
Inflammation and Bone Metabolism in Rheumatoid Arthritis: Molecular Mechanisms of Joint Destruction and Pharmacological Treatments

Abstract
:1. Introduction
2. Physiological Bone Metabolism
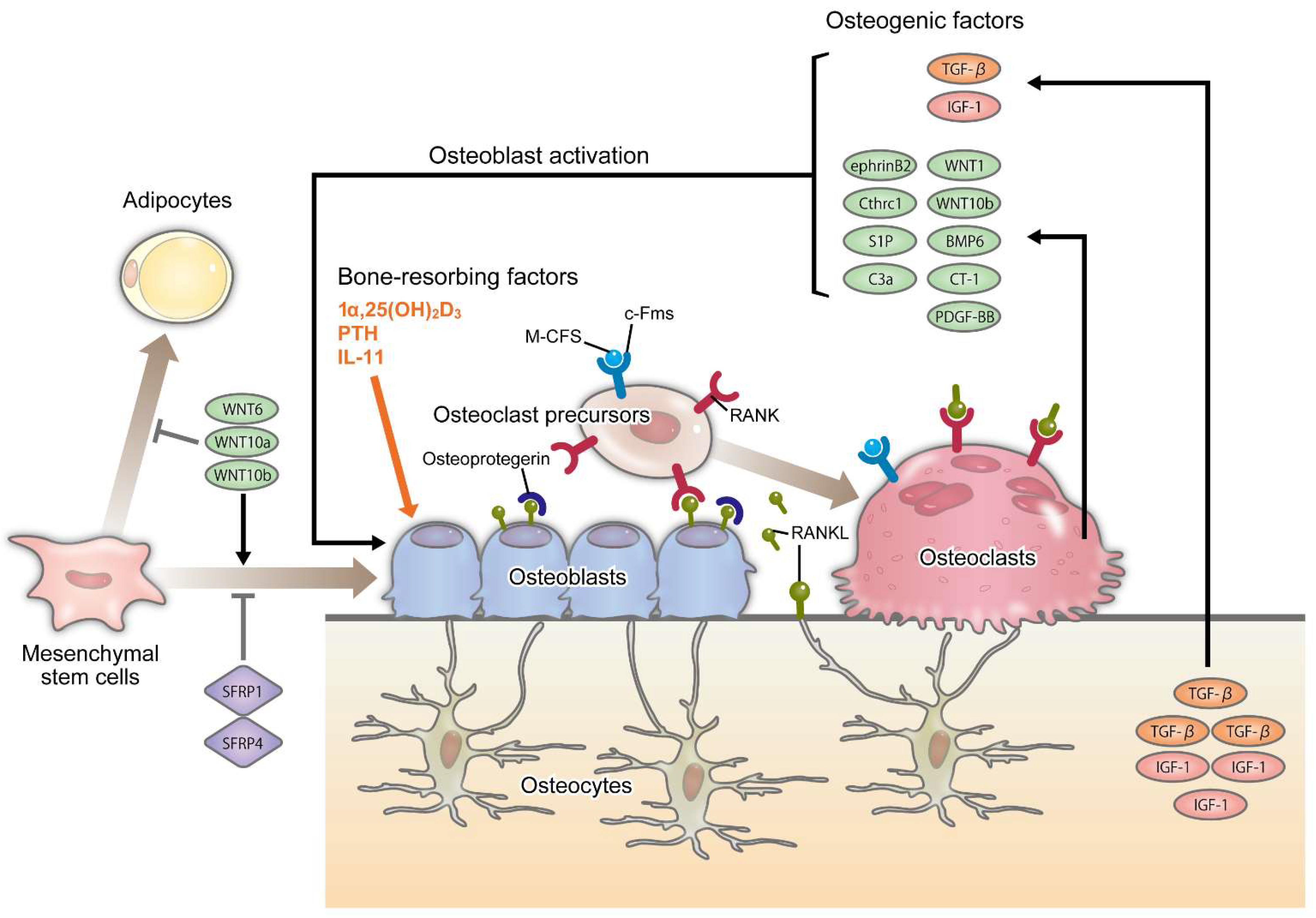
2.1. Bone Remodeling
2.2. Osteoblasts and Osteocytes
2.3. Osteoclasts and Bone Resorption
2.4. Coupling Factors in Bone Metabolism
3. Pathological Bone Metabolism in RA
3.1. Predisposing Factors and Associated Systemic Effects
3.2. Factors Associated with Local Effects on Joints
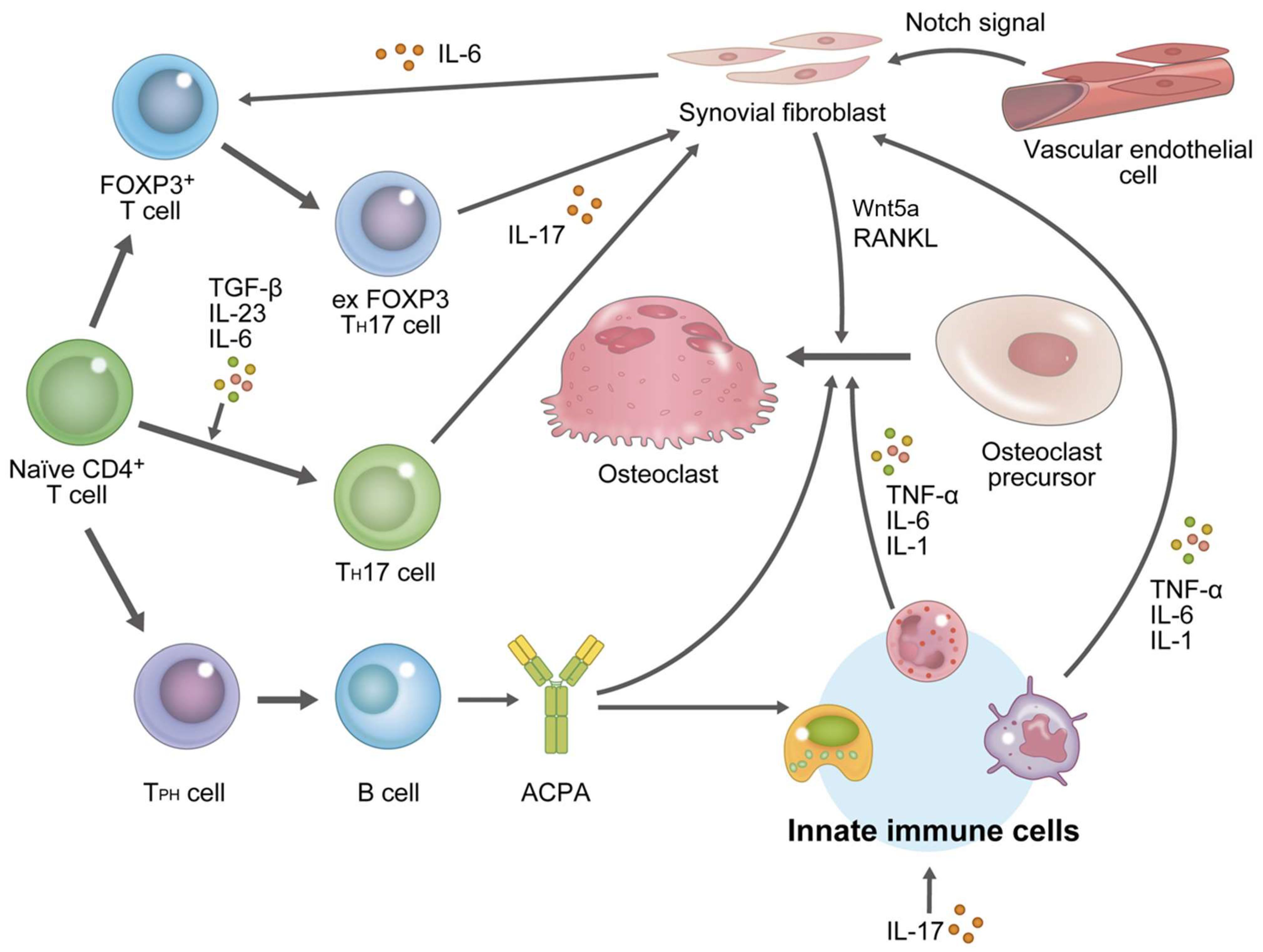
3.2.1. Differentiation of Osteoclasts under RA Condition
3.2.2. Effects of T Cells on Osteoarticular Destruction
3.2.3. Effects of B Cells on Osteoarticular Destruction
3.2.4. Effects of Synovial Fibroblasts on Osteoarticular Destruction
4. Effects of RA Treatment on Bone Metabolism
4.1. csDMARDs (Conventional Synthetic Disease-Modifying Antirheumatic Drugs)
4.1.1. Preclinical Studies
4.1.2. Clinical Studies
4.2. TNF-α Inhibitors
4.2.1. Preclinical Studies
4.2.2. Clinical Studies
4.3. IL-6 Inhibitors
4.3.1. Preclinical Studies
4.3.2. Clinical Studies
4.4. CTLA-4 Ig
4.4.1. Preclinical Studies
4.4.2. Clinical Studies
4.5. JAK Inhibitors
4.5.1. Preclinical Studies
4.5.2. Clinical Studies
4.6. Other Treatments
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 1α,25(OH)2D3 | 1α,25-dihydroxy-vitamin D3 |
| ABT | abatacept |
| ACPA | anticitrullinated protein antibody |
| ADA | adalimumab |
| AtoM | arthritis-associated osteoclastogenic macrophages |
| BAR | baricitinib |
| bDMARDs | biological disease-modifying antirheumatic drugs |
| CIA | collagen-induced arthritis |
| csDMARDs | conventional synthetic disease-modifying antirheumatic drugs |
| CTLA-4 | cytotoxic T-lymphocyte-associated protein-4 |
| CZP | certolizumab pegol |
| DC-OC | dendritic cell-derived osteoclast |
| DKK1 | dickkopf1 |
| ETN | etanercept |
| FIL | filgotinib |
| FLS | fibroblast-like synoviocyte |
| Fox | forkhead box protein |
| IFX | infliximab |
| M-CSF | macrophage colony-stimulating factor |
| Mo-OC | monocyte-derived osteoclast |
| mTORC1 | mammalian target of rapamycin complex 1 |
| MTX | methotrexate |
| NFATc1 | nuclear factor of activated T cell c1 |
| OPG | osteoprotegerin |
| PEF | peficitinib |
| RA | rheumatoid arthritis |
