
The Role of Endothelial Progenitor Cells in Atherosclerosis and Impact of Anti-Lipemic Treatments on Endothelial Repair


Abstract
:1. Introduction
2. Endothelium Biology and Function
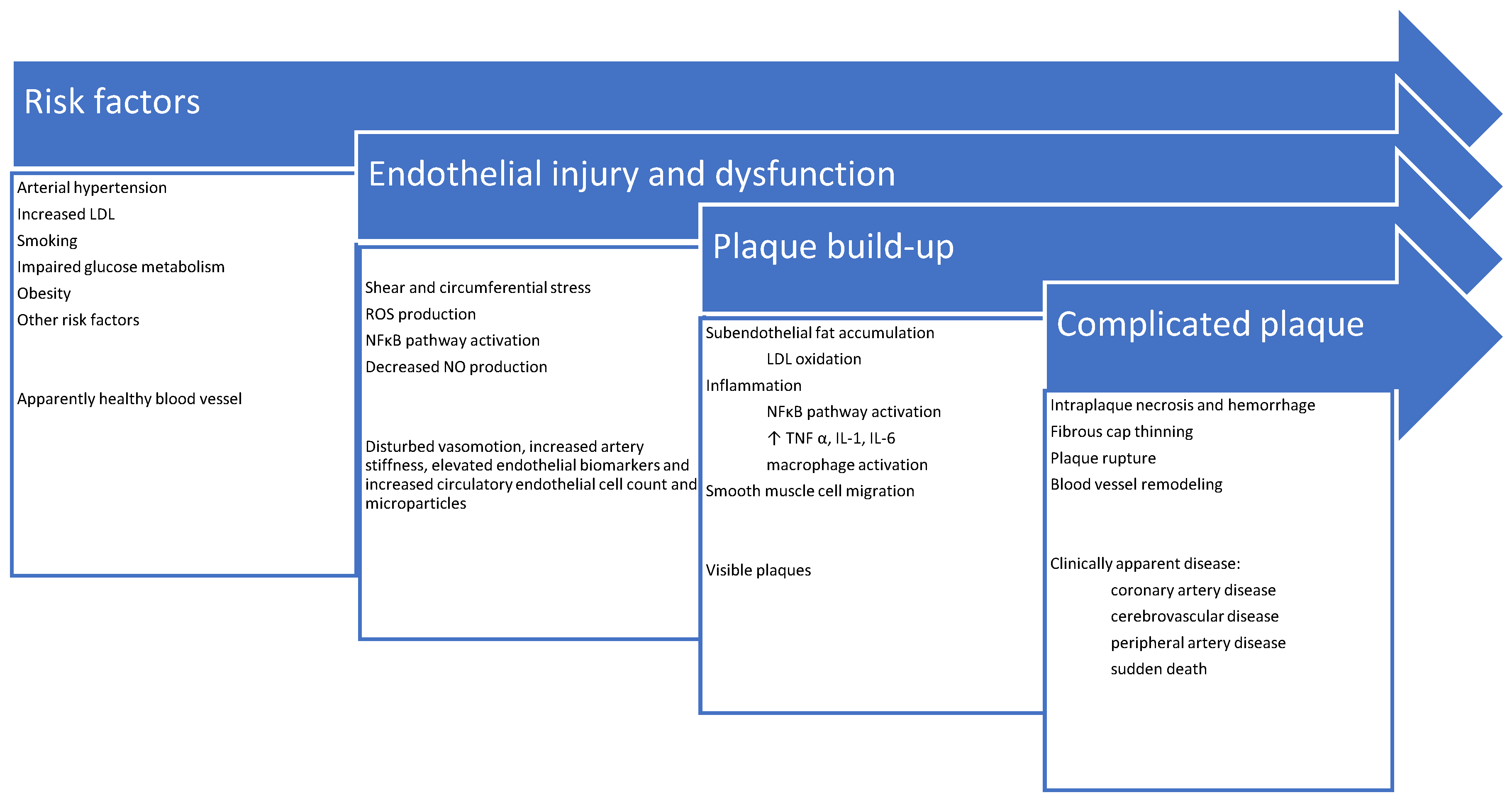
3. Pathophysiology of Atherosclerosis
4. Endothelial Repair
5. Endothelial Repair in Patients with Lipid Disorders
6. Standard Treatment for Lipid Disorders: Statins, Ezetimibe and Fibrates
7. Novel Treatments for Lipid Disorders: Proprotein Convertase Subtilisin/Kexin Type 9 (PCSK9) Modulating Agents and Angiopoietin-like Proteins (Angtpl3) Inhibitors
8. Plasma Apheresis
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Milutinović, A.; Šuput, D.; Zorc-Pleskovič, R. Pathogenesis of Atherosclerosis in the Tunica Intima, Media, and Adventitia of Coronary Arteries: An Updated Review. Bosn. J. Basic Med. Sci. 2020, 20, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Rosenblit, P.D. Extreme Atherosclerotic Cardiovascular Disease (ASCVD) Risk Recognition. Curr. Diab. Rep. 2019, 19, 61. [Google Scholar] [CrossRef] [PubMed]
- Silveira Rossi, J.L.; Barbalho, S.M.; Reverete de Araujo, R.; Bechara, M.D.; Sloan, K.P.; Sloan, L.A. Metabolic Syndrome and Cardiovascular Diseases: Going beyond Traditional Risk Factors. Diabetes Metab. Res. Rev. 2021, e3502. [Google Scholar] [CrossRef] [PubMed]
- Polovina, M.M.; Potpara, T.S. Endothelial Dysfunction in Metabolic and Vascular Disorders. Postgrad. Med. 2014, 126, 38–53. [Google Scholar] [CrossRef] [PubMed]
- Golledge, J. Update on the Pathophysiology and Medical Treatment of Peripheral Artery Disease. Nat. Rev. Cardiol. 2022, 19, 663–669. [Google Scholar] [CrossRef]
- Fonarow, G.C.; Kosiborod, M.N.; Rane, P.B.; Nunna, S.; Villa, G.; Habib, M.; Arellano, J.; Mues, K.E.; Sun, K.; Wade, R.L. Patient Characteristics and Acute Cardiovascular Event Rates among Patients with Very High-Risk and Non-Very High-Risk Atherosclerotic Cardiovascular Disease. Clin. Cardiol. 2021, 44, 1457–1466. [Google Scholar] [CrossRef]
- Aplin, M.; Andersen, A.; Brandes, A.; Dominguez, H.; Dahl, J.S.; Damgaard, D.; Iversen, H.K.; Iversen, K.K.; Nielsen, E.; Risum, N.; et al. Assessment of Patients with a Suspected Cardioembolic Is-chemic Stroke. A National Consensus Statement. Scand. Cardiovasc. J. 2021, 55, 315–325. [Google Scholar] [CrossRef]
- Feig, J.E. Regression of Atherosclerosis: Insights from Animal and Clinical Studies. Ann. Glob. Health 2014, 80, 13–23. [Google Scholar] [CrossRef]
- Chopra, H.; Hung, M.K.; Kwong, D.L.; Zhang, C.F.; Pow, E.H.N. Insights into Endothelial Progenitor Cells: Origin, Classification, Potentials, and Prospects. Stem Cells Int. 2018, 2018, 9847015. [Google Scholar] [CrossRef]
- Yang, J.-X.; Pan, Y.-Y.; Wang, X.-X.; Qiu, Y.-G.; Mao, W. Endothelial Progenitor Cells in Age-Related Vascular Remodeling. Cell Transpl. 2018, 27, 786–795. [Google Scholar] [CrossRef]
- Rajendran, P.; Rengarajan, T.; Thangavel, J.; Nishigaki, Y.; Sakthisekaran, D.; Sethi, G.; Nishigaki, I. The Vascular Endothelium and Human Diseases. Int. J. Biol. Sci. 2013, 9, 1057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fortini, F.; Vieceli Dalla Sega, F.; Marracino, L.; Severi, P.; Rapezzi, C.; Rizzo, P.; Ferrari, R. Well-Known and Novel Players in Endothelial Dysfunction: Updates on a Notch(Ed) Landscape. Biomedicines 2021, 9, 997. [Google Scholar] [CrossRef] [PubMed]
- Sancheti, S.; Shah, P.; Phalgune, D.S. Correlation of Endothelial Dysfunction Measured by Flow-Mediated Vasodilatation to Severity of Coronary Artery Disease. Indian Heart J. 2018, 70, 622–626. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.F. An Investigation of the Underlying Mechanisms of Arterial Vasomotion. Ph.D. Thesis, University of Oxford, Oxford, UK, 2020. [Google Scholar]
- Cyr, A.R.; Huckaby, L.V.; Shiva, S.S.; Zuckerbraun, B.S. Nitric Oxide and Endothelial Dysfunction. Crit. Care Clin. 2020, 36, 307–321. [Google Scholar] [CrossRef]
- Kwaifa, I.K.; Bahari, H.; Yong, Y.K.; Noor, S.M. Endothelial Dysfunction in Obesity-Induced Inflammation: Molecular Mechanisms and Clinical Implications. Biomolecules 2020, 10, 291. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, K.; De Wit, C. Endothelium-Derived Hyperpolarizing Factor and Myoendothelial Coupling: The In Vivo Perspective. Front. Physiol. 2020, 11, 602930. [Google Scholar] [CrossRef]
- Altabas, V.; Altabas, K.; Kirigin, L. Endothelial Progenitor Cells (EPCs) in Ageing and Age-Related Diseases: How Currently Available Treatment Modalities Affect EPC Biology, Atherosclerosis, and Cardiovascular Outcomes. Mech. Ageing Dev. 2016, 159, 49–62. [Google Scholar] [CrossRef]
- Grover, S.P.; Mackman, N. Tissue Factor in Atherosclerosis and Atherothrombosis. Atherosclerosis 2020, 307, 80–86. [Google Scholar] [CrossRef]
- Poston, R.N. Atherosclerosis: Integration of Its Pathogenesis as a Self-Perpetuating Propagating Inflammation: A Review. Cardiovasc. Endocrinol. Metab. 2019, 8, 51–61. [Google Scholar] [CrossRef]
- Haverich, A. A Surgeon’s View on the Pathogenesis of Atherosclerosis. Circulation 2017, 135, 205–207. [Google Scholar] [CrossRef]
