
Stereoselective Promiscuous Reactions Catalyzed by Lipases



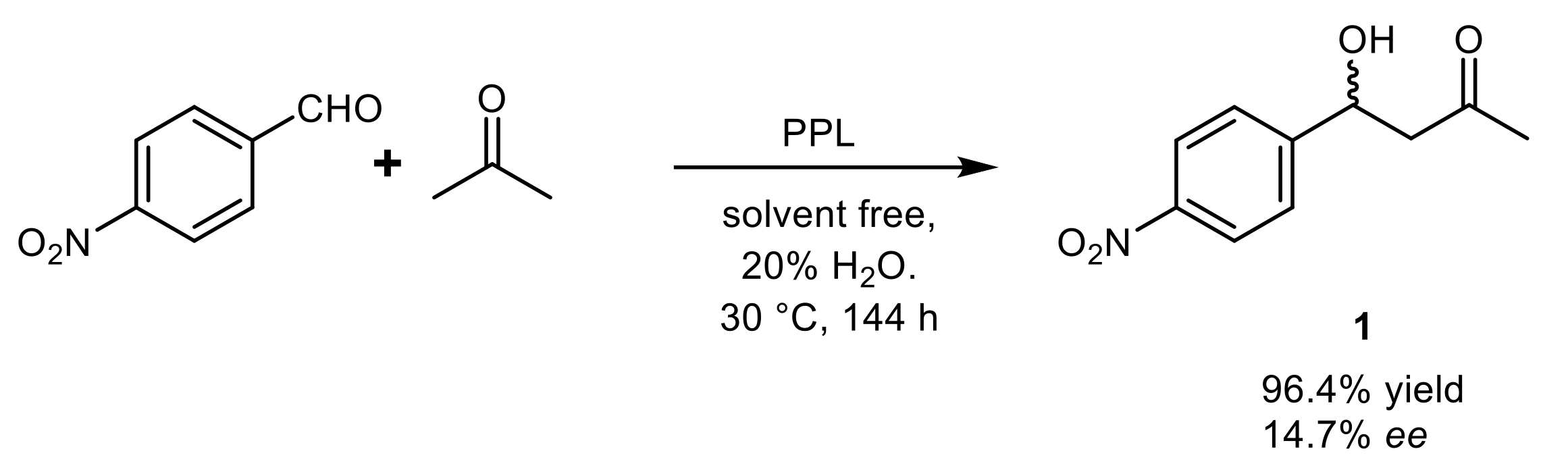{kind=link}
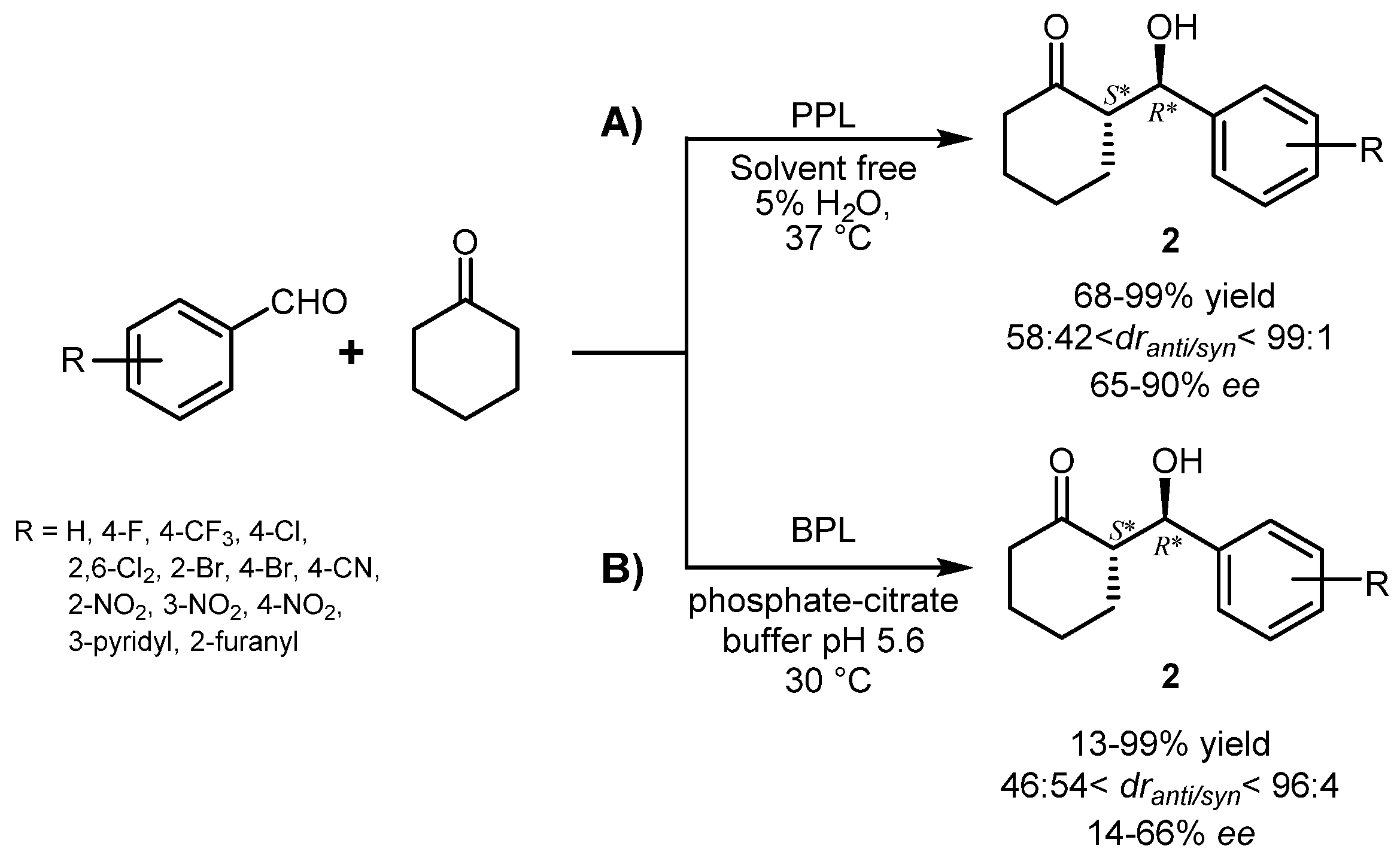{kind=link}
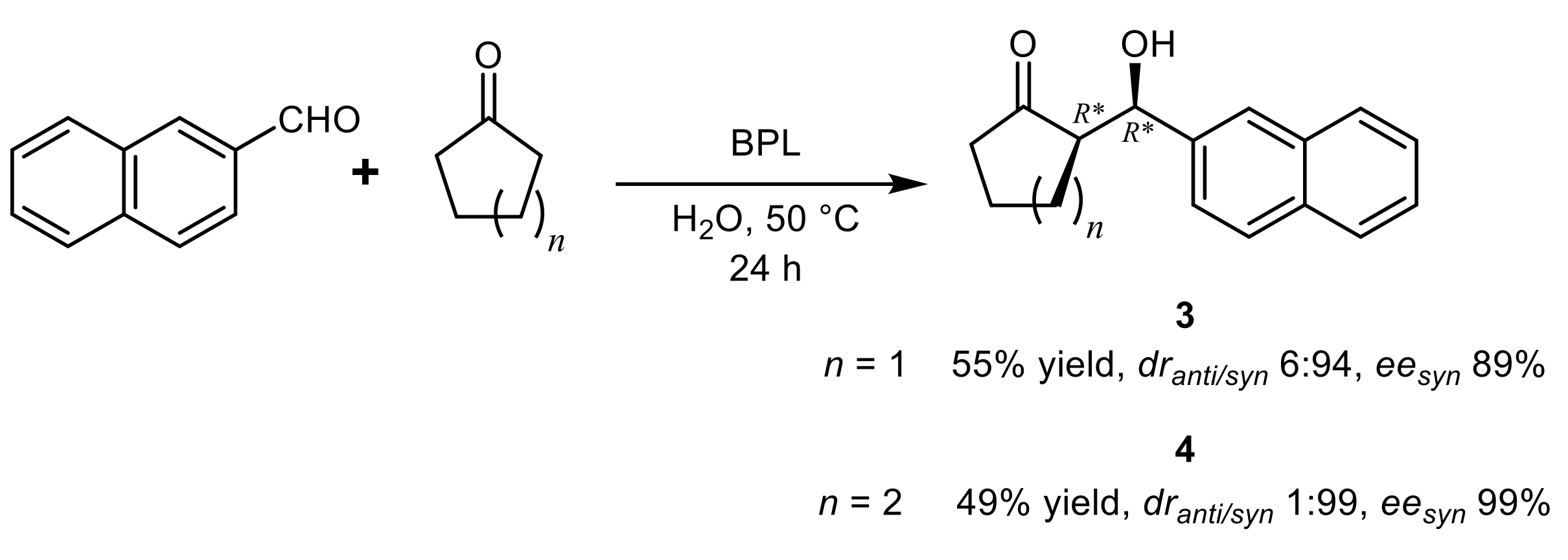{kind=link}
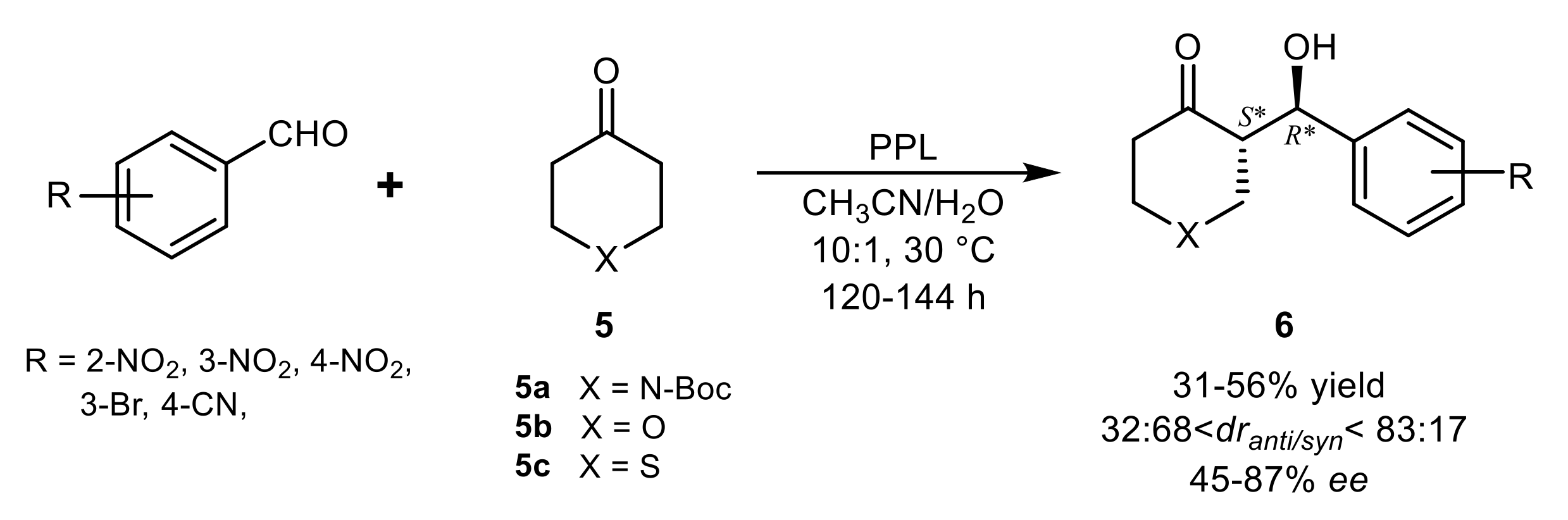{kind=link}
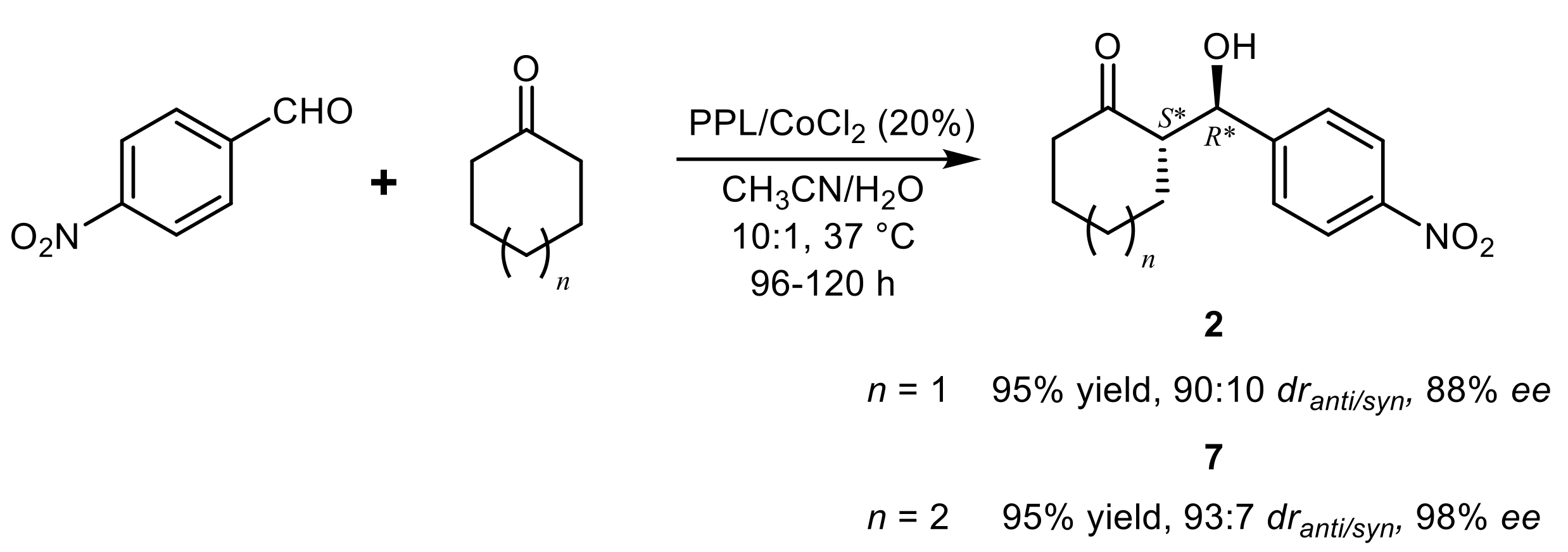{kind=link}
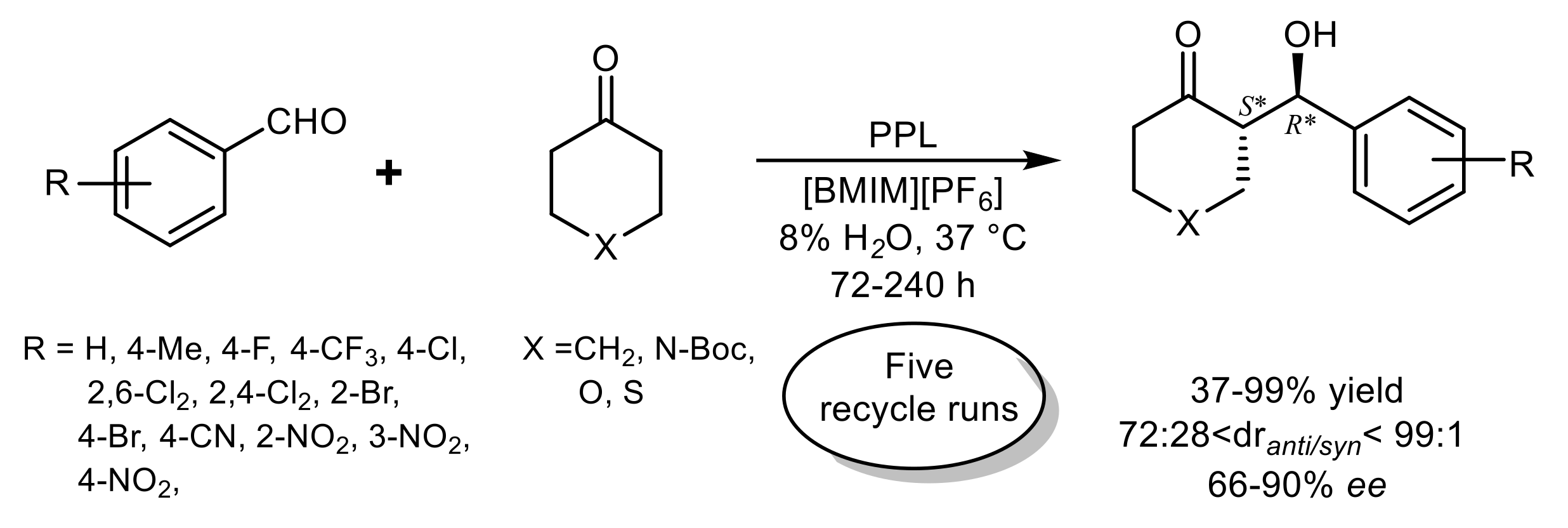{kind=link}
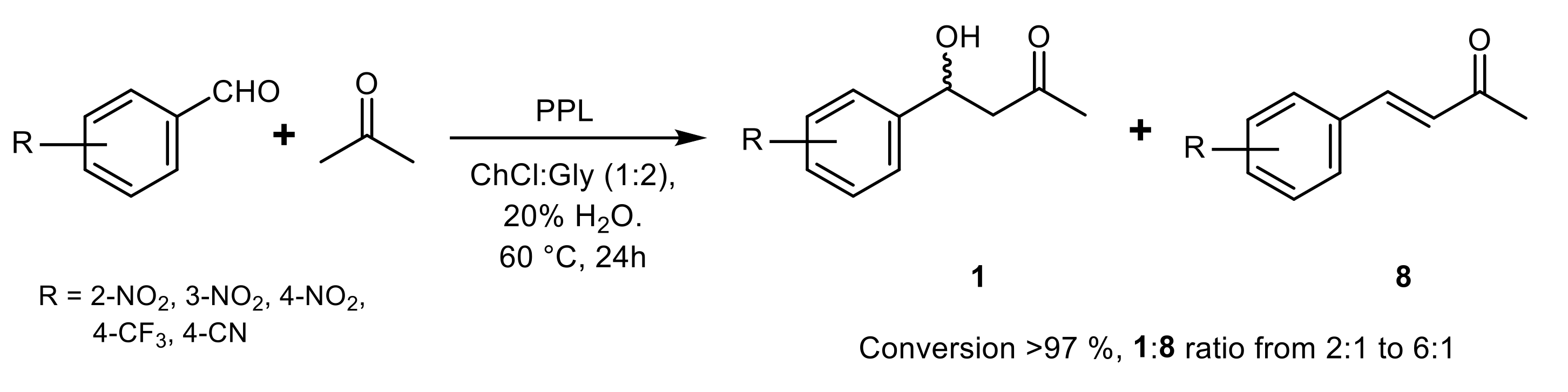{kind=link}
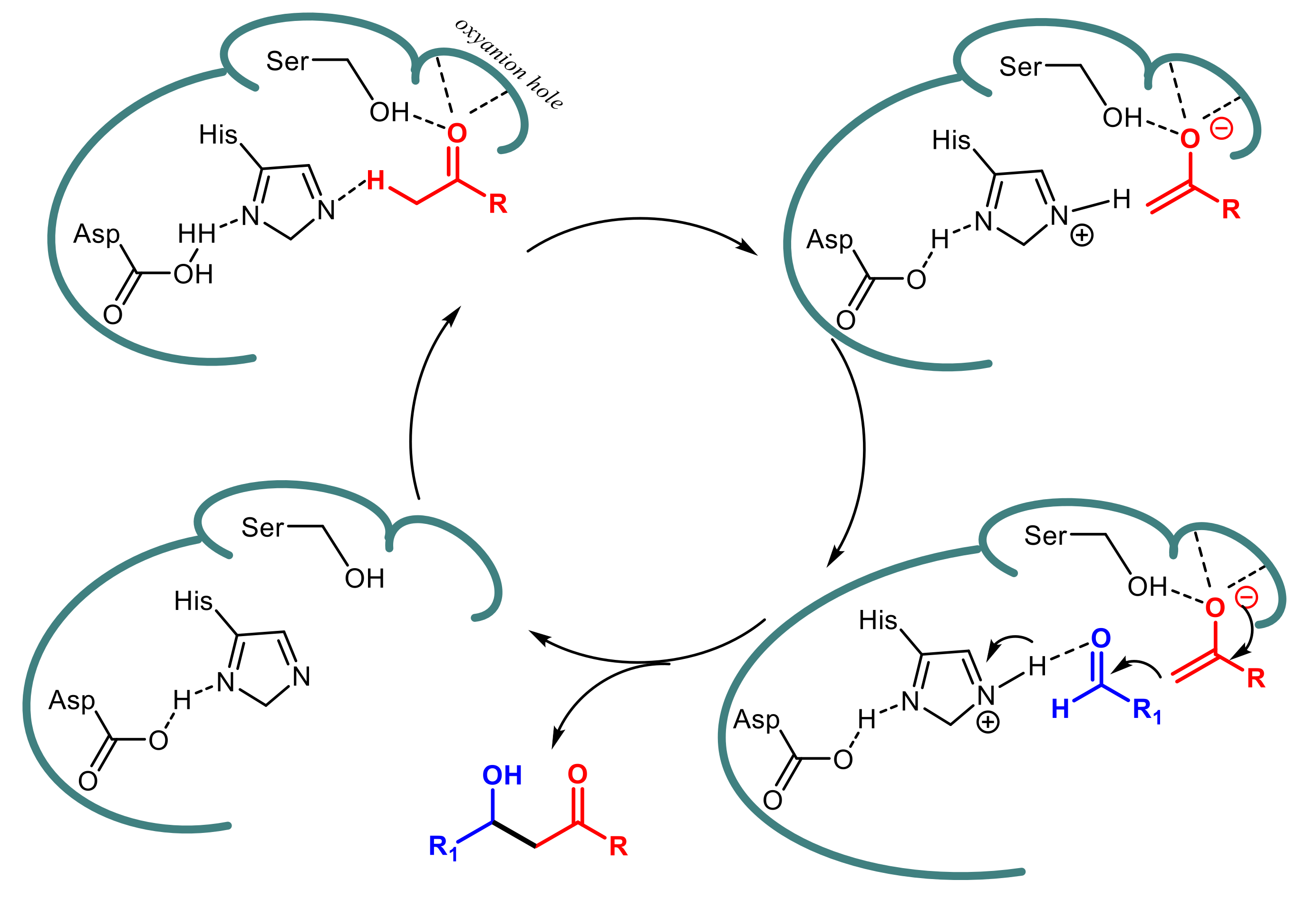{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
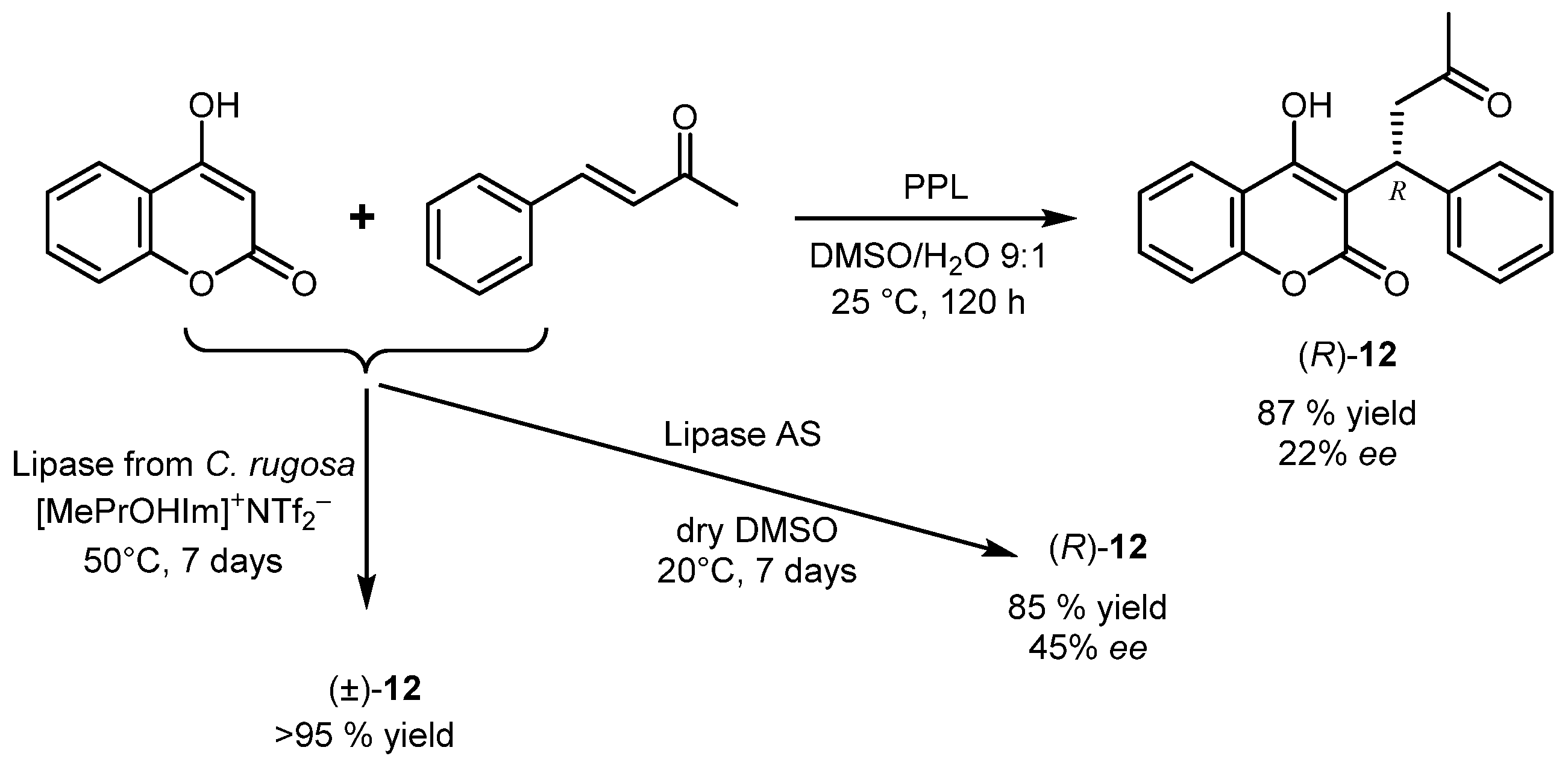
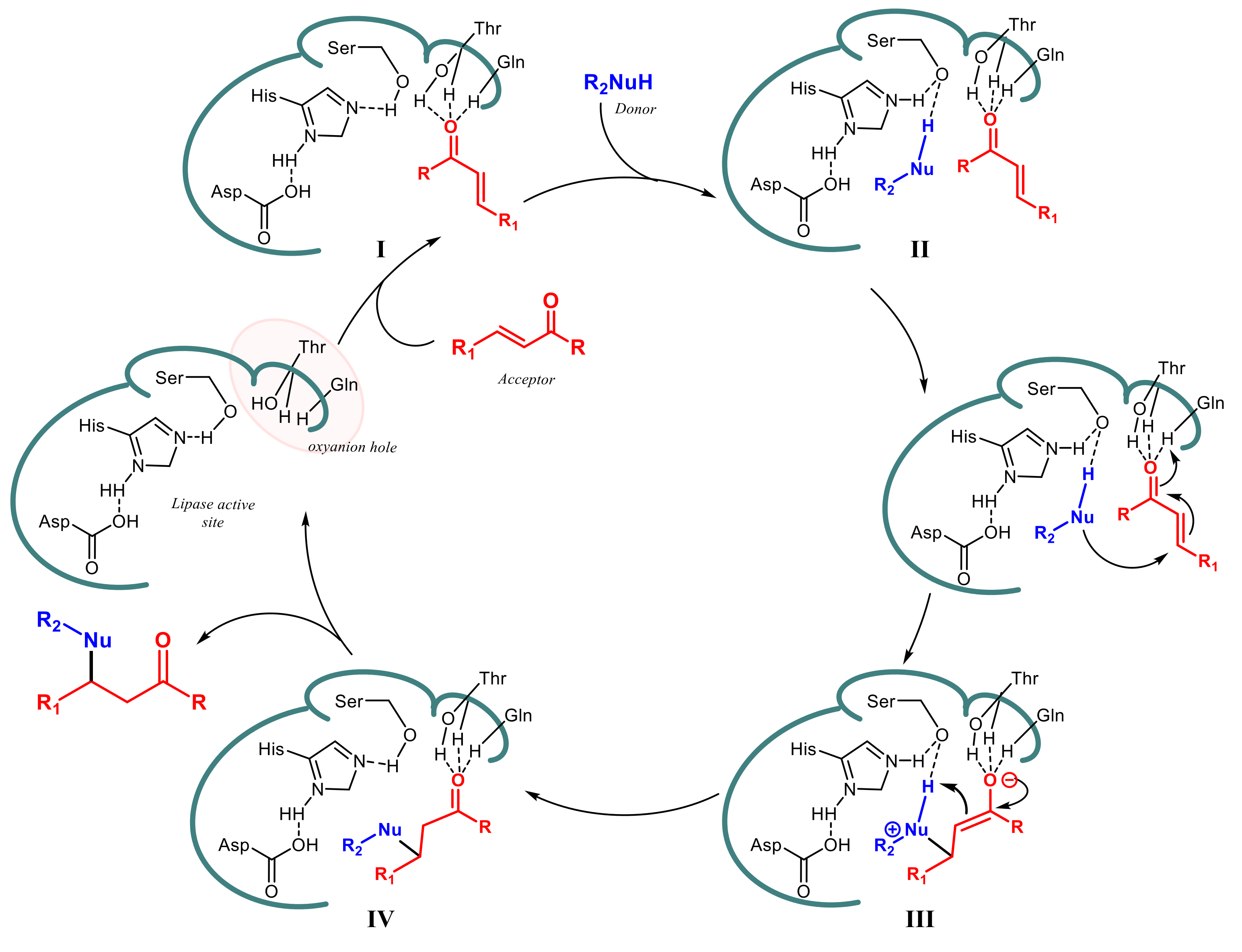
2. Lipase-Catalyzed Aldol Reactions
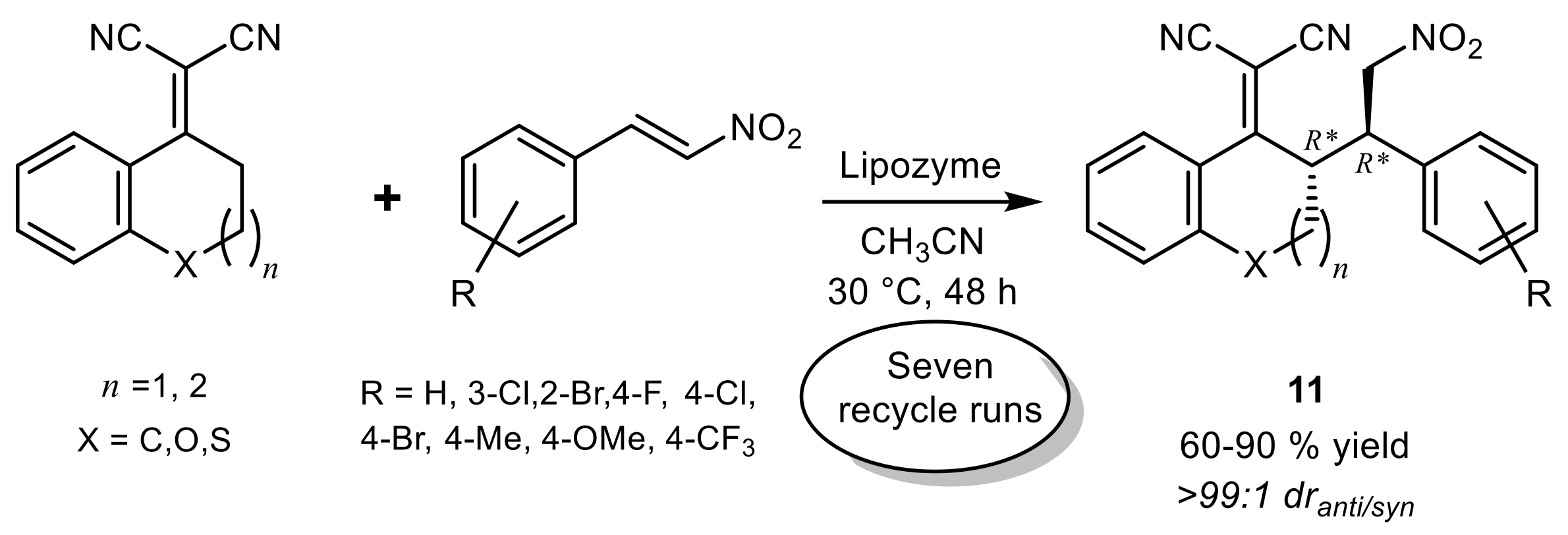
3. Lipase-Catalyzed Michael Reactions
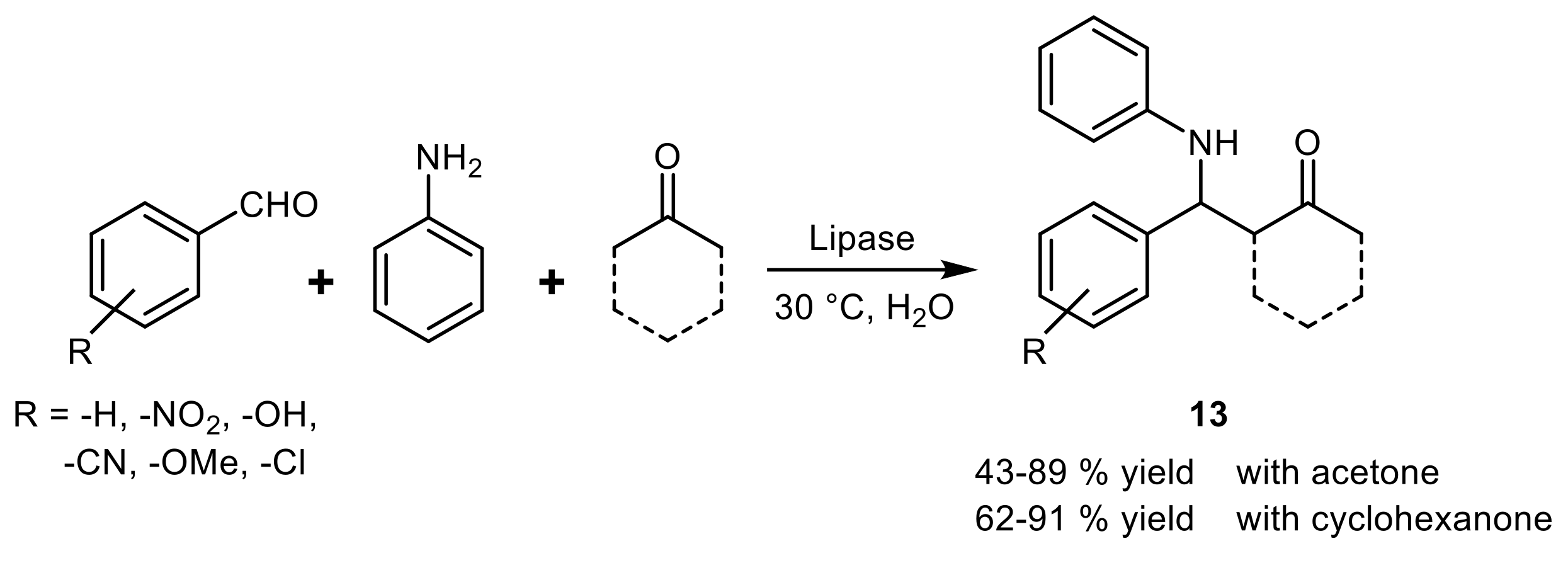
4. Lipase-Catalyzed Multicomponent Reactions
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tao, J.A.; Kazlauskas, R.J. Biocatalysis for Green Chemistry and Chemical Process Development, 1st ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2011. [Google Scholar]
- Sheldon, R.A.; Brady, D. Broadening the scope of biocatalysis in sustainable organic synthesis. ChemSusChem 2019, 12, 2859–2881. [Google Scholar] [CrossRef]
- Turner, N.J. Directed evolution drives the next generation of biocatalysts. Nat. Chem. Biol. 2009, 5, 567–573. [Google Scholar] [CrossRef] [PubMed]
- Muñoz Solano, D.; Hoyos, P.; Hernáiz, M.J.; Alcántara, A.R.; Sánchez-Montero, J.M. Industrial biotransformations in the synthesis of building blocks leading to enantiopure drugs. Bioresour. Technol. 2012, 115, 196–207. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Snajdrova, R.; Moore, J.C.; Baldenius, K.; Bornscheuer, U.T. Biocatalysis: Enzymatic Synthesis for Industrial Applications. Angew. Chem. Int. Ed. 2021, 60, 88–119. [Google Scholar] [CrossRef] [PubMed]