| RANK | receptor activator of nuclear factor-κB |
| RANKL | receptor activator of nuclear factor-κB ligand |
| Runx | runt-related transcription factor |
| S1P | sphingosine-1-phosphate |
| SAR | sarilumab |
| sFRP | secreted Frizzled-related protein |
| TCZ | tocilizumab |
| TGF-b | transforming growth factor-beta |
| TNF-a | tumor necrosis factor-alpha |
| TOF | tofacitinib |
| tsDMARDs | targeted synthetic disease-modifying antirheumatic drugs |
| UPA | upadacitinib |
| Wnt | wingless-related MMTV integration site |
References
- Smolen, J.S.; Aletaha, D.; Barton, A.; Burmester, G.R.; Emery, P.; Firestein, G.S.; Kavanaugh, A.; McInnes, I.B.; Solomon, D.H.; Strand, V.; et al. Rheumatoid arthritis. Nat. Rev. Dis. Primers 2018, 4, 18001. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Wang, Y.; Xu, D.; Nossent, J.; Pavlos, N.J.; Xu, J. Rheumatoid arthritis: Pathological mechanisms and modern pharmacologic therapies. Bone Res. 2018, 6, 15. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, A.; Sakai, R.; Inoue, E.; Harigai, M. Prevalence of patients with rheumatoid arthritis and age-stratified trends in clinical characteristics and treatment, based on the National Database of Health Insurance Claims and Specific Health Checkups of Japan. Int. J. Rheum. Dis. 2020, 23, 1676–1684. [Google Scholar] [CrossRef] [PubMed]
- Goldring, S.R. Inflammatory signaling induced bone loss. Bone 2015, 80, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, K.; Takayanagi, H. Osteoimmunology. Cold Spring Harb. Perspect. Med. 2019, 9, a031245. [Google Scholar] [CrossRef]
- Emery, P.; Smolen, J.S.; Ganguli, A.; Meerwein, S.; Bao, Y.; Kupper, H.; Chen, N.; Kavanaugh, A. Effect of adalimumab on the work-related outcomes scores in patients with early rheumatoid arthritis receiving methotrexate. Rheumatology 2016, 55, 1458–1465. [Google Scholar] [CrossRef] [Green Version]
- Takeuchi, T.; Nakajima, R.; Komatsu, S.; Yamazaki, K.; Nakamura, T.; Agata, N.; Igarashi, A.; Tango, T.; Tanaka, Y. Impact of Adalimumab on Work Productivity and Activity Impairment in Japanese Patients with Rheumatoid Arthritis: Large-Scale, Prospective, Single-Cohort ANOUVEAU Study. Adv. Ther. 2017, 34, 686–702. