
Coenzyme A Restriction as a Factor Underlying Pre-Eclampsia with Polycystic Ovary Syndrome as a Risk Factor

Abstract
:1. Introduction
2. Results
2.1. Metabolomic Data Indicate Restricted Coenzyme A Levels and Signalling Lipid Formation
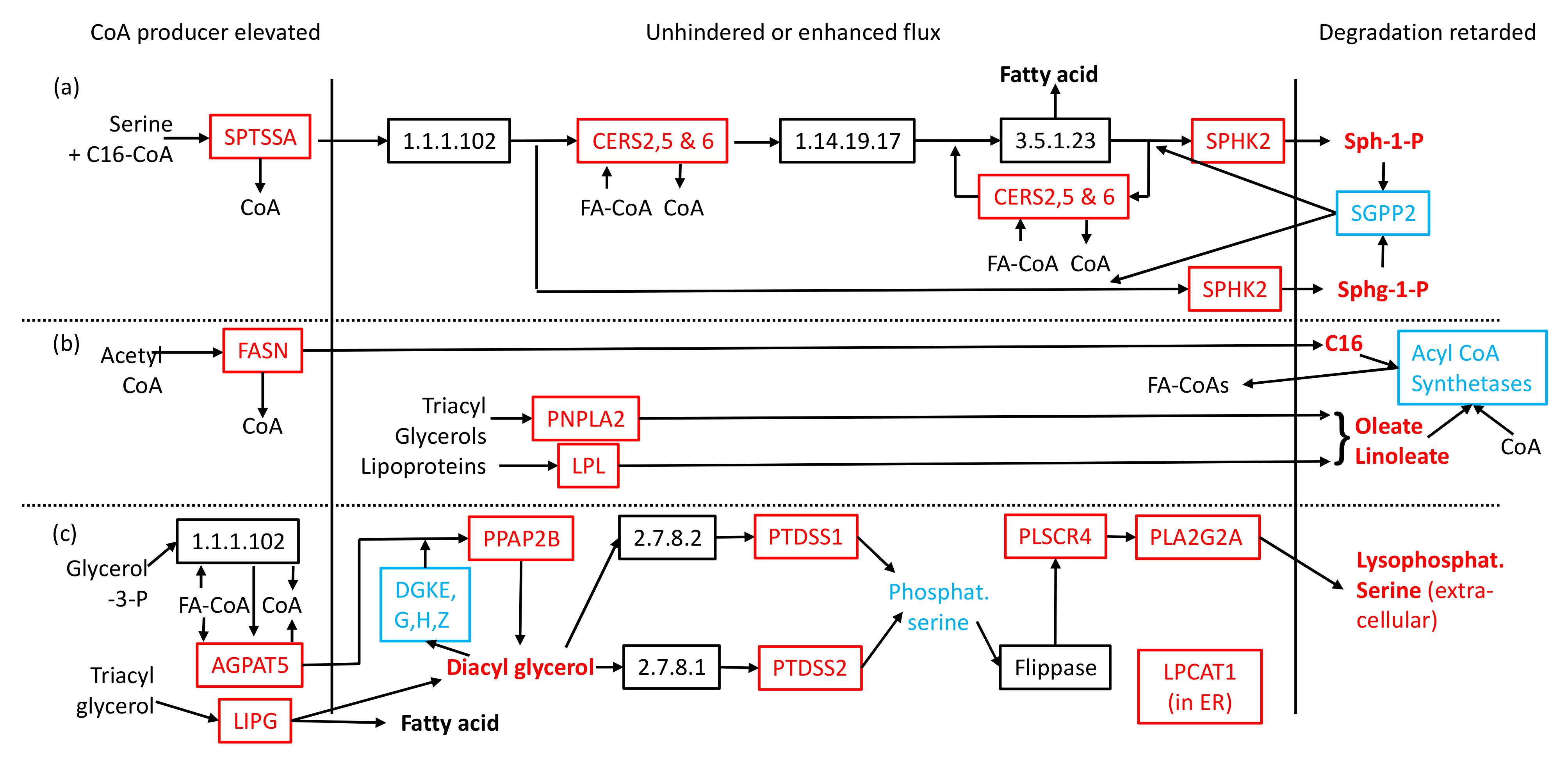
2.2. Transcriptomic Data in Metabolic Context Can Account for Reduction in Coenzyme A and Observed Lipid Perturbations
2.3. CoA Release and Changes to mRNA Levels Explain Elevated Signalling Lipids and Toxic Compounds
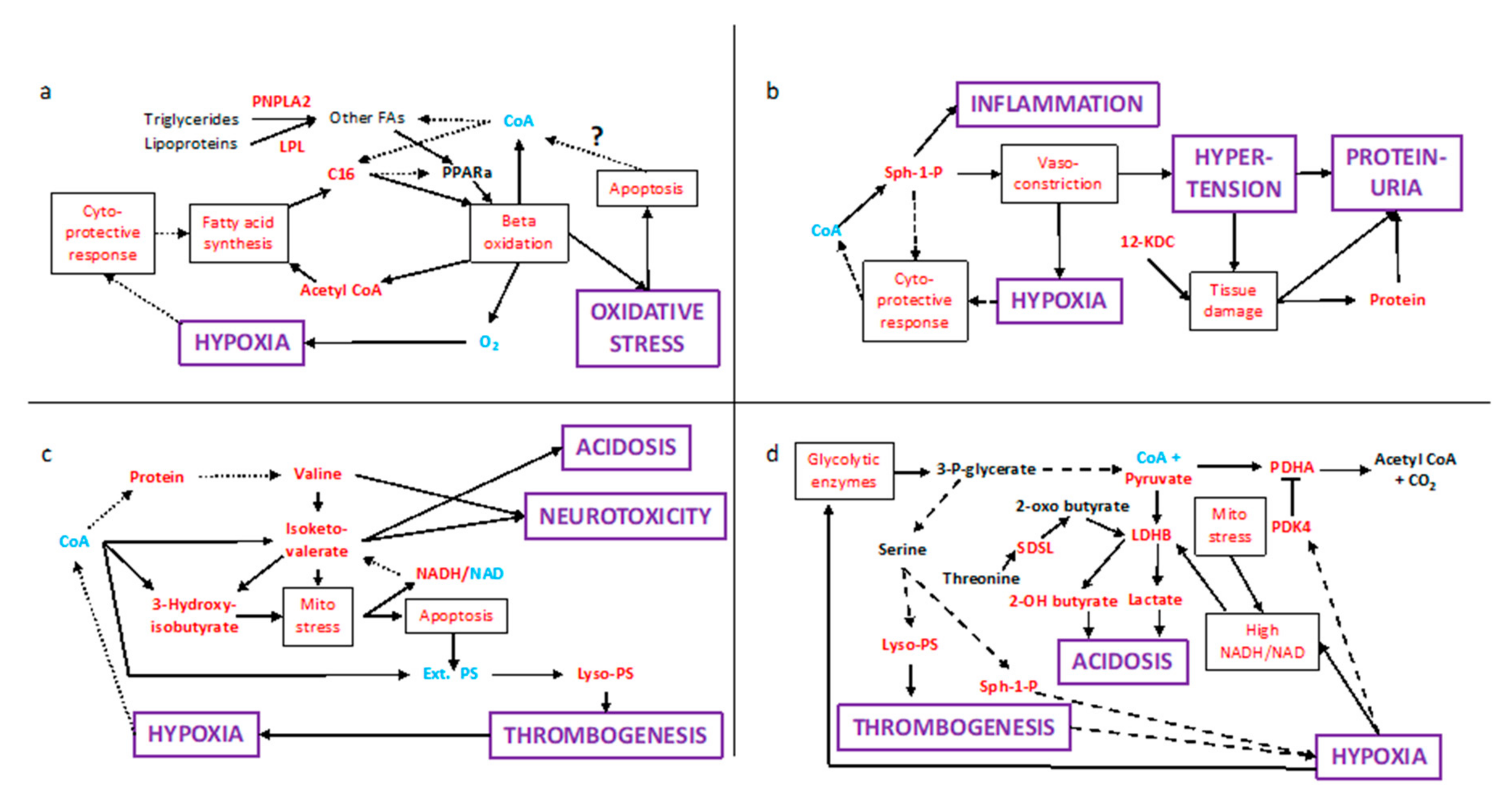
2.4. Multiple Conflicting Regulatory Pathways Arise from the Elevated Signalling Lipids
2.5. Population Variation
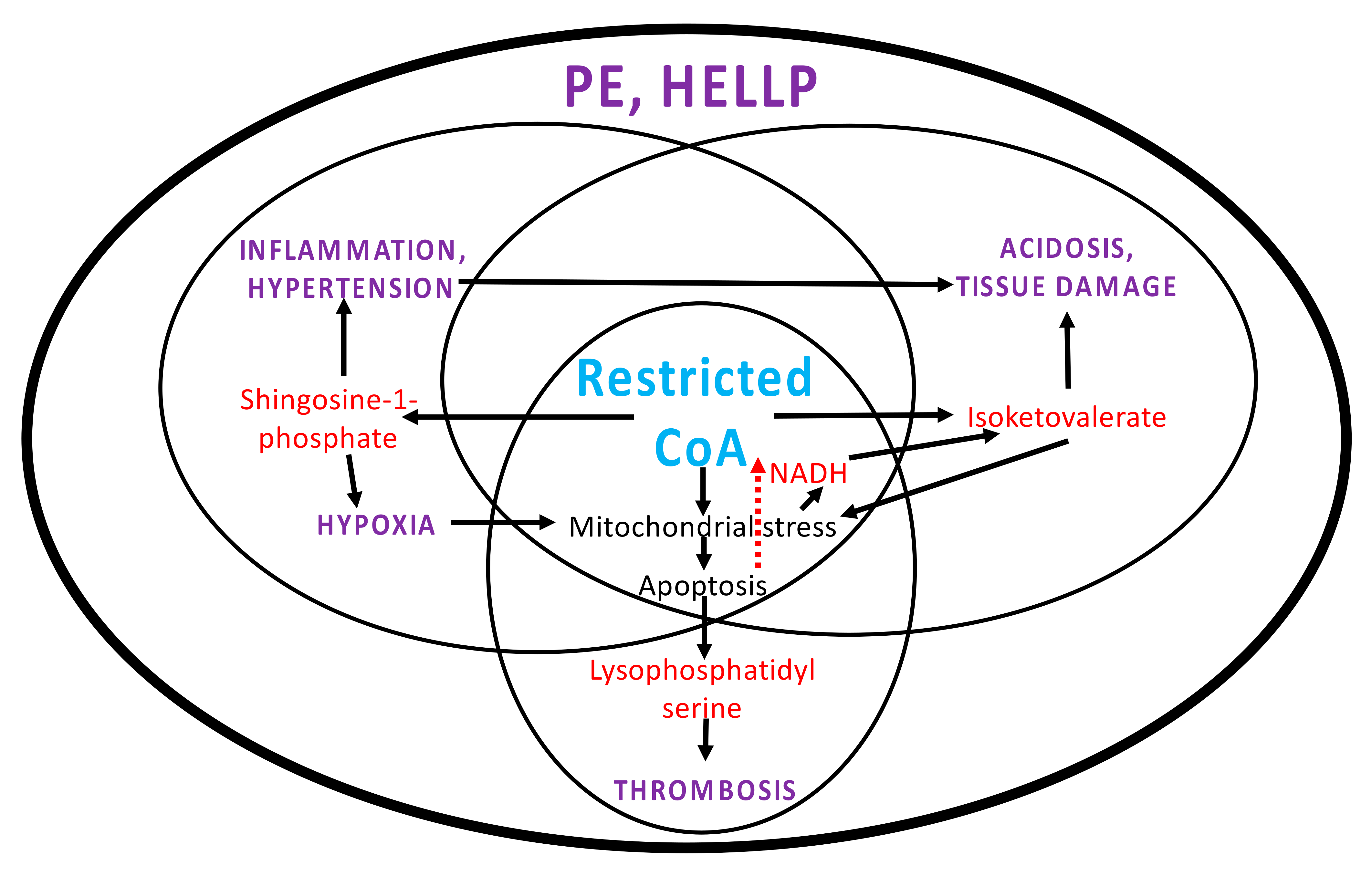
3. Discussion
3.1. Systems Pathology in PE
3.2. PCOS and Other Risk Factors
3.3. Apoptosis and CoA
3.4. Potential Avenues for Translation, Prevention and Treatment
4. Data and Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Burton, G.J.; Redman, C.W.; Roberts, J.M.; Moffett, A. Pre-eclampsia: Pathophysiology and clinical implications. BMJ 2019, 366, I2381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brosens, I.; Pijnenborg, R.; Vercruysse, L.; Romero, R. The “great Obstetrical Syndromes” are associated with disorders of deep placentation. Am. J. Obstet. Gynecol. 2011, 204, 193–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hubel, C.A. Oxidative Stress in the Pathogenesis of Preeclampsia. Proc. Soc. Exp. Biol. Med. 1999, 222, 222–235. [Google Scholar] [CrossRef] [PubMed]
- Tranquilli, A.L.; Brown, M.A.; Zeeman, G.G.; Dekker, G.; Sibai, B.M. The definition of severe and early-onset preeclampsia. Statements from the International Society for the Study of Hypertension in Pregnancy (ISSHP). Pregnancy Hypertens. 2012, 3, 44–47. [Google Scholar] [CrossRef] [PubMed]
- Brew, O.; Sullivan, M.H.F.; Woodman, A. Comparison of normal and pre-eclamptic placental gene expression: A systematic review with meta-analysis. PLoS ONE 2016, 11, e0161504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, G.H.; Galazis, N.; Docheva, N.; Layfield, R.; Atiomo, W. Overlap of proteomics biomarkers between women with pre-eclampsia and PCOS: A systematic review and biomarker database integration. Hum. Reprod. 