- Hult, K.; Berglund, P. Enzyme promiscuity: Mechanism and applications. Trends in Biotechnol. 2007, 25, 231–238. [Google Scholar] [CrossRef]
- Busto, E.; Gotor-Fernandez, V.; Gotor, V. Hydrolases: Catalytically promiscuous enzymes for non-conventional reactions in organic synthesis. Chem. Soc. Rev. 2010, 39, 4504–4523. [Google Scholar] [CrossRef] [PubMed]
- Humble, M.S.; Berglund, P. Biocatalytic promiscuity. Eur. J. Org. Chem. 2011, 3391–3401. [Google Scholar] [CrossRef]
- Miao, Y.; Rahimi, M.; Geertsema, E.M.; Poelarends, G.J. Recent developments in enzyme promiscuity for carbon–carbon bond-forming reactions. Curr. Opin. Chem. Biol. 2015, 25, 115–123. [Google Scholar] [CrossRef]
- Dwivedee, B.P.; Soni, S.; Sharma, M.; Bhaumik, J.; Laha, J.K.; Banerjee, U.C. Promiscuity of lipase-catalyzed reactions for organic synthesis: A recent update. ChemistrySelect 2018, 3, 2441–2466. [Google Scholar] [CrossRef]
- Khersonsky, O.; Tawfik, D.S. Enzyme promiscuity: A mechanistic and evolutionary perspective. Annu. Rev. Biochem. 2010, 79, 471–505. [Google Scholar] [CrossRef]
- Zaks, A.; Klibanov, A.M. Enzyme-catalyzed processes in organic solvents. Proc. Natl. Acad. Sci. USA 1985, 82, 3192–3196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elgharbawya, A.A.; Riyadi, F.A.; Alama, M.d.Z.; Moniruzzaman, M. Ionic liquids as a potential solvent for lipase-catalysed reactions: A review. J. Mol. Liq. 2018, 251, 150–166. [Google Scholar] [CrossRef]
- Tan, J.-N.; Dou, Y. Deep eutectic solvents for biocatalytic transformations: Focused lipase-catalyzed organic reactions. Appl. Microbiol. Biotechnol. 2020, 104, 1481–1496. [Google Scholar] [CrossRef] [PubMed]
- Ghanem, A.; Aboul-Enein, H.Y. Application of lipases in kinetic resolution of racemates. Chirality 2005, 17, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Seddigi, Z.S.; Malik, M.S.; Ahmed, S.A.; Babalghith, A.O.; Kamal, A. Lipases in asymmetric transformations: Recent advances in classical kinetic resolution and lipase–metal combinations for dynamic processes. Coordin. Chem. Rev. 2017, 348, 54–70. [Google Scholar] [CrossRef]