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, Y.; Kameda, H.; Saito, K.; Kaneko, Y.; Tanaka, E.; Yasuda, S.; Tamura, N.; Fujio, K.; Fujii, T.; Kojima, T.; et al. Effect of subcutaneous tocilizumab treatment on work/housework status in biologic-naïve rheumatoid arthritis patients using inverse probability of treatment weighting: FIRST ACT-SC study. Arthritis Res. Ther. 2018, 20, 151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michaud, K.; Pope, J.E.; Emery, P.; Zhu, B.; Gaich, C.L.; DeLozier, A.M.; Zhang, X.; Dickson, C.L.; Smolen, J.S. Relative Impact of Pain and Fatigue on Work Productivity in Patients with Rheumatoid Arthritis from the RA-BEAM Baricitinib Trial. Rheumatol. Ther. 2019, 6, 409–419. [Google Scholar] [CrossRef] [Green Version]
- Smolen, J.S.; Landewé, R.B.M.; Bijlsma, J.W.J.; Burmester, G.R.; Dougados, M.; Kerschbaumer, A.; McInnes, I.B.; Sepriano, A.; van Vollenhoven, R.F.; de Wit, M.; et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2019 update. Ann. Rheum. Dis. 2020, 79, 685–699. [Google Scholar] [CrossRef] [Green Version]
- Van der Heijde, D.M. Plain X-rays in rheumatoid arthritis: Overview of scoring methods, their reliability and applicability. Bailliere’s Clin. Rheumatol. 1996, 10, 435–453. [Google Scholar] [CrossRef]
- Fries, J.F.; Spitz, P.; Kraines, R.G.; Holman, H.R. Measurement of patient outcome in arthritis. Arthritis Rheum. 1980, 23, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Danks, L.; Komatsu, N.; Guerrini, M.M.; Sawa, S.; Armaka, M.; Kollias, G.; Nakashima, T.; Takayanagi, H. RANKL expressed on synovial fibroblasts is primarily responsible for bone erosions during joint inflammation. Ann. Rheum. Dis. 2016, 75, 1187–1195. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Xia, S.; Fu, B. RNA-seq analysis of synovial fibroblasts in human rheumatoid arthritis. Mol. Med. Rep. 2014, 10, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Malemud, C.J. Matrix Metalloproteinases and Synovial Joint Pathology. Prog. Mol. Biol. Transl. Sci. 2017, 148, 305–325. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, R.; Hata, K.; Takahata, Y.; Murakami, T.; Nakamura, E.; Ohkawa, M.; Ruengsinpinya, L. Role of Signal Transduction Pathways and Transcription Factors in Cartilage and Joint Diseases. Int. J. Mol. Sci. 2020, 21, 1340. [Google Scholar] [CrossRef] [Green Version]