2015, 30, 133–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acharjee, A.; Ament, Z.; West, J.A.; Stanley, E.; Griffin, J.L. Integration of metabolomics, lipidomics and clinical data using a machine learning method. BMC Bioinform. 2016, 17 (Suppl. S15), 37–49. [Google Scholar] [CrossRef] [Green Version]
- Teede, H.J.; Misso, M.L.; Costello, M.F.; Dokras, A.; Laven, J.; Moran, L.; Piltonen, T.; Norman, R.J. Recommendations from the international evidence-based guideline for the assessment and management of polycystic ovary syndrome. Hum. Reprod. 2018, 33, 1602–1618. [Google Scholar] [CrossRef] [Green Version]
- Boomsma, C.M.; Eijkemans, M.J.C.; Hughes, E.G.; Visser, G.H.A.; Fauser, B.C.J.M.; Macklon, N.S. A meta-analysis of pregnancy outcomes in women with polycystic ovary syndrome. Hum. Reprod. Update 2006, 12, 673–683. [Google Scholar] [CrossRef]
- Qin, J.Z.; Pang, L.H.; Li, M.J.; Fan, X.J.; Huang, R.D.; Chen, H.Y. Obstetric complications in women with polycystic ovary syndrome: A systematic review and meta-analysis. Reprod. Biol. Endocrinol. 2013, 11, 56. [Google Scholar] [CrossRef] [Green Version]
- Pan, H.; Xian, P.; Yang, D.; Zhang, C.; Tang, H.; He, X.; Lin, H.; Wen, X.; Ma, H.; Lai, M. Polycystic ovary syndrome is an independent risk factor for hypertensive disorders of pregnancy: A systematic review, meta-analysis, and meta-regression. Endocrine 2021, 74, 518–529. [Google Scholar] [CrossRef]
- Kelley, A.S.; Smith, Y.R.; Padmanabhan, V. A Narrative Review of Placental Contribution to Adverse Pregnancy Outcomes in Women with Polycystic Ovary Syndrome. J. Clin. Endocrinol. Metab. 2019, 104, 5299–5315. [Google Scholar] [CrossRef]
- Wishart, D.S.; Feunang, Y.D.; Marcu, A.; Guo, A.C.; Liang, K.; Vázquez-Fresno, R.; Sajed, T.; Johnson, D.; Li, C.; Karu, N.; et al. HMDB 4.0: The human metabolome database for 2018. Nucleic. Acids Res. 2018, 45, D608–D617. [Google Scholar] [CrossRef]
- Issemann, I.; Prince, R.; Tugwood, J.; Green, S. A role for fatty acids and liver fatty acid binding protein in peroxisome proliferation? Biochem. Soc. Trans. 1992, 20, 824–827. [Google Scholar] [CrossRef] [Green Version]