- Patti, A.; Sanfilippo, C. Breaking molecular symmetry through biocatalytic reactions to gain access to valuable chiral synthons. Symmetry 2020, 12, 1454. [Google Scholar] [CrossRef]
- Thangaraj, B.; Solomon, P.R. Immobilization of lipases–A review Part II: Carrier Materials. ChemBioEng. Rev. 2019, 6, 67–194. [Google Scholar] [CrossRef]
- Chandra, P.; Enespa, R.; Singh Arora, P.K. Microbial lipases and their industrial applications: A comprehensive review. Microb. Cell. Fact. 2020, 19, 169. [Google Scholar] [CrossRef]
- Kitazume, T.; Ikeya, T.; Murata, K. Synthesis of optically active trifluorinated compounds: Asymmetric Michael addition with hydrolytic enzymes. J. Chem. Soc. Chem. Commun. 1986, 1331–1333. [Google Scholar] [CrossRef]
- Branneby, C.; Carlqvist, P.; Magnusson, A.; Hult, K.; Brinck, T.; Berglund, P. Carbon-carbon bonds by hydrolytic enzymes. J. Am. Chem. Soc. 2003, 125, 874–875. [Google Scholar] [CrossRef]
- Yamashita, Y.; Yasukawa, T.; Yoo, W.-J.; Kitanosono, T.; Kobayashi, S. Catalytic enantioselective aldol reactions. Chem. Soc. Rev. 2018, 47, 4388–4480. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Feng, X.-W.; Wang, N.; Zhou, Y.-J.; Yu, X.-Q. Biocatalytic promiscuity: The first lipase-catalysed asymmetric aldol reaction. Green Chem. 2008, 10, 616–618. [Google Scholar] [CrossRef]
- Xie, Z.-B.; Wang, N.; Zhou, L.-H.; Wan, F.; He, T.; Le, Z.-G.; Yu, X.-Q. Lipase-catalyzed stereoselective cross-aldol reaction promoted by water. ChemCatChem 2013, 5, 1935–1940. [Google Scholar] [CrossRef]
- Xie, Z.-B.; Wang, N.; Jiang, G.-F.; Yu, X.-Q. Biocatalytic asymmetric aldol reaction in buffer solution. Tetrahedron Lett. 2013, 54, 945–948. [Google Scholar] [CrossRef]