- Robling, A.G.; Bonewald, L.F. The Osteocyte: New Insights. Annu. Rev. Physiol. 2020, 82, 485–506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takashi, Y.; Fukumoto, S. Phosphate-sensing and regulatory mechanism of FGF23 production. J. Endocrinol. Investig. 2020, 43, 877–883. [Google Scholar] [CrossRef]
- Manolagas, S.C.; Parfitt, A.M. What old means to bone. Trends Endocrinol. Metab. 2010, 21, 369–374. [Google Scholar] [CrossRef]
- Sims, N.A.; Martin, T.J. Osteoclasts Provide Coupling Signals to Osteoblast Lineage Cells Through Multiple Mechanisms. Annu. Rev. Physiol. 2020, 82, 507–529. [Google Scholar] [CrossRef]
- Udagawa, N.; Koide, M.; Nakamura, M.; Nakamichi, Y.; Yamashita, T.; Uehara, S.; Kobayashi, Y.; Furuya, Y.; Yasuda, H.; Fukuda, C.; et al. Osteoclast differentiation by RANKL and OPG signaling pathways. J. Bone Miner. Metab. 2021, 39, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Zeng, H.C.; Bae, Y.; Dawson, B.C.; Chen, Y.; Bertin, T.; Munivez, E.; Campeau, P.M.; Tao, J.; Chen, R.; Lee, B.H. MicroRNA miR-23a cluster promotes osteocyte differentiation by regulating TGF-β signalling in osteoblasts. Nat. Commun. 2017, 8, 15000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ducy, P.; Zhang, R.; Geoffroy, V.; Ridall, A.L.; Karsenty, G. Osf2/Cbfa1: A transcriptional activator of osteoblast differentiation. Cell 1997, 89, 747–754. [Google Scholar] [CrossRef] [Green Version]
- Komori, T.; Yagi, H.; Nomura, S.; Yamaguchi, A.; Sasaki, K.; Deguchi, K.; Shimizu, Y.; Bronson, R.T.; Gao, Y.H.; Inada, M.; et al. Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell 1997, 89, 755–764. [Google Scholar] [CrossRef] [Green Version]
- Komori, T. Molecular Mechanism of Runx2-Dependent Bone Development. Mol. Cells 2020, 43, 168–175. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, K.; Zhou, X.; Kunkel, G.; Zhang, Z.; Deng, J.M.; Behringer, R.R.; de Crombrugghe, B. The novel zinc finger-containing transcription factor osterix is required for osteoblast differentiation and bone formation. Cell 2002, 108, 17–29. [Google Scholar] [CrossRef] [Green Version]
- Baron, R.; Kneissel, M. WNT signaling in bone homeostasis and disease: From human mutations to treatments. Nat. Med. 2013, 19, 179–192. [Google Scholar] [CrossRef] [PubMed]
- Maeda, K.; Takahashi, N.; Kobayashi, Y. Roles of Wnt signals in bone resorption during physiological and pathological states. J. Mol. Med. 2013, 91, 15–23. [Google Scholar] [CrossRef]