- Subbotin, V.M. Excessive Intimal Hyperplasia in Human Coronary Arteries before Intimal Lipid Depositions Is the Initiation of Coronary Atherosclerosis and Constitutes a Therapeutic Target. Drug Discov. Today 2016, 21, 1578–1595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otsuka, F.; Kramer, M.C.A.; Woudstra, P.; Yahagi, K.; Ladich, E.; Finn, A.V.; De Winter, R.J.; Kolodgie, F.D.; Wight, T.N.; Davis, H.R.; et al. Natural Progression of Atherosclerosis from Pathologic Intimal Thickening to Late Fibroatheroma in Human Coronary Arteries: A Pathology Study. Atherosclerosis 2015, 241, 772–782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chistiakov, D.A.; Orekhov, A.N.; Bobryshev, Y.V. Effects of Shear Stress on Endothelial Cells: Go with the Flow. Acta Physiol. 2017, 219, 382–408. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.-Z.; Jiménez, J.M.; Ou, K.; McCormick, M.E.; Zhang, L.-D.; Davies, P.F. Hemodynamic Disturbed Flow Induces Differential DNA Methylation of Endothelial Kruppel-Like Factor 4 Promoter In Vitro and In Vivo. Circ. Res. 2014, 115, 32–43. [Google Scholar] [CrossRef] [Green Version]
- Staarmann, B.; Smith, M.; Prestigiacomo, C.J. Shear Stress and Aneurysms: A Review. Neurosurg. Focus 2019, 47, E2. [Google Scholar] [CrossRef] [Green Version]
- Zhong, S.; Li, L.; Shen, X.; Li, Q.; Xu, W.; Wang, X.; Tao, Y.; Yin, H. An Update on Lipid Oxidation and Inflammation in Cardiovascular Diseases. Free Radic. Biol. Med. 2019, 144, 266–278. [Google Scholar] [CrossRef] [PubMed]
- Libby, P. Inflammation during the Life Cycle of the Atherosclerotic Plaque. Cardiovasc. Res. 2021, 117, 2525–2536. [Google Scholar] [CrossRef]
- Basu, D.; Hu, Y.; Huggins, L.-A.; Mullick, A.E.; Graham, M.J.; Wietecha, T.; Barnhart, S.; Mogul, A.; Pfeiffer, K.; Zirlik, A.; et al. Novel Reversible Model of Atherosclerosis and Regression Using Oligonucleotide Regulation of the LDL Receptor. Circ. Res. 2018, 122, 560–567. [Google Scholar] [CrossRef]
- Sorokin, V.; Vickneson, K.; Kofidis, T.; Woo, C.C.; Lin, X.Y.; Foo, R.; Shanahan, C.M. Role of Vascular Smooth Muscle Cell Plasticity and Interactions in Vessel Wall Inflammation. Front. Immunol. 2020, 11, 599415. [Google Scholar] [CrossRef]
- Katsuumi, G.; Shimizu, I.; Yoshida, Y.; Minamino, T. Vascular Senescence in Cardiovascular and Metabolic Diseases. Front. Cardiovasc. Med. 2018, 5, 18. [Google Scholar] [CrossRef] [Green Version]
- Erbel, C.; Wolf, A.; Lasitschka, F.; Linden, F.; Domschke, G.; Akhavanpoor, M.; Doesch, A.O.; Katus, H.A.; Gleissner, C.A. Prevalence of M4 Macrophages within Human Coronary Atherosclerotic Plaques Is Associated with Features of Plaque Instability. Int. J. Cardiol. 2015, 186, 219–225. [Google Scholar] [CrossRef] [PubMed]
- Shioi, A.; Ikari, Y. Plaque Calcification During Atherosclerosis Progression and Regression. J. Atheroscler. Thromb. 2018, 25, 294–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canet-Soulas, E.; Bessueille, L.; Mechtouff, L.; Magne, D. The Elusive Origin of Atherosclerotic Plaque Calcification. Front. Cell Dev. Biol. 2021, 9, 390. [Google Scholar] [CrossRef]
- Wang, D.; Wang, Z.; Zhang, L.; Wang, Y. Roles of Cells from the Arterial Vessel Wall in Atherosclerosis. Mediat. Inflamm. 2017, 2017, 8135934. [Google Scholar] [CrossRef] [PubMed]
- Yousufuddin, M.; Young, N. Aging and Ischemic Stroke. Aging 2019, 11, 2542–2544. [Google Scholar] [CrossRef] [PubMed]
- White, S.J.; Newby, A.C.; Johnson, T.W. Endothelial Erosion of Plaques as a Substrate for Coronary Thrombosis. Thromb. Haemost. 2016, 115, 509–519. [Google Scholar]
- Gunawardena, T.; Merinopoulos, I.; Wickramarachchi, U.; Vassiliou, V.; Eccleshall, S. Endothelial Dysfunction and Coronary Vasoreactivity—A Review of the History, Physiology, Diagnostic Techniques, and Clinical Relevance. Curr. Cardiol. Rev. 2021, 17, 85–100. [Google Scholar] [CrossRef]
- Rocha, H.N.; Garcia, V.P.; Batista, G.M.; Silva, G.M.; Mattos, J.D.; Campos, M.O.; Nóbrega, A.C.; Fernandes, I.A.; Rocha, N.G. Disturbed Blood Flow Induces Endothelial Apoptosis without Mobilizing Repair Mechanisms in Hypertension. Life Sci. 2018, 209, 103–110. [Google Scholar] [CrossRef]
- Altabas, V. Diabetes, Endothelial Dysfunction, and Vascular Repair: What Should a Diabetologist Keep His Eye On? Int. J. Endocrinol. 2015, 2015, 848272. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Rehman, J.; Malik, A.B. Endothelial Progenitor Cells and Vascular Repair. Curr. Opin. Hematol. 2014, 21, 224. [Google Scholar] [CrossRef] [Green Version]
- Basile, D.P.; Yoder, M.C. Circulating and Tissue Resident Endothelial Progenitor Cells. J. Cell. Physiol. 2014, 229, 10–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrone, D.; Picoi, M.E.L.; Felice, F.; De Martino, A.; Scatena, C.; Spontoni, P.; Naccarato, A.G.; Di Stefano, R.; Bortolotti, U.; Dal Monte, M. Endothelial Progenitor Cells: An Appraisal of Relevant Data from Bench to Bedside. Int. J. Mol. Sci. 2021, 22, 12874. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.; Kang, L.; Wang, Z.; Chen, A.; Zhao, Q.; Li, H. 17β-Estradiol Promotes Recovery after Myocardial Infarction by Enhancing Homing and Angiogenic Capacity of Bone Marrow-Derived Endothelial Progenitor Cells through ERα-SDF-1/CXCR4 Crosstalking. Acta Biochim. Biophys. Sin. 2018, 50, 1247–1256. [Google Scholar] [CrossRef]