- Wang, Y.; Liang, X.-Y.; Chen, X.-Y.; Liang, Z.-H.; Cheng, H.; Li, X.; Li, L.-L. Promotion of water in BPL-catalyzed asymmetric cross aldol reaction. Biomass Convers. Biorefinery 2020. [Google Scholar] [CrossRef]
- Wang, Y.; Cheng, H.; Li, X.; Li, L.-L.; Liang, Z.-H.; Liang, X.-Y.; Chen, X.-Y. MML-catalyzed direct aldol reaction in green solvents. Biomass Convers. Biorefinery 2020. [Google Scholar] [CrossRef]
- Guan, Z.; Fu, J.-P.; He, Y.-H. Biocatalytic promiscuity: Lipase-catalyzed asymmetric aldol reaction of heterocyclic ketones with aldehydes. Tetrahedron Lett. 2012, 53, 4959–4961. [Google Scholar] [CrossRef]
- Yıldız, T.; Yaşa, H.; Hasdemir, B.; Yusufoğlu, A.S. Different bio/Lewis acid-catalyzed stereoselective aldol reactions in various mediums. Monatsh. Chem. 2017, 148, 1445–1452. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, N.; Xie, Z.-B.; Zhou, L.-H.; Yu, X.-Q. Ionic liquid as a recyclable and efficient medium for lipase-catalyzed asymmetric cross aldol reaction. J. Mol. Catal. B Enzym. 2014, 110, 100–110. [Google Scholar] [CrossRef]
- González-Martínez, D.; Gotor, V.; Gotor-Fernández, V. Application of deep eutectic solvents in promiscuous lipase-catalysed aldol reactions. Eur. J. Org. Chem. 2016, 1513–1519. [Google Scholar] [CrossRef]
- Branneby, C.; Carlqvist, P.; Hult, K.; Brinck, T.; Berglund, P. Aldol additions with mutant lipase: Analysis by experiments and theoretical calculations. J. Mol. Catal. B Enzym. 2004, 31, 123–128. [Google Scholar] [CrossRef]
- Reznikov, A.N.; Klimochkin, Y.N. Recent developments in highly stereoselective Michael addition reactions catalyzed by metal complexes. Synthesis 2020, 52, 781–795. [Google Scholar] [CrossRef]
- Malkar, R.S.; Jadhav, A.L.; Yadav, G.D. Innovative catalysis in Michael addition reactions for C-X bond formation. Mol. Catal. 2020, 485, 110814. [Google Scholar] [CrossRef]