- Katagiri, T.; Watabe, T. Bone Morphogenetic Proteins. Cold Spring Harb. Perspect. Biol. 2016, 8, a021899. [Google Scholar] [CrossRef] [Green Version]
- Jann, J.; Gascon, S.; Roux, S.; Faucheux, N. Influence of the TGF-β Superfamily on Osteoclasts/Osteoblasts Balance in Physiological and Pathological Bone Conditions. Int. J. Mol. Sci. 2020, 21, 7597. [Google Scholar] [CrossRef]
- Wang, F.; Tarkkonen, K.; Nieminen-Pihala, V.; Nagano, K.; Majidi, R.A.; Puolakkainen, T.; Rummukainen, P.; Lehto, J.; Roivainen, A.; Zhang, F.P.; et al. Mesenchymal Cell-Derived Juxtacrine Wnt1 Signaling Regulates Osteoblast Activity and Osteoclast Differentiation. J. Bone Miner. Res. 2019, 34, 1129–1142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, F.; Rummukainen, P.; Heino, T.J.; Kiviranta, R. Osteoblastic Wnt1 regulates periosteal bone formation in adult mice. Bone 2021, 143, 115754. [Google Scholar] [CrossRef] [PubMed]
- Bennett, C.N.; Longo, K.A.; Wright, W.S.; Suva, L.J.; Lane, T.F.; Hankenson, K.D.; MacDougald, O.A. Regulation of osteoblastogenesis and bone mass by Wnt10b. Proc. Natl. Acad. Sci. USA 2005, 102, 3324–3329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bennett, C.N.; Ouyang, H.; Ma, Y.L.; Zeng, Q.; Gerin, I.; Sousa, K.M.; Lane, T.F.; Krishnan, V.; Hankenson, K.D.; MacDougald, O.A. Wnt10b increases postnatal bone formation by enhancing osteoblast differentiation. J. Bone Miner. Res. 2007, 22, 1924–1932. [Google Scholar] [CrossRef] [PubMed]
- Huybrechts, Y.; Mortier, G.; Boudin, E.; Van Hul, W. WNT Signaling and Bone: Lessons From Skeletal Dysplasias and Disorders. Front. Endocrinol. 2020, 11, 165. [Google Scholar] [CrossRef] [Green Version]
- Cawthorn, W.P.; Bree, A.J.; Yao, Y.; Du, B.; Hemati, N.; Martinez-Santibanez, G.; MacDougald, O.A. Wnt6, Wnt10a and Wnt10b inhibit adipogenesis and stimulate osteoblastogenesis through a beta-catenin-dependent mechanism. Bone 2012, 50, 477–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Y.; Patil, S.; Jia, J. The Development of Molecular Biology of Osteoporosis. Int. J. Mol. Sci. 2021, 22, 8182. [Google Scholar] [CrossRef]
