 12.71 3: Bile salt and organic anion SLC transporters 60.31, Catalytic cycle of mammalian FMOs 49.72, Kit receptor transcriptional targets 49.72, Second messenger role in netrin-1 signaling 37.78, Tetrahydrobiopterin (BH4) biosynthesis, recycling, salvage and regulation 37.78, Cell cycle negative regulation by p75 neurotrophin receptor 33.66 | negative regulation of T cell migration 199.52, blood vessel endothelial cell proliferation involved in sprouting angiogenesis 120.22, basement membrane assembly 116.02, dolichyl diphosphate biosynthetic process 82.52, tetrahydrobiopterin metabolic process 73.01 | 1: SUZ12 12.02, THRA 10.23, SOX9 8.81, SRY 6.88, MTF2 5.96 2: n.s. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bielmeier, C.B.; Schmitt, S.I.; Kleefeldt, N.; Boneva, S.K.; Schlecht, A.; Vallon, M.; Tamm, E.R.; Hillenkamp, J.; Ergün, S.; Neueder, A.; et al. Deficiency in Retinal TGFβ Signaling Aggravates Neurodegeneration by Modulating Pro-Apoptotic and MAP Kinase Pathways. Int. J. Mol. Sci. 2022, 23, 2626. https://doi.org/10.3390/ijms23052626
Bielmeier CB, Schmitt SI, Kleefeldt N, Boneva SK, Schlecht A, Vallon M, Tamm ER, Hillenkamp J, Ergün S, Neueder A, et al. Deficiency in Retinal TGFβ Signaling Aggravates Neurodegeneration by Modulating Pro-Apoptotic and MAP Kinase Pathways. International Journal of Molecular Sciences. 2022; 23(5):2626. https://doi.org/10.3390/ijms23052626
Chicago/Turabian StyleBielmeier, Christina B., Sabrina I. Schmitt, Nikolai Kleefeldt, Stefaniya K. Boneva, Anja Schlecht, Mario Vallon, Ernst R. Tamm, Jost Hillenkamp, Süleyman Ergün, Andreas Neueder, and et al. 2022. "Deficiency in Retinal TGFβ Signaling Aggravates Neurodegeneration by Modulating Pro-Apoptotic and MAP Kinase Pathways" International Journal of Molecular Sciences 23, no. 5: 2626. https://doi.org/10.3390/ijms23052626
APA StyleBielmeier, C. B., Schmitt, S. I., Kleefeldt, N., Boneva, S. K., Schlecht, A., Vallon, M., Tamm, E. R., Hillenkamp, J., Ergün, S., Neueder, A., & Braunger, B. M. (2022). Deficiency in Retinal TGFβ Signaling Aggravates Neurodegeneration by Modulating Pro-Apoptotic and MAP Kinase Pathways. International Journal of Molecular Sciences, 23(5), 2626. https://doi.org/10.3390/ijms23052626