- Jozefczuk, E.; Guzik, T.J.; Siedlinski, M. Significance of sphingosine-1-phosphate in cardiovascular physiology and pathology. Pharmacol. Res. 2020, 156, 104793. [Google Scholar] [CrossRef]
- Stone, M.D.; Nelsestuen, G.L. Efficacy of soluble phospholipids in the prothrombinase reaction. Biochemistry 2005, 44, 4037–4041. [Google Scholar] [CrossRef]
- Davis, R.A.; Miyake, J.H.; Hui, T.Y.; Spann, N.J. Regulation of cholesterol-7α-hydroxylase: BAREly missing a SHP. J. Lipid Res. 2002, 43, 533–543. [Google Scholar] [CrossRef]
- Sousa, A.P.; Cunha, D.M.; Franco, C.; Teixeira, C.; Gojon, F.; Baylina, P.; Fernandes, R. Which Role Plays 2-Hydroxybutyric Acid on Insulin Resistance? Metabolites 2021, 11, 835. [Google Scholar] [CrossRef]
- Herold, A.H.; Sneed, K.B. Treatment of a Young Adult Taking Gamma-Butyrolactone (GBL) in a Primary Care Clinic. J. Am. Board Fam. Pract. 2002, 15, 161–163. [Google Scholar] [PubMed]
- Chalmers, R.A. Organic acids in urine of patients with congenital lactic acidoses: An aid to differential diagnosis. J. Inherit. Metab. Dis. 1984, 7, 79–89. [Google Scholar] [CrossRef]
- McGinnis, R.; Steinthorsdottir, V.; Williams, N.O.; Thorleifsson, G.; Shooter, S.; Hjartardottir, S.; Bumpstead, S.; Stefansdottir, L.; Hildyard, L.; Sigurdsson, J.K.; et al. Variants in the fetal genome near FLT1 are associated with risk of preeclampsia. Nat. Genet. 2017, 49, 1255–1260. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.P.; Brennecke, S.P.; East, C.E.; Göring, H.H.; Kent Jr, J.W.; Dyer, T.D.; Said, J.M.; Roten, L.T.; Iversen, A.C.; Abraham, L.J.; et al. Genome-wide association scan identifies a risk locus for preeclampsia on 2q14, near the inhibin, beta B gene. PLoS ONE 2012, 7, e33666. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Bracken, M.B.; DeWan, A.T. Genome-Wide Association Study of Pre-Eclampsia Detects Novel Maternal Single Nucleotide Polymorphisms and Copy-Number Variants in Subsets of the Hyperglycemia and Adverse Pregnancy Outcome (HAPO) Study Cohort. Ann. Hum. Genet. 