- Nakanishi, R.; Akiyama, H.; Kimura, H.; Otsuki, B.; Shimizu, M.; Tsuboyama, T.; Nakamura, T. Osteoblast-targeted expression of Sfrp4 in mice results in low bone mass. J. Bone Miner. Res. 2008, 23, 271–277. [Google Scholar] [CrossRef]
- Yao, W.; Cheng, Z.; Shahnazari, M.; Dai, W.; Johnson, M.L.; Lane, N.E. Overexpression of secreted frizzled-related protein 1 inhibits bone formation and attenuates parathyroid hormone bone anabolic effects. J. Bone Miner. Res. 2010, 25, 190–199. [Google Scholar] [CrossRef] [Green Version]
- Yamada, A.; Iwata, T.; Yamato, M.; Okano, T.; Izumi, Y. Diverse functions of secreted frizzled-related proteins in the osteoblastogenesis of human multipotent mesenchymal stromal cells. Biomaterials 2013, 34, 3270–3278. [Google Scholar] [CrossRef]
- Nakashima, T.; Hayashi, M.; Fukunaga, T.; Kurata, K.; Oh-Hora, M.; Feng, J.Q.; Bonewald, L.F.; Kodama, T.; Wutz, A.; Wagner, E.F.; et al. Evidence for osteocyte regulation of bone homeostasis through RANKL expression. Nat. Med. 2011, 17, 1231–1234. [Google Scholar] [CrossRef] [PubMed]
- Xiong, J.; Onal, M.; Jilka, R.L.; Weinstein, R.S.; Manolagas, S.C.; O’Brien, C.A. Matrix-embedded cells control osteoclast formation. Nat. Med. 2011, 17, 1235–1241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Powell, W.F., Jr.; Barry, K.J.; Tulum, I.; Kobayashi, T.; Harris, S.E.; Bringhurst, F.R.; Pajevic, P.D. Targeted ablation of the PTH/PTHrP receptor in osteocytes impairs bone structure and homeostatic calcemic responses. J. Endocrinol. 2011, 209, 21–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delgado-Calle, J.; Tu, X.; Pacheco-Costa, R.; McAndrews, K.; Edwards, R.; Pellegrini, G.G.; Kuhlenschmidt, K.; Olivos, N.; Robling, A.; Peacock, M.; et al. Control of Bone Anabolism in Response to Mechanical Loading and PTH by Distinct Mechanisms Downstream of the PTH Receptor. J. Bone Miner. Res. 2017, 32, 522–535. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, N.; Conway, S.J.; Ferrari, S.L. Regulation of beta catenin signaling and parathyroid hormone anabolic effects in bone by the matricellular protein periostin. Proc. Natl. Acad. Sci. USA 2012, 109, 15048–15053. [Google Scholar] [CrossRef] [Green Version]
- Bonnet, N.; Standley, K.N.; Bianchi, E.N.; Stadelmann, V.; Foti, M.; Conway, S.J.; Ferrari, S.L. The matricellular protein periostin is required for sost inhibition and the anabolic response to mechanical loading and physical activity. J. Biol. Chem. 2009, 284, 35939–35950. [Google Scholar] [CrossRef] [Green Version]