- Naito, T.; Shun, M.; Nishimura, H.; Gibo, T.; Tosaka, M.; Kawashima, M.; Ando, A.; Ogawa, T.; Sanaka, T.; Nitta, K. Pleiotropic Effect of Erythropoiesis-Stimulating Agents on Circulating Endothelial Progenitor Cells in Dialysis Patients. Clin. Exp. Nephrol. 2021, 25, 1111–1120. [Google Scholar] [CrossRef] [PubMed]
- Mogharbel, B.F.; Abdelwahid, E.; Irioda, A.C.; Francisco, J.C.; Simeoni, R.B.; De Souza, D.; De Souza, C.M.C.O.; Beltrame, M.P.; Ferreira, R.J.; Guarita-Souza, L.C.; et al. Bone Marrow-Derived Stem Cell Populations Are Differentially Regulated by Thyroid or/and Ovarian Hormone Loss. Int. J. Mol. Sci. 2017, 18, 2139. [Google Scholar] [CrossRef] [Green Version]
- Balistreri, C.R.; Buffa, S.; Pisano, C.; Lio, D.; Ruvolo, G.; Mazzesi, G. Are Endothelial Progenitor Cells the Real Solution for Cardiovascular Diseases? Focus on Controversies and Perspectives. BioMed Res. Int. 2015, 2015, 835934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mobarrez, F.; Antoniewicz, L.; Bosson, J.A.; Kuhl, J.; Pisetsky, D.S.; Lundbäck, M. The Effects of Smoking on Levels of Endothelial Progenitor Cells and Microparticles in the Blood of Healthy Volunteers. PLoS ONE 2014, 9, e90314. [Google Scholar] [CrossRef] [Green Version]
- Berezin, A.E. Endothelial Progenitor Cells Dysfunction and Impaired Tissue Reparation: The Missed Link in Diabetes Mellitus Development. Diabetes Metab. Syndr. Clin. Res. Rev. 2017, 11, 215–220. [Google Scholar] [CrossRef]
- Seijkens, T.; Hoeksema, M.A.; Beckers, L.; Smeets, E.; Meiler, S.; Levels, J.; Tjwa, M.; De Winther, M.P.; Lutgens, E. Hypercholesterolemia-Induced Priming of Hematopoietic Stem and Progenitor Cells Aggravates Atherosclerosis. FASEB J. 2014, 28, 2202–2213. [Google Scholar] [CrossRef] [Green Version]
- Mandraffino, G.; Imbalzano, E.; Sardo, M.A.; D’ascola, A.; Mamone, F.; Gullo, A.L.; Alibrandi, A.; Loddo, S.; Mormina, E.; David, A. Circulating Progenitor Cells in Hypertensive Patients with Different Degrees of Cardiovascular Involvement. J. Hum. Hypertens. 2014, 28, 543–550. [Google Scholar] [CrossRef]
- Massot, A.; Navarro-Sobrino, M.; Penalba, A.; Arenillas, J.F.; Giralt, D.; Ribo, M.; Molina, C.A.; Alvarez-Sabín, J.; Montaner, J.; Rosell, A. Decreased Levels of Angiogenic Growth Factors in Intracranial Atherosclerotic Disease despite Severity-Related Increase in Endothelial Progenitor Cell Counts. Cerebrovasc. Dis. 2013, 35, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Ji, K.; Qian, L.; Nan, J.; Xue, Y.; Zhang, S.; Wang, G.; Yin, R.; Zhu, Y.; Wang, L.; Ma, J.; et al. Ox-LDL Induces Dysfunction of Endothelial Progenitor Cells via Activation of NF-B. BioMed Res. Int. 2015, 2015, 175291. [Google Scholar] [CrossRef] [PubMed]
- Hermsdorff, H.H.M.; Barbosa, K.B.; Volp, A.C.P.; Puchau, B.; Bressan, J.; Zulet, M.Á.; Martínez, J.A. Gender-Specific Relationships between Plasma Oxidized Low-Density Lipoprotein Cholesterol, Total Antioxidant Capacity, and Central Adiposity Indicators. Eur. J. Prev. Cardiol. 2014, 21, 884–891. [Google Scholar] [CrossRef] [PubMed]
- Zhuo, X.; Bu, H.; Hu, K.; Si, Z.; Chen, L.; Chen, Y.; Yang, L.; Jiang, Y.; Xu, Y.; Zhao, P.; et al. Differences in the Reaction of Hyperlipidemia on Different Endothelial Progenitor Cells Based on Sex. Biomed. Rep. 2021, 15, 64. [Google Scholar] [CrossRef] [PubMed]
- Schwertani, A.; Choi, H.Y.; Genest, J. HDLs and the Pathogenesis of Atherosclerosis. Curr. Opin. Cardiol. 2018, 33, 311–316. [Google Scholar] [CrossRef]
- Lucchesi, D.; Popa, S.G.; Sancho, V.; Giusti, L.; Garofolo, M.; Daniele, G.; Pucci, L.; Miccoli, R.; Penno, G.; Del Prato, S. Influence of High Density Lipoprotein Cholesterol Levels on Circulating Monocytic Angiogenic Cells Functions in Individuals with Type 2 Diabetes Mellitus. Cardiovasc. Diabetol. 2018, 17, 78. [Google Scholar] [CrossRef] [Green Version]
- Shih, C.-M.; Lin, F.-Y.; Yeh, J.-S.; Lin, Y.-W.; Loh, S.-H.; Tsao, N.-W.; Nakagami, H.; Morishita, R.; Sawamura, T.; Li, C.-Y.; et al. Dysfunctional High Density Lipoprotein Failed to Rescue the Function of Oxidized Low Density Lipoprotein-Treated Endothelial Progenitor Cells: A Novel Index for the Prediction of HDL Functionality. Transl. Res. 2019, 205, 17–32. [Google Scholar] [CrossRef]
- Peterson, S.J.; Shapiro, J.I.; Thompson, E.; Singh, S.; Liu, L.; Weingarten, J.A.; O’Hanlon, K.; Bialczak, A.; Bhesania, S.R.; Abraham, N.G. Oxidized HDL, Adipokines, and Endothelial Dysfunction: A Potential Biomarker Profile for Cardiovascular Risk in Women with Obesity. Obesity 2019, 27, 87–93. [Google Scholar] [CrossRef] [Green Version]
- Ren, Z.; Guo, J.; Xiao, X.; Huang, J.; Li, M.; Cai, R.; Zeng, H. The Effect of Sex Differences on Endothelial Function and Circulating Endothelial Progenitor Cells in Hypertriglyceridemia. Cardiol. Res. Pract. 2020, 2020, 2132918. [Google Scholar] [CrossRef]