2013, 77, 277–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, G.; Songdej, N.; Voora, D.; Goldfinger, L.E.; Del Carpio-Cano, F.E.; Myers, R.A.; Rao, A.K. Transcription factor RUNX1 regulates Platelet PCTP (phosphatidyl transfer protein): Implications for cardiovascular events. Circulation 2017, 136, 927–939. [Google Scholar] [CrossRef]
- Ramaswamy, G.; Karim, M.A.; Murti, K.G.; Jackowski, S. PPARalpha controls the intracellular Coenzyme A concentration via regulation of PANK1alpha gene expression. J. Lipid Res. 2004, 45, 17–31. [Google Scholar] [CrossRef] [Green Version]
- Yoon, M. PPARα in obesity: Sex difference and estrogen involvement. PPAR Res. 2010, 2010, 584296. [Google Scholar] [CrossRef] [Green Version]
- Jensen-Urstad, A.P.L.; Song, H.; Lodhi, I.J.; Funai, K.; Yin, L.; Coleman, T.; Semenkovich, C.F. Nutrient-dependent phosphorylation channels lipid synthesis to regulate PPARalpha. J. Lipid Res. 2013, 54, 1848–1859. [Google Scholar] [CrossRef] [Green Version]
- Kawano, Y.; Ersoy, B.A.; Li, Y.; Nishiumi, S.; Yoshida, M.; Cohen, D.E. Thioesterase superfamily member 2 (Them2) and Phosphatidylcholine Transfer Protein (PC-TP) Interact To Promote Fatty Acid Oxidation and Control Glucose Utilization. Mol. Cell. Biol. 2014, 34, 2396–2408. [Google Scholar] [CrossRef] [Green Version]
- Nie, C.; He, T.; Zhang, W.; Zhang, G.; Ma, X. Branched-chain amino acids: Beyond nutrition metabolism. Int. J. Mol. Sci. 2018, 19, 954. [Google Scholar] [CrossRef] [Green Version]
- Brosnan, J.T.; Brosnan, M.E. Branched-chain amino acids: Enzyme and substrate regulation. J. Nutr. 2006, 137, 207S–211S. [Google Scholar] [CrossRef]
- Kodigepalli, K.M.; Bowers, K.; Sharp, A.; Nanjundan, M. Roles and regulation of scramblases. FEBS Lett. 2015, 589, 3–14. [Google Scholar] [CrossRef] [Green Version]
- UniProt Annotation for PDK1_HUMAN. Available online: https://www.uniprot.org/uniprot/Q15118 (accessed on 24 January 2022).
- Connaughton, S.; Chowdhury, F.; Attia, R.R.; Song, S.; Zhang, Y.; Elam, M.B.; Cook, G.A.; Park, E.A. Regulation of pyruvate dehydrogenase kinase isoform 4 (PDK4) gene expression by glucocorticoids and insulin. Mol. Cell. Endocrinol. 2011, 315, 159. [Google Scholar] [CrossRef] [Green Version]