- Walker, E.C.; McGregor, N.E.; Poulton, I.J.; Solano, M.; Pompolo, S.; Fernandes, T.J.; Constable, M.J.; Nicholson, G.C.; Zhang, J.G.; Nicola, N.A.; et al. Oncostatin M promotes bone formation independently of resorption when signaling through leukemia inhibitory factor receptor in mice. J. Clin. Investig. 2010, 120, 582–592. [Google Scholar] [CrossRef] [Green Version]
- Koide, M.; Kobayashi, Y.; Yamashita, T.; Uehara, S.; Nakamura, M.; Hiraoka, B.Y.; Ozaki, Y.; Iimura, T.; Yasuda, H.; Takahashi, N.; et al. Bone Formation Is Coupled to Resorption Via Suppression of Sclerostin Expression by Osteoclasts. J. Bone Min. Res. 2017, 32, 2074–2086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koide, M.; Kobayashi, Y. Regulatory mechanisms of sclerostin expression during bone remodeling. J. Bone Min. Metab. 2019, 37, 9–17. [Google Scholar] [CrossRef]
- Udagawa, N.; Takahashi, N.; Akatsu, T.; Tanaka, H.; Sasaki, T.; Nishihara, T.; Koga, T.; Martin, T.J.; Suda, T. Origin of osteoclasts: Mature monocytes and macrophages are capable of differentiating into osteoclasts under a suitable microenvironment prepared by bone marrow-derived stromal cells. Proc Natl. Acad. Sci. USA 1990, 87, 7260–7264. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, N.; Akatsu, T.; Udagawa, N.; Sasaki, T.; Yamaguchi, A.; Moseley, J.M.; Martin, T.J.; Suda, T. Osteoblastic cells are involved in osteoclast formation. Endocrinology 1988, 123, 2600–2602. [Google Scholar] [CrossRef] [PubMed]
- Suda, T.; Takahashi, N.; Udagawa, N.; Jimi, E.; Gillespie, M.T.; Martin, T.J. Modulation of osteoclast differentiation and function by the new members of the tumor necrosis factor receptor and ligand families. Endocr. Rev. 1999, 20, 345–357. [Google Scholar] [CrossRef] [PubMed]
- Naito, A.; Azuma, S.; Tanaka, S.; Miyazaki, T.; Takaki, S.; Takatsu, K.; Nakao, K.; Nakamura, K.; Katsuki, M.; Yamamoto, T.; et al. Severe osteopetrosis, defective interleukin-1 signalling and lymph node organogenesis in TRAF6-deficient mice. Genes Cells Devoted Mol. Cell. Mech. 1999, 4, 353–362. [Google Scholar] [CrossRef]