- Wils, J.; Favre, J.; Bellien, J. Modulating Putative Endothelial Progenitor Cells for the Treatment of Endothelial Dysfunction and Cardiovascular Complications in Diabetes. Pharmacol. Ther. 2017, 170, 98–115. [Google Scholar] [CrossRef]
- Chia, P.Y.; Teo, A.; Yeo, T.W. Overview of the Assessment of Endothelial Function in Humans. Front. Med. 2020, 7, 542567. [Google Scholar] [CrossRef] [PubMed]
- Maruhashi, T.; Kihara, Y.; Higashi, Y. Assessment of Endothelium-Independent Vasodilation: From Methodology to Clinical Perspectives. J. Hypertens. 2018, 36, 1460–1467. [Google Scholar] [CrossRef] [PubMed]
- De Macedo, A.R.; Machado, J.C.; Luz, L.M.S.; Da Nobrega, A.C.L.; De Souza, M.N. A Novel Model to Simulate Venous Occlusion Plethysmography Data and to Estimate Arterial and Venous Parameters. Res. Biomed. Eng. 2020, 36, 463–473. [Google Scholar] [CrossRef]
- Masi, S.; Rizzoni, D.; Taddei, S.; Widmer, R.J.; Montezano, A.C.; Lüscher, T.F.; Schiffrin, E.L.; Touyz, R.M.; Paneni, F.; Lerman, A. Assessment and Pathophysiology of Microvascular Disease: Recent Progress and Clinical Implications. Eur. Heart J. 2021, 42, 2590–2604. [Google Scholar] [CrossRef] [PubMed]
- Williams, B.; Mancia, G.; Spiering, W.; Agabiti Rosei, E.; Azizi, M.; Burnier, M.; Clement, D.; Coca, A.; De Simone, G.; Dominiczak, A.; et al. 2018 Practice Guidelines for the Management of Arterial Hypertension of the European Society of Cardiology and the European Society of Hypertension. Blood Press. 2018, 27, 314–340. [Google Scholar] [CrossRef] [PubMed]
- Bochenek, M.L.; Schäfer, K. Role of Endothelial Cells in Acute and Chronic Thrombosis. Hämostaseologie 2019, 39, 128–139. [Google Scholar] [CrossRef] [Green Version]
- Gendron, N.; Smadja, D.M. Circulating Endothelial Cells: A New Biomarker of Endothelial Dysfunction in Hematological Diseases. Ann. Biol. Clin. 2016, 74, 395–404. [Google Scholar] [CrossRef]
- Chen, S.; Sun, Y.; Neoh, K.H.; Chen, A.; Li, W.; Yang, X.; Han, R.P. Microfluidic Assay of Circulating Endothelial Cells in Coronary Artery Disease Patients with Angina Pectoris. PLoS ONE 2017, 12, e0181249. [Google Scholar]
- Vítková, V.; Živný, J.; Janota, J. Endothelial Cell-Derived Microvesicles: Potential Mediators and Biomarkers of Pathologic Processes. Biomark. Med. 2018, 12, 161–175. [Google Scholar] [CrossRef]
- Byrne, P.; Cullinan, J.; Smith, A.; Smith, S.M. Statins for the Primary Prevention of Cardiovascular Disease: An Overview of Systematic Reviews. BMJ Open 2019, 9, e023085. [Google Scholar] [CrossRef] [Green Version]
- Virani, S.S.; Smith, S.C., Jr.; Stone, N.J.; Grundy, S.M. Secondary Prevention for Atherosclerotic Cardiovascular Disease: Comparing Recent US and European Guidelines on Dyslipidemia. Circulation 2020, 141, 1121–1123. [Google Scholar] [CrossRef] [PubMed]
- Mortensen, M.B.; Nordestgaard, B.G.; Afzal, S.; Falk, E. ACC/AHA Guidelines Superior to ESC/EAS Guidelines for Primary Prevention with Statins in Non-Diabetic Europeans: The Copenhagen General Population Study. Eur. Heart J. 2017, 38, 586–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorabi, A.M.; Kiaie, N.; Hajighasemi, S.; Banach, M.; Penson, P.E.; Jamialahmadi, T.; Sahebkar, A. Statin-Induced Nitric Oxide Signaling: Mechanisms and Therapeutic Implications. J. Clin. Med. 2019, 8, 2051. [Google Scholar] [CrossRef] [Green Version]
- Sandhu, K.; Mamas, M.; Butler, R. Endothelial Progenitor Cells: Exploring the Pleiotropic Effects of Statins. World J. Cardiol. 2017, 9, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Ricottini, E.; Madonna, R.; Grieco, D.; Zoccoli, A.; Stampachiacchiere, B.; Patti, G.; Tonini, G.; De Caterina, R.; Di Sciascio, G. Effect of High-Dose Atorvastatin Reload on the Release of Endothelial Progenitor Cells in Patients on Long-Term Statin Treatment Who Underwent Percutaneous Coronary Intervention (from the ARMYDA-EPC Study). Am. J. Cardiol. 2016, 117, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Eisen, A.; Leshem-Lev, D.; Yavin, H.; Orvin, K.; Mager, A.; Rechavia, E.; Bental, T.; Dadush, O.; Battler, A.; Kornowski, R. Effect of High Dose Statin Pretreatment on Endothelial Progenitor Cells after Percutaneous Coronary Intervention (HIPOCRATES Study). Cardiovasc. Drugs Ther. 2015, 29, 129–135. [Google Scholar] [CrossRef]
- Hibbert, B.; Ma, X.; Pourdjabbar, A.; Simard, T.; Rayner, K.; Sun, J.; Chen, Y.-X.; Filion, L.; O’Brien, E.R. Pre-Procedural Atorvastatin Mobilizes Endothelial Progenitor Cells: Clues to the Salutary Effects of Statins on Healing of Stented Human Arteries. PLoS ONE 2011, 6, e16413. [Google Scholar] [CrossRef]
- Oikonomou, E.; Siasos, G.; Zaromitidou, M.; Hatzis, G.; Mourouzis, K.; Chrysohoou, C.; Zisimos, K.; Mazaris, S.; Tourikis, P.; Athanasiou, D. Atorvastatin Treatment Improves Endothelial Function through Endothelial Progenitor Cells Mobilization in Ischemic Heart Failure Patients. Atherosclerosis 2015, 238, 159–164. [Google Scholar] [CrossRef]
- Ansheles, A.A.; Rvacheva, A.V.; Sergienko, I.V. Effect of Atorvastatin Therapy on the Level of CD34+ CD133+ CD309+ Endothelial Progenitor Cells in Patients with Coronary Heart Disease. Bull. Exp. Biol. Med. 2017, 163, 133–136. [Google Scholar] [CrossRef]