- Wu, P.; Peters, J.M.; Harris, R.A. Adaptive increase in pyruvate dehydrogenase kinase 4 during starvation is mediated by peroxisome proliferator-activated receptor alpha. Biochem. Biophys. Res. Commun. 2001, 287, 391–396. [Google Scholar] [CrossRef]
- Lee, J.H.; Kim, E.J.; Kim, D.K.; Lee, J.M.; Park, S.B.; Lee, I.K.; Harris, R.A.; Lee, M.O.; Choi, H.S. Hypoxia induces PDK4 gene expression through induction of the orphan nuclear receptor ERR γ. PLoS ONE 2012, 7, e46324. [Google Scholar]
- Lynch, C.J.; Adams, S.H. Branched-chain amino acids in metabolic signalling and insulin resistance. Nat. Rev. Endocrinol. 2014, 10, 723–726. [Google Scholar]
- Nelson, D.B.; Duraiswamy, S.; McIntire, D.D.; Mayo, M.J.; Leveno, K.J. Does preeclampsia involve the pancreas? A report of original research. J. Matern. Fetal Neonatal Med. 2015, 28, 836–838. [Google Scholar] [CrossRef]
- Haukland, H.H.; Florholmen, J.; Øian, P.; Maltau, J.M.; Burhol, P. The effect of pre-eclampsia on the pancreas: Changes in serum cationic trypsinogen and pancreatic amylase. Br. J. Obstet. Gynaecol. 1987, 97, 765–767. [Google Scholar] [CrossRef]
- Zaki, M.; Basha, W.; El-Bassyouni, H.T.; El-Toukhy, S.; Hussein, T. Evaluation of DNA damage profile in obese women and its association to risk of metabolic syndrome, polycystic ovary syndrome and recurrent preeclampsia. Genes Diseases 2018, 5, 367–373. [Google Scholar] [CrossRef]
- Lokeswara, A.W.; Hiksas, R.; Irwinda, R.; Wibowo, N. Preeclampsia: From Cellular Wellness to Inappropriate Cell Death, and the Roles of Nutrition. Front. Cell Dev. Biol. 2021, 9, 726513. [Google Scholar] [CrossRef]
- Wojtczak, L.; Slyshenkov, V.S. Protection by pantothenic acid against apoptosis and cell damage by oxygen free radicals—The role of glutathione. BioFactors 2003, 17, 61–73. [Google Scholar] [CrossRef]