- Lomaga, M.A.; Yeh, W.C.; Sarosi, I.; Duncan, G.S.; Furlonger, C.; Ho, A.; Morony, S.; Capparelli, C.; Van, G.; Kaufman, S.; et al. TRAF6 deficiency results in osteopetrosis and defective interleukin-1, CD40, and LPS signaling. Genes Dev. 1999, 13, 1015–1024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walsh, M.C.; Lee, J.; Choi, Y. Tumor necrosis factor receptor- associated factor 6 (TRAF6) regulation of development, function, and homeostasis of the immune system. Immunol. Rev. 2015, 266, 72–92. [Google Scholar] [CrossRef]
- Ishida, N.; Hayashi, K.; Hoshijima, M.; Ogawa, T.; Koga, S.; Miyatake, Y.; Kumegawa, M.; Kimura, T.; Takeya, T. Large scale gene expression analysis of osteoclastogenesis in vitro and elucidation of NFAT2 as a key regulator. J. Biol. Chem. 2002, 277, 41147–41156. [Google Scholar] [CrossRef] [Green Version]
- Takayanagi, H.; Kim, S.; Koga, T.; Nishina, H.; Isshiki, M.; Yoshida, H.; Saiura, A.; Isobe, M.; Yokochi, T.; Inoue, J.; et al. Induction and activation of the transcription factor NFATc1 (NFAT2) integrate RANKL signaling in terminal differentiation of osteoclasts. Dev. Cell 2002, 3, 889–901. [Google Scholar] [CrossRef] [Green Version]
- Kukita, T.; Wada, N.; Kukita, A.; Kakimoto, T.; Sandra, F.; Toh, K.; Nagata, K.; Iijima, T.; Horiuchi, M.; Matsusaki, H.; et al. RANKL-induced DC-STAMP is essential for osteoclastogenesis. J. Exp. Med. 2004, 200, 941–946. [Google Scholar] [CrossRef] [Green Version]
- Yagi, M.; Miyamoto, T.; Sawatani, Y.; Iwamoto, K.; Hosogane, N.; Fujita, N.; Morita, K.; Ninomiya, K.; Suzuki, T.; Miyamoto, K.; et al. DC-STAMP is essential for cell-cell fusion in osteoclasts and foreign body giant cells. J. Exp. Med. 2005, 202, 345–351. [Google Scholar] [CrossRef] [Green Version]
- Inaoka, T.; Bilbe, G.; Ishibashi, O.; Tezuka, K.; Kumegawa, M.; Kokubo, T. Molecular cloning of human cDNA for cathepsin K: Novel cysteine proteinase predominantly expressed in bone. Biochem. Biophys. Res. Commun. 1995, 206, 89–96. [Google Scholar] [CrossRef]
- Koga, T.; Inui, M.; Inoue, K.; Kim, S.; Suematsu, A.; Kobayashi, E.; Iwata, T.; Ohnishi, H.; Matozaki, T.; Kodama, T.; et al. Costimulatory signals mediated by the ITAM motif cooperate with RANKL for bone homeostasis. Nature 2004, 428, 758–763. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, M.; Udagawa, N.; Matsuura, S.; Mogi, M.; Nakamura, H.; Horiuchi, H.; Saito, N.; Hiraoka