- Ye, H.; He, F.; Fei, X.; Lou, Y.; Wang, S.; Yang, R.; Hu, Y.; Chen, X. High-Dose Atorvastatin Reloading before Percutaneous Coronary Intervention Increased Circulating Endothelial Progenitor Cells and Reduced Inflammatory Cytokine Expression during the Perioperative Period. J. Cardiovasc. Pharmacol. Ther. 2014, 19, 290–295. [Google Scholar] [CrossRef]
- Huang, B.; Cheng, Y.; Xie, Q.; Lin, G.; Wu, Y.; Feng, Y.; Gao, J.; Xu, D. Effect of 40 Mg versus 10 Mg of Atorvastatin on Oxidized Low-Density Lipoprotein, High-Sensitivity C-Reactive Protein, Circulating Endothelial-Derived Microparticles, and Endothelial Progenitor Cells in Patients with Ischemic Cardiomyopathy. Clin. Cardiol. 2012, 35, 125–130. [Google Scholar] [CrossRef]
- Niu, H.; Wei, Z.; Zhang, Y.; He, J.; Jia, D. Atorvastatin Improves Coronary Flow and Endothelial Function in Patients with Coronary Slow Flow. Exp. Ther. Med. 2018, 15, 904–908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baran, Ç.; Durdu, S.; Dalva, K.; Zaim, Ç.; Dogan, A.; Ocakoglu, G.; Gürman, G.; Arslan, Ö.; Akar, A.R. Effects of Preoperative Short Term Use of Atorvastatin on Endothelial Progenitor Cells after Coronary Surgery: A Randomized, Controlled Trial. Stem Cell Rev. Rep. 2012, 8, 963–971. [Google Scholar] [CrossRef] [PubMed]
- Spadaccio, C.; Pollari, F.; Casacalenda, A.; Alfano, G.; Genovese, J.; Covino, E.; Chello, M. Atorvastatin Increases the Number of Endothelial Progenitor Cells after Cardiac Surgery: A Randomized Control Study. J. Cardiovasc. Pharmacol. 2010, 55, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Leone, A.M.; Rutella, S.; Giannico, M.B.; Perfetti, M.; Zaccone, V.; Brugaletta, S.; Garramone, B.; Niccoli, G.; Porto, I.; Liuzzo, G.; et al. Effect of Intensive vs. Standard Statin Therapy on Endothelial Progenitor Cells and Left Ventricular Function in Patients with Acute Myocardial Infarction: Statins for Regeneration after Acute Myocardial Infarction and PCI (STRAP) Trial. Int. J. Cardiol. 2008, 130, 457–462. [Google Scholar] [CrossRef]
- Liu, H.; Qi, X.; Long-le Ma, D.-K.Y.; Wang, L. Atorvastatin Improves Endothelial Progenitor Cell Function and Reduces Pulmonary Hypertension in Patients with Chronic Pulmonary Heart Disease. Exp. Clin. Cardiol. 2013, 18, e40. [Google Scholar]
- Sobrino, T.; Blanco, M.; Pérez-Mato, M.; Rodríguez-Yáñez, M.; Castillo, J. Increased Levels of Circulating Endothelial Progenitor Cells in Patients with Ischaemic Stroke Treated with Statins during Acute Phase. Eur. J. Neurol. 2012, 19, 1539–1546. [Google Scholar] [CrossRef]
- Chantzichristos, V.G.; Agouridis, A.P.; Moutzouri, E.; Stellos, K.; Elisaf, M.S.; Tselepis, A.D. Effect of Rosuvastatin or Its Combination with Omega-3 Fatty Acids on Circulating CD34+ Progenitor Cells and on Endothelial Colony Formation in Patients with Mixed Dyslipidaemia. Atherosclerosis 2016, 251, 240–247. [Google Scholar] [CrossRef]
- Tousoulis, D.; Andreou, I.; Tsiatas, M.; Miliou, A.; Tentolouris, C.; Siasos, G.; Papageorgiou, N.; Papadimitriou, C.A.; Dimopoulos, M.-A.; Stefanadis, C. Effects of Rosuvastatin and Allopurinol on Circulating Endothelial Progenitor Cells in Patients with Congestive Heart Failure: The Impact of Inflammatory Process and Oxidative Stress. Atherosclerosis 2011, 214, 151–157. [Google Scholar] [CrossRef]
- Erbs, S.; Beck, E.B.; Linke, A.; Adams, V. High-Dose Rosuvastatin in Chronic Heart Failure Promotes Vasculogenesis, Corrects Endothelial Function, and Improves Cardiac Remodeling—Results from a Randomized, Double-Blind, and Placebo-Controlled Study. Int. J. Cardiol. 2011, 146, 56–63. [Google Scholar] [CrossRef]
- Pirro, M.; Schillaci, G.; Romagno, P.F.; Mannarino, M.R.; Bagaglia, F.; Razzi, R.; Pasqualini, L.; Vaudo, G.; Mannarino, E. Influence of Short-Term Rosuvastatin Therapy on Endothelial Progenitor Cells and Endothelial Function. J. Cardiovasc. Pharmacol. Ther. 2009, 14, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Pesaro, A.E.P.; Serrano, C.V., Jr.; Katz, M.; Marti, L.; Fernandes, J.L.; Parra, P.R.; Campos, A.H. Increasing Doses of Simvastatin versus Combined Ezetimibe/Simvastatin: Effect on Circulating Endothelial Progenitor Cells. J. Cardiovasc. Pharmacol. Ther. 2013, 18, 447–452. [Google Scholar] [CrossRef] [PubMed]
- Westerweel, P.E.; Visseren, F.L.; Hajer, G.R.; Olijhoek, J.K.; Hoefer, I.E.; De Bree, P.; Rafii, S.; Doevendans, P.A.; Verhaar, M.C. Endothelial Progenitor Cell Levels in Obese Men with the Metabolic Syndrome and the Effect of Simvastatin Monotherapy vs. Simvastatin/Ezetimibe Combination Therapy. Eur. Heart J. 2008, 29, 2808–2817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paradisi, G.; Bracaglia, M.; Basile, F.; Di’Ipolito, S.; Di Nicuolo, F. Effect of Pravastatin on Endothelial Function and Endothelial Progenitor Cells in Healthy Postmenopausal Women. Clin. Exp. Obstet. Gynecol. 2012, 39, 153–159. [Google Scholar] [PubMed]
- Higashi, Y.; Matsuoka, H.; Umei, H.; Sugano, R.; Fujii, Y.; Soga, J.; Kihara, Y.; Chayama, K.; Imaizumi, T. Endothelial Function in Subjects with Isolated Low HDL Cholesterol: Role of Nitric Oxide and Circulating Progenitor Cells. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E202–E209. [Google Scholar] [CrossRef]