- Cao, S.; Xu, W.; Zhang, N.; Wang, Y.; Luo, Y.; He, X.; Huang, K. A mitochondria-dependent pathway mediates the apoptosis of gse-induced yeast. PLoS ONE 2012, 7, e32943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shurubor, Y.I.; D’Aurelio, M.; Clark-Matott, J.; Isakova, E.P.; Deryabina, Y.I.; Beal, M.F.; Cooper, A.J.L.; Krasnikov, B.F. Determination of Coenzyme A and acetyl-Coenzyme A in biological samples using HPLC with UV detection. Molecules 2017, 22, 1388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Catov, J.M.; Nohr, E.A.; Bodnar, L.M.; Knudson, V.K.; Olsen, S.F.; Olsen, J. Association of periconceptional multivitamin use with reduced risk of preeclampsia among normal-weight women in the Danish National Birth Cohort. Am. J. Epidemiol. 2009, 169, 1304–1311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, L.K.; Subramanian, C.; Yun, M.K.; Frank, M.W.; White, S.W.; Rock, C.O.; Lee, R.E.; Jackowski, S. A therapeutic approach to pantothenate kinase associated neurodegeneration. Nat. Commun. 2018, 9, 4399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Czumaj, A.; Szrok-Jurga, S.; Hebanowska, A.; Turyn, J.; Swierczynski, J.; Sledzinski, T.; Stelmanska, E. The pathophysiological role of CoA. Int. J. Mol. Sci. 2020, 21, 9057. [Google Scholar] [CrossRef]
- Ishimwe, J.A. Maternal microbiome in preeclampsia pathophysiology and implications on offspring health. Physiol. Rep. 2021, 9, e14875. [Google Scholar] [CrossRef]
- Nordqvist, M.; Jacobsson, B.; Brantsæter, A.L.; Myhre, R.; Nilsson, S.; Sengpiel, V. Timing of probiotic milk consumption during pregnancy and effects on the incidence of preeclampsia and preterm delivery: A prospective observational cohort study in Norway. BMJ Open 2018, 8, e018021. [Google Scholar] [CrossRef] [Green Version]
- Kenny, L.C.; Broadhurst, D.I.; Dunn, W.; Brown, M.; North, R.A.; McCowan, L.; Roberts, C.; Cooper, G.J.S.; Kell, D.B.; Baker, P.N. Robust early pregnancy prediction of later preeclampsia using metabolomic biomarkers. Hypertension 2010, 56, 741–749. [Google Scholar] [CrossRef] [Green Version]
- Bahado-Singh, R.O.; Syngelaki, A.; Mandal, R.; Graham, S.F.; Akolekar, R.; Han, B.; Bjondahl, T.C.; Dong, E.; Bauer, S.; Alpay-Savasan, Z.; et al. Metabolic determination of pathogenesis of late-onset preeclampsia. J. Matern. Fetal Neonatal Med. 2017, 30, 658–664. [Google Scholar] [CrossRef]