- Torre, O.; Alfonso, I.; Gotor, V. Lipase catalysed Michael addition of secondary amines to acrylonitrile. Chem. Commun. 2004, 1724–1725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.-M.; Zhang, F.; Wu, Q.; Zhang, Q.-Y.; Lin, X.-F. Hydrolase-catalyzed Michael addition of 1,3-dicarbonyl compounds to α,β-unsaturated compounds in organic solvent. J. Mol. Catal. B Enzym. 2007, 49, 50–54. [Google Scholar] [CrossRef]
- Steunenberg, P.; Sijm, M.; Zuilhof, H.; Sanders, J.P.M.; Scott, E.L.; Franssen, M.C.R. Lipase-catalyzed aza-Michael reaction on acrylate derivatives. J. Org. Chem. 2013, 78, 3802–3813. [Google Scholar] [CrossRef]
- Rivera-Ramírez, J.D.; López-Munguía, J.E.A.; Castillo, A.M.E. Thermodynamically controlled chemoselectivity in lipase-catalyzed aza-Michael additions. J. Mol. Catal. B: Enzym. 2015, 112, 76–82. [Google Scholar] [CrossRef]
- Strohmeier, G.A.; Sović, T.; Steinkellner, G.; Hartner, F.S.; Andryushkova, A.; Purkarthofer, T.; Glieder, A.; Gruber, K.; Griengl, H. Investigation of lipase-catalyzed Michael-type carbon–carbon bond formations. Tetrahedron 2009, 65, 5663–5668. [Google Scholar] [CrossRef]
- Cai, J.-F.; Guan, Z.; He, Y.-H. The lipase-catalyzed asymmetric C–C Michael addition. J. Mol. Catal. B Enzym. 2011, 68, 240–244. [Google Scholar] [CrossRef]
- Rizzo, P.V.S.; Boarin, L.A.; Freitas, I.O.M.; Gomes, R.S.; Beatriz, A.; Rinaldi, A.W.; Domingues, N.L.C. The study of biocatalyzed thio-Michael reaction: A greener and multi-gram protocol. Tetrahedron Lett. 2014, 55, 430–434. [Google Scholar] [CrossRef]
- Zhou, L.-H.; Wang, N.; Chen, G.-N.; Yang, Q.; Yang, S.-Y.; Zhang, W.; Zhang, Y.; Yu, X.-Q. Lipase-catalyzed highly diastereoselective direct vinylogous Michael addition reaction of α,α-dicyanoolefins to nitroalkenes. J. Mol. Catal. B Enzym. 2014, 109, 170–177. [Google Scholar] [CrossRef]