- Lin, L.-Y.; Huang, C.-C.; Chen, J.-S. Effects of Pitavastatin versus Atorvastatin on the Peripheral Endothelial Progenitor Cells and Vascular Endothelial Growth Factor in High-Risk Patients: A Pilot Prospective, Double-Blind, Randomized Study. Cardiovasc. Diabetol. 2014, 13, 111. [Google Scholar] [CrossRef]
- Chiang, K.-H.; Cheng, W.-L.; Shih, C.-M.; Lin, Y.-W.; Tsao, N.-W.; Kao, Y.-T.; Lin, C.-T.; Wu, S.-C.; Huang, C.-Y.; Lin, F.-Y. Statins, HMG-CoA Reductase Inhibitors, Improve Neovascularization by Increasing the Expression Density of CXCR4 in Endothelial Progenitor Cells. PLoS ONE 2015, 10, e0136405. [Google Scholar] [CrossRef]
- Cerda, A.; Fajardo, C.M.; Basso, R.G.; Hirata, M.H.; Hirata, R.D.C. Role of MicroRNAs 221/222 on Statin Induced Nitric Oxide Release in Human Endothelial Cells. Arq. Bras. Cardiol. 2014, 104, 195–201. [Google Scholar] [CrossRef]
- António, N.; Fernandes, R.; Soares, A.; Soares, F.; Lopes, A.; Carvalheiro, T.; Paiva, A.; Pêgo, G.M.; Providência, L.A.; Gonçalves, L.; et al. Impact of Prior Chronic Statin Therapy and High-Intensity Statin Therapy at Discharge on Circulating Endothelial Progenitor Cell Levels in Patients with Acute Myocardial Infarction: A Prospective Observational Study. Eur. J. Clin. Pharm. 2014, 70, 1181–1193. [Google Scholar] [CrossRef]
- Ricottini, E.; Madonna, R.; Patti, G.; Grieco, D.; Zoccoli, A.; Stampachiacchiere, B.; D’Ambrosio, A.; Tonini, G.; Caterina, R.D.; Sciascio, G.D. Benefit of Atorvastatin Reload on Endothelial Progenitor Cells in Patients on Chronic Statin Treatment Undergoing PCI. J. Am. Coll. Cardiol. 2013, 61, E1635. [Google Scholar] [CrossRef] [Green Version]
- Di Sciascio, G.; Patti, G.; Pasceri, V.; Gaspardone, A.; Colonna, G.; Montinaro, A. Efficacy of Atorvastatin Reload in Patients on Chronic Statin Therapy Undergoing Percutaneous Coronary Intervention: Results of the ARMYDA-RECAPTURE (Atorvastatin for Reduction of Myocardial Damage During Angioplasty) Randomized Trial. J. Am. Coll. Cardiol. 2009, 54, 558–565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newman, C.B.; Preiss, D.; Tobert, J.A.; Jacobson, T.A.; Page, R.L., II; Goldstein, L.B.; Chin, C.; Tannock, L.R.; Miller, M.; Raghuveer, G.; et al. Statin Safety and As-sociated Adverse Events: A Scientific Statement from the American Heart Association. Arter. Thromb. Vasc. Biol. 2019, 39, e38–e81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, R.; Liu, W.; Zeng, J.; Meng, J.; Jiang, H.; Wang, J.; Xing, D. Niemann-Pick C1-Like 1 Inhibitors for Reducing Cholesterol Absorption. Eur. J. Med. Chem. 2022, 230, 114111. [Google Scholar] [CrossRef] [PubMed]
- Lins, L.C.A.; França, C.N.; Fonseca, F.A.H.; Barbosa, S.P.M.; Matos, L.N.; Aguirre, A.C.; Bianco, H.T.; Do Amaral, J.B.; Izar, M.C. Effects of Ezetimibe on Endothelial Progenitor Cells and Microparticles in High-Risk Patients. Cell Biochem. Biophys. 2014, 70, 687–696. [Google Scholar] [CrossRef]
- Huang, W.-P.; Yin, W.-H.; Chen, J.-S.; Huang, P.-H.; Chen, J.-W.; Lin, S.-J. Fenofibrate Reverses Dysfunction of EPCs Caused by Chronic Heart Failure. J. Cardiovasc. Transl. Res. 2020, 13, 158–170. [Google Scholar] [CrossRef]
- Bardolia, C.; Amin, N.S.; Turgeon, J. Emerging Non-Statin Treatment Options for Lowering Low-Density Lipoprotein Cholesterol. Front. Cardiovasc. Med. 2021, 8, 789931. [Google Scholar] [CrossRef]
- Tang, Y.; Li, S.-L.; Hu, J.-H.; Sun, K.-J.; Liu, L.-L.; Xu, D.-Y. Research Progress on Alternative Non-Classical Mechanisms of PCSK9 in Atherosclerosis in Patients with and without Diabetes. Cardiovasc. Diabetol. 2020, 19, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaudhary, R.; Garg, J.; Shah, N.; Sumner, A. PCSK9 Inhibitors: A New Era of Lipid Lowering Therapy. World J. Cardiol. 2017, 9, 76–91. [Google Scholar] [CrossRef]
- Nicholls, S.J.; Puri, R.; Anderson, T.; Ballantyne, C.M.; Cho, L.; Kastelein, J.J.P.; Koenig, W.; Somaratne, R.; Kassahun, H.; Yang, J.; et al. Effect of Evolocumab on Progression of Coronary Disease in Statin-Treated Patients: The GLAGOV Randomized Clinical Trial. JAMA 2016, 316, 2373–2384. [Google Scholar] [CrossRef]
- Lepor, N.E.; Sun, J.; Canton, G.; Contreras, L.; Hippe, D.S.; Isquith, D.A.; Balu, N.; Kedan, I.; Simonini, A.A.; Yuan, C. Regression in Carotid Plaque Lipid Content and Neovasculature with PCSK9 Inhibition: A Time Course Study. Atherosclerosis 2021, 327, 31–38. [Google Scholar] [CrossRef]
- Schwartz, G.G.; Steg, P.G.; Szarek, M.; Bhatt, D.L.; Bittner, V.A.; Diaz, R.; Edelberg, J.M.; Goodman, S.G.; Hanotin, C.; Harrington, R.A.; et al. Alirocumab and Cardiovascular Outcomes after Acute Coronary Syndrome. N. Engl. J. Med. 2018, 379, 2097–2107. [Google Scholar] [CrossRef] [PubMed]
- Sabatine, M.S.; De Ferrari, G.M.; Giugliano, R.P.; Huber, K.; Lewis, B.S.; Ferreira, J.; Kuder, J.F.; Murphy, S.A.; Wiviott, S.D.; Kurtz, C.E. Clinical Benefit of Evolocumab by Severity and Extent of Coronary Artery Disease: Analysis from FOURIER. Circulation 2018, 138, 756–766. [Google Scholar] [CrossRef] [PubMed]