- Odibo, A.O.; Goetzinger, K.R.; Odibo, L.; Cahill, A.G.; Macones, G.A.; Nelson, D.M.; Dietzen, D.J. First-trimester prediction of preeclampsia using metabolomic biomarkers: A discovery phase study. Pregnat. Diagn. 2011, 31, 990–994. [Google Scholar] [CrossRef] [Green Version]
- Dunn, W.B.; Brown, M.; Worton, S.A.; Crocker, I.P.; Broadhurst, D.; Horgan, R.; Kenny, L.C.; Baker, P.N.; Kell, D.B.; Heazell, A.E.P. Changes in the metabolic footprint of placental explant-conditioned culture medium identifies metabolic disturbances related to hypoxia and pre-eclampsia. Placenta 2009, 30, 974–980. [Google Scholar] [CrossRef]
- Uniprot Consortium. UniProt: The universal protein knowledgebase in 2021. Nucleic. Acids Res. 2021, 49, D1. [Google Scholar]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic. Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Molecule. ** Denotes Membership of the Kenny PE-Predictive Set (14–16 wks.) | Comment | Human Metabolome DataBase ID [13]/LipidMaps ID |
|---|---|---|
| Linoleic acid Oleic acid ** Hexadecanoic acid | Linoleate and oleate are activating ligands of peroxisome-proliferator activating receptor-alpha (PPARa), which stimulates peroxisome formation and beta-oxidation [14] | HMDB0000673 HMDB0000207 HMDB0000220 |
| Sphingosine-1-phosphate ** Sphinganine-1-phosphate ** | Through their receptors mediate a wide range of responses including vasoconstriction, inflammation and cytoprotection [15] | HMDB0000277 HMDB0001383 |
| Eicosatrienoic acid | Can be synthesised in the endoplasmic reticulum by fatty-acyl elongation. Competes with arachidonic acid for cytochrome oxidase and lipoxygenase activities. | HMDB0002925 |
| Octadecenoyl-sn-glycero-3-phosphoserine Dioctanoyl-sn-glycero-3-phosphocholine | Lysophosphatidyl serines and phosphatidyl cholines promote thrombogenesis in the context of lipid bilayers [16] | HMDB0240603 LMGP01011251 |
| Hexadecenoyl-eicosatetraenoyl-sn-glycerol ** Di-(octadecadienoyl_sn-glycerol ** | Diacyl glycerols allosterically activate Protein Kinase C, exerting diverse physiological effects | HMDB0007141 LMGL02010063 |