- Xie, B.-H.; Guan, Z.; He, Y.-H. Promiscuous enzyme-catalyzed Michael addition: Synthesis of warfarin and derivatives. J. Chem. Technol. Biotechnol. 2012, 87, 1709–1714. [Google Scholar] [CrossRef]
- Sano, K.; Saito, S.; Hirose, Y.; Kohari, Y.; Nakano, H.; Seki, C.; Tokiwa, M.; Takeshita, M.; Uwai, K. Development of a novel method for warfarin synthesis via lipase-catalyzed steroselective Michael reaction. Heterocycles 2013, 87, 1269–1278. [Google Scholar] [CrossRef]
- Fan, Y.; Cai, D.; Wang, X.; Yang, L. Ionic liquids: Efficient media for the lipase-catalyzed Michael addition. Molecules 2018, 23, 2154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carlqvist, P.; Svedendahl, M.; Branneby, C.; Hult, K.; Brinck, T.; Berglund, P. Exploring the active-site of a rationally redesigned lipase for catalysis of Michael-type additions. ChemBioChem 2005, 6, 331–336. [Google Scholar] [CrossRef] [PubMed]
- Gu, B.; Hu, Z.-E.; Yang, Z.-J.; Li, J.; Zhou, Z.-W.; Wang, N.; Yu, X.-Q. Probing the mechanism of CAL-B-catalyzed aza-Michael addition of aniline compounds with acrylates using mutation and molecular docking simulations. Chem. Sel. 2019, 4, 3848–3854. [Google Scholar] [CrossRef]
- Li, K.; He, T.; Li, C.; Feng, X.-W.; Wang, N.; Yu, X.-Q. Lipase-catalysed direct Mannich reaction in water: Utilization of biocatalytic promiscuity for C–C bond formation in a “one-pot” synthesis. Green Chem. 2009, 11, 777–779. [Google Scholar] [CrossRef]
- He, T.; Li, K.; Wu, M.-Y.; Feng, X.-W.; Wang, N.; Wang, H.-Y.; Li, C.; Yu, X.-Q. Utilization of biocatalytic promiscuity for direct Mannich reaction. J. Mol. Catal. B Enzym. 2010, 67, 189–194. [Google Scholar] [CrossRef]
- Wu, L.-L.; Xiang, Y.; Yang, D.-C.; Guan, Z.; He, Y.-H. Biocatalytic asymmetric Mannich reaction of ketimines using wheat germ lipase. Catal. Sci. Technol. 2016, 6, 3963–3970. [Google Scholar] [CrossRef]