- Tripaldi, R.; Lanuti, P.; Simeone, P.G.; Liani, R.; Bologna, G.; Ciotti, S.; Simeone, P.; Di Castelnuovo, A.; Marchisio, M.; Cipollone, F. Endogenous PCSK9 May Influence Circulating CD45 Neg/CD34 Bright and CD45 Neg/CD34 Bright/CD146 Neg Cells in Patients with Type 2 Diabetes Mellitus. Sci. Rep. 2021, 11, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Zadok, O.I.B.; Mager, A.; Leshem-Lev, D.; Lev, E.; Kornowski, R.; Eisen, A. The Effect of Proprotein Convertase Subtilisin Kexin Type 9 Inhibitors on Circulating Endothelial Progenitor Cells in Patients with Cardiovascular Disease. Cardiovasc. Drugs Ther. 2021, 36, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Guedeney, P.; Giustino, G.; Sorrentino, S.; Claessen, B.E.; Camaj, A.; Kalkman, D.N.; Vogel, B.; Sartori, S.; De Rosa, S.; Baber, U. Efficacy and Safety of Alirocumab and Evolocumab: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. Eur. Heart J. 2019, 43, e17–e25. [Google Scholar] [CrossRef] [PubMed]
- Santulli, G. Angiopoietin-Like Proteins: A Comprehensive Look. Front. Endocrinol. 2014, 5, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oike, Y.; Tian, Z.; Miyata, K.; Morinaga, J.; Endo, M.; Kadomatsu, T. ANGPTL2—A New Causal Player in Accelerating Heart Disease Development in the Aging. Circ. J. 2017, 81, 1379–1385. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.K.; Chaube, B.; Zhang, X.; Sun, J.; Citrin, K.M.; Canfrán-Duque, A.; Aryal, B.; Rotllan, N.; Varela, L.; Lee, R.G. Hepatocyte-Specific Suppression of ANGPTL4 Improves Obesity-Associated Diabetes and Mitigates Atherosclerosis in Mice. J. Clin. Investig. 2021, 131, e140989. [Google Scholar] [CrossRef]
- Robciuc, M.R.; Maranghi, M.; Lahikainen, A.; Rader, D.; Bensadoun, A.; Öörni, K.; Metso, J.; Minicocci, I.; Ciociola, E.; Ceci, F. Angptl3 Deficiency Is Associated with Increased Insulin Sensitivity, Lipoprotein Lipase Activity, and Decreased Serum Free Fatty Acids. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1706–1713. [Google Scholar] [CrossRef] [Green Version]
- Dewey, F.E.; Gusarova, V.; Dunbar, R.L.; O’Dushlaine, C.; Schurmann, C.; Gottesman, O.; McCarthy, S.; Van Hout, C.V.; Bruse, S.; Dansky, H.M. Genetic and Pharmacologic Inactivation of ANGPTL3 and Cardiovascular Disease. N. Engl. J. Med. 2017, 377, 211–221. [Google Scholar] [CrossRef]
- Graham, M.J.; Lee, R.G.; Brandt, T.A.; Tai, L.-J.; Fu, W.; Peralta, R.; Yu, R.; Hurh, E.; Paz, E.; McEvoy, B.W. Cardiovascular and Metabolic Effects of ANGPTL3 Antisense Oligonucleotides. N. Engl. J. Med. 2017, 377, 222–232. [Google Scholar] [CrossRef] [PubMed]
- Quagliarini, F.; Wang, Y.; Kozlitina, J.; Grishin, N.V.; Hyde, R.; Boerwinkle, E.; Valenzuela, D.M.; Murphy, A.J.; Cohen, J.C.; Hobbs, H.H. Atypical Angiopoietin-like Protein That Regulates ANGPTL3. Proc. Natl. Acad. Sci. USA 2012, 109, 19751–19756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schinzari, F.; Vizioli, G.; Campia, U.; Tesauro, M.; Cardillo, C. Variable Changes of Circulating ANGPTL3 and ANGPTL4 in Different Obese Phenotypes: Relationship with Vasodilator Dysfunction. Biomedicines 2021, 9, 1037. [Google Scholar] [CrossRef] [PubMed]
- Raal, F.J.; Rosenson, R.S.; Reeskamp, L.F.; Hovingh, G.K.; Kastelein, J.J.; Rubba, P.; Ali, S.; Banerjee, P.; Chan, K.-C.; Gipe, D.A. Evinacumab for Homozygous Familial Hypercholesterolemia. N. Engl. J. Med. 2020, 383, 711–720. [Google Scholar] [CrossRef]
- Camenisch, G.; Pisabarro, M.T.; Sherman, D.; Kowalski, J.; Nagel, M.; Hass, P.; Xie, M.-H.; Gurney, A.; Bodary, S.; Liang, X.H.; et al. ANGPTL3 Stimulates Endothelial Cell Adhesion and Migration via Integrin Alpha Vbeta 3 and Induces Blood Vessel Formation In Vivo. J. Biol. Chem. 2002, 277, 17281–17290. [Google Scholar] [CrossRef] [Green Version]
- Caiado, F.; Dias, S. Endothelial Progenitor Cells and Integrins: Adhesive Needs. Fibrogenes. Tissue Repair 2012, 5, 4. [Google Scholar] [CrossRef] [Green Version]
- Luo, F.; Wu, P.; Chen, J.; Guo, Y.; Wang, J.; Li, X.; Fang, Z. ANGPTL3 Possibly Promotes Cardiac Angiogenesis through Improving Proangiogenic Ability of Endothelial Progenitor Cells after Myocardial Infarction. Lipids Health Dis. 2018, 17, 1–3. [Google Scholar] [CrossRef] [Green Version]
- Lui, M.; Garberich, R.; Strauss, C.; Davin, T.; Knickelbine, T. Usefulness of Lipid Apheresis in the Treatment of Familial Hypercholesterolemia. J. Lipids 2014, 2014, 864317. [Google Scholar] [CrossRef] [Green Version]
- Thompson, G.; Parhofer, K.G. Current Role of Lipoprotein Apheresis. Curr. Atheroscler. Rep. 2019, 21, 26. [Google Scholar] [CrossRef] [Green Version]
- Wang, A.; Richhariya, A.; Gandra, S.R.; Calimlim, B.; Kim, L.; Quek, R.G.; Nordyke, R.J.; Toth, P.P. Systematic Review of Low-Density Lipoprotein Cholesterol Apheresis for the Treatment of Familial Hypercholesterolemia. J. Am. Heart Assoc. 2016, 5, e003294. [Google Scholar] [CrossRef] [Green Version]