| 12-Ketodeoxycholic acid | Ketodeoxycholates are membrane disruptors. Elevated levels presumably due to dysregulation by liver [17] | HMDB0000328 |
| Oxo-methyl butanoic acid ** | Valine degradation product also known as isoketovaleric acid: Neurotoxic, acidogen and metabotoxic | HMDB0000019 |
| 3-hydroxybutanoic acid | Comprises 2 isomers. One is a ketone body. The other (3-hydroxy-isobutanoic acid) is a valine degradation product causing mitochondrial stress and lactic acidaemia | HMDB0000442 HMDB0000023 |
| 3-Hydroxyisovaleric acid | Leucine degradation product; can cause mitochondrial stress, acidosis and metabotoxicity | HMDB0000754 |
| 2-hydroxybutanoic acid | Product of threonine degradation and glutathione synthesis. Marker for insulin resistance and acidosis, indicative of high NADH/NAD ratio [18] | HMDB0000008 |
| Gamma-butyrolactone ** | Hydrolyses to gamma-hydroxybutyrate (HMDB0000710) which affects the nervous system, and at high concentrations is an acidogen and neurotoxin [19] | HMDB0000549 |
| Oxolan-3-one ** | Marker for lactic acid acidosis [20] | HMDB0002523 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hodgman, C.; Khan, G.H.; Atiomo, W. Coenzyme A Restriction as a Factor Underlying Pre-Eclampsia with Polycystic Ovary Syndrome as a Risk Factor. Int. J. Mol. Sci. 2022, 23, 2785. https://doi.org/10.3390/ijms23052785
Hodgman C, Khan GH, Atiomo W. Coenzyme A Restriction as a Factor Underlying Pre-Eclampsia with Polycystic Ovary Syndrome as a Risk Factor. International Journal of Molecular Sciences. 2022; 23(5):2785. https://doi.org/10.3390/ijms23052785
Chicago/Turabian StyleHodgman, Charlie, Gulafshana Hafeez Khan, and William Atiomo. 2022. "Coenzyme A Restriction as a Factor Underlying Pre-Eclampsia with Polycystic Ovary Syndrome as a Risk Factor" International Journal of Molecular Sciences 23, no. 5: 2785. https://doi.org/10.3390/ijms23052785
APA StyleHodgman, C., Khan, G. H., & Atiomo, W. (2022). Coenzyme A Restriction as a Factor Underlying Pre-Eclampsia with Polycystic Ovary Syndrome as a Risk Factor. International Journal of Molecular Sciences, 23(5), 2785. https://doi.org/10.3390/ijms23052785