- Ding, X.; Dong, C.-L.; Guan, Z.; He, Y.-H. Concurrent asymmetric reactions combining photocatalysis and enzyme catalysis: Direct enantioselective synthesis of 2,2-disubstituted indol-3-ones from 2-arylindoles. Angew. Chem. 2019, 131, 124–130. [Google Scholar] [CrossRef]
- Xu, J.-C.; Li, W.-M.; Zheng, H.; Lai, Y.-F.; Zhang, P.-F. One-pot synthesis of tetrahydrochromene derivatives catalyzed by lipase. Tetrahedron 2011, 67, 9582–9587. [Google Scholar] [CrossRef]
- Chen, X.; Zhang, W.; Yang, F.; Guo, C.; Zhao, Z.; Ji, D.; Zhou, F.; Wang, Z.; Zhao, R.; Wang, L. Synthesis of dihydropyrano[4,3-b]pyranes via a multi-component reaction catalyzed by lipase. Green Chem. Lett. Rev. 2017, 10, 54–58. [Google Scholar] [CrossRef] [Green Version]
- Dalal, K.S.; Padvi, S.A.; Wagh, Y.B.; Dalal, D.S.; Chaudhari, B.L. Lipase from porcine pancreas: An efficient biocatalyst for the synthesis of ortho-aminocarbonitriles. Chem. Sel. 2018, 3, 10378–10382. [Google Scholar] [CrossRef]
- Yang, Z.-J.; Gong, Q.-T.; Wang, Y.; Yu, Y.; Liu, Y.-H.; Wang, N.; Yu, X.-Q. Biocatalytic tandem multicomponent reactions for one-pot synthesis of 2-amino-4H-pyran library and in vitro biological evaluation. Mol. Catal. 2020, 491, 110983. [Google Scholar] [CrossRef]
- Dalal, K.S.; Chaudhari, M.A.; Dalal, D.S.; Chaudhari, B.L. The first efficient biocatalytic route for the synthesis of Kojic acid derivatives in aqueous media. Catal. Comm. 2021, 152, 106289. [Google Scholar] [CrossRef]
- Yin, D.-H.; Liu, W.; Wang, Z.-X.; Huang, X.; Zhang, J.; Huang, D.-C. Enzyme-catalyzed direct three-component aza-Diels–Alder reaction using lipase from Candida sp. 99–125. Chin. Chem. Lett. 2017, 28, 153–158. [Google Scholar] [CrossRef]