- Tavori, H.; Giunzioni, I.; Linton, M.F.; Fazio, S. Loss of Plasma Proprotein Convertase Subtilisin/Kexin 9 (PCSK9) after Lipoprotein Apheresis. Circ. Res. 2013, 113, 1290–1295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuzaki, M.; Hiramori, K.; Imaizumi, T.; Kitabatake, A.; Hishida, H.; Nomura, M.; Fujii, T.; Sakuma, I.; Fukami, K.; Honda, T.; et al. Intravascular Ultrasound Evaluation of Coronary Plaque Regression by Low Density Lipoprotein-Apheresis in Familial Hypercholesterolemia: The Low Density Lipoprotein-Apheresis Coronary Morphology and Reserve Trial (LACMART). J. Am. Coll. Cardiol. 2002, 40, 220–227. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, S.; Luo, P.; Reda, D.J.; Latif, F.; Hastings, J.L.; Armstrong, E.J.; Bagai, J.; Abu-Fadel, M.; Baskar, A.; Kamath, P. Plaque Regression and Endothelial Progenitor Cell Mobilization with Intensive Lipid Elimination Regimen (PREMIER). Circ. Cardiovasc. Interv. 2020, 13, e008933. [Google Scholar] [CrossRef] [PubMed]
- Mörtzell Henriksson, M.; Newman, E.; Witt, V.; Derfler, K.; Leitner, G.; Eloot, S.; Dhondt, A.; Deeren, D.; Rock, G.; Ptak, J.; et al. Adverse Events in Apheresis: An Update of the WAA Registry Data. Transfus. Apher. Sci. 2016, 54, 2–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
| Substance | Study Population | Major Findings in Study Groups |
|---|---|---|
| Atorvastatin reloading with 80 mg [76,77] | 53 patients on long-term statin treatment who underwent percutaneous coronary interventions (PCI) | ↑ EPC count ↑ EPC-CFU |
| Atorvastatin 80 mg vs. atorvastatin 10 mg preloading [78] | 20 statin-naïve male patients undergoing angiography | 3.5-fold increase in EPC levels in the 80 mg group |
| Atorvastatin 40 mg vs. atorvastatin 10 mg [79] | 26 patients with ischemic heart failure | ↑ EPC ↑ FMD ↓ TNF-α |
| Atorvastatin 40 mg vs. atorvastatin 10 mg vs. placebo [80] | 58 patients with coronary heart disease | ↑ EPC in both atorvastatin groups ↓ VEGF and CRP |
| Atorvastatin 80 mg reloading vs. 40 mg vs. no statin [81] | 45 patients undergoing coronary angioplasty | ↑ early EPCs in 80 mg group ↓ increase in cardiac troponin |
| Atorvastatin 40 mg vs. 10 mg [82] | 100 patients with ischemic cardiomyopathy | ↑ EPC ↓ hsCRP, oxLDL |
| Atorvastatin 40 mg vs. control group [83] | 108 patients with coronary slow flow | ↑ EPCs ↑ EPC adhesion, migration and proliferation ↑ NO ↓ hs-CRP, ET-1 and IL-6 |
| Atorvastatin 40 mg vs. placebo [84] | 60 consecutive patients who underwent isolated, first-time CABG | ↑ early EPCs ↓ hsCRP Less atrial fibrillation |
| Atorvastatin 20 mg vs. placebo [85] | 50 patients undergoing elective coronary surgery | ↑ EPCs |
| Atorvastatin 80 mg vs. atorvastatin 20 mg [86] | 40 ST-segment elevation myocardial infarction (STEMI) patients undergoing PCI | ↑ EPCs |
| Atorvastatin 20 mg vs. placebo [87] | 68 patients with chronic pulmonary heart disease | ↑ EPCs |
| Atorvastatin 20 mg vs. no statin [88] | 48 patients with a first-time non-lacunar ischaemic stroke | ↑ EPC increment EPC increment ≥4 CFU-EC predicted favorable clinical outcome |
| Rosuvastatin 40 mg [89] | 26 patients with mixed dyslipidaemia | ↑ EPC count ↑ EPC-CFU |
| Rosuvastatin 10 mg vs. placebo [90] | 60 patients with systolic heart failure | ↑ EPC FMD, VEGF, fibrinogen, MMP-9, IL-6, IL-1β, oxLDL, PerOx, NT-proBNP, and uric acid levels did not correlate with EPC level |
| Rosuvastatin 40 mg vs. placebo [91] | 42 patients with chronic heart failure (CHF) | ↑ EPC ↑ FMD |
| Rosuvastatin 10 mg vs. no treatment [92] | 32 hypercholesterolemic patients | ↑ EPC ↑ FMD |
| Simvastatin 80 mg vs. simvastatin 20/10 mg ezetimibe [93] | 68 patients with coronary artery disease | no effect on EPC |
| Simvastatin 80 mg mono-treatment with combination treatment of 10 mg simvastatin and 10 mg ezetimibe [94] | 19 obese men with the metabolic syndrome | ↑ EPCs regardless of study group |
| Pravastatin 40 mg vs. placebo [95] | 20 healthy postmenopausal women | ↑ EPC-CFU |
| Pravastatin 10 mg vs. placebo [96] | 29 patients with isolated low HDL cholesterol | ↑ EPC ↑ FMD |
| Pitavastatin 2 mg vs. atorvastatin 10 mg [97] | 26 patients at high cardiovascular risk | ↑ EPC ↑ eNOS expression ↑ adhesion ability of early EPCs ↑ migration and tube formation capacities of late EPCs |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Altabas, V.; Biloš, L.S.K. The Role of Endothelial Progenitor Cells in Atherosclerosis and Impact of Anti-Lipemic Treatments on Endothelial Repair. Int. J. Mol. Sci. 2022, 23, 2663. https://doi.org/10.3390/ijms23052663
Altabas V, Biloš LSK. The Role of Endothelial Progenitor Cells in Atherosclerosis and Impact of Anti-Lipemic Treatments on Endothelial Repair. International Journal of Molecular Sciences. 2022; 23(5):2663. https://doi.org/10.3390/ijms23052663
Chicago/Turabian StyleAltabas, Velimir, and Lora Stanka Kirigin Biloš. 2022. "The Role of Endothelial Progenitor Cells in Atherosclerosis and Impact of Anti-Lipemic Treatments on Endothelial Repair" International Journal of Molecular Sciences 23, no. 5: 2663. https://doi.org/10.3390/ijms23052663
APA StyleAltabas, V., & Biloš, L. S. K. (2022). The Role of Endothelial Progenitor Cells in Atherosclerosis and Impact of Anti-Lipemic Treatments on Endothelial Repair. International Journal of Molecular Sciences, 23(5), 2663. https://doi.org/10.3390/ijms23052663