- Wang, J.-L.; Chen, X.-Y.; Wu, Q.; Lin, X.-F. One-pot synthesis of spirooxazino derivatives via enzyme- initiated multicomponent reactions. Adv. Synth. Catal. 2014, 356, 999–1005. [Google Scholar] [CrossRef]
- Chen, X.-Y.; Wang, J.-L.; Lin, X.-F.; Wu, Q. Lipase-initiated one-pot synthesis of spirooxazino derivatives: Redesign of multicomponent reactions to expand substrate scope and application potential. Tetrahedron 2016, 72, 3318–3323. [Google Scholar] [CrossRef]
- Chen, X.-Y.; Hu, Y.-J.; Lin, X.-F.; Wu, Q. Asymmetric synthesis of strongly fluorescent spirooxazino derivatives via multi-enzymatic telescopic reactions. J. Mol. Catal. B Enzym. 2016, 133, S277–S280. [Google Scholar] [CrossRef]
- Wu, L.; Qin, L.; Nie, Y.; Xu, Y.; Zhao, Y.L. Computer-aided understanding and engineering of enzymatic selectivity. Biotechnol. Adv. 2022, 54, 107793. [Google Scholar] [CrossRef]
- Qu, G.; Li, A.; Acevedo-Rocha, C.G.; Sun, Z.; Reetz, M.T. The crucial role of methodology development in directed evolution of selective enzymes. Angew. Chem. Int. Ed. 2020, 59, 13204–13231. [Google Scholar] [CrossRef] [PubMed]





















Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Patti, A.; Sanfilippo, C. Stereoselective Promiscuous Reactions Catalyzed by Lipases. Int. J. Mol. Sci. 2022, 23, 2675. https://doi.org/10.3390/ijms23052675
Patti A, Sanfilippo C. Stereoselective Promiscuous Reactions Catalyzed by Lipases. International Journal of Molecular Sciences. 2022; 23(5):2675. https://doi.org/10.3390/ijms23052675
Chicago/Turabian StylePatti, Angela, and Claudia Sanfilippo. 2022. "Stereoselective Promiscuous Reactions Catalyzed by Lipases" International Journal of Molecular Sciences 23, no. 5: 2675. https://doi.org/10.3390/ijms23052675
APA StylePatti, A., & Sanfilippo, C. (2022). Stereoselective Promiscuous Reactions Catalyzed by Lipases. International Journal of Molecular Sciences, 23(5), 2675. https://doi.org/10.3390/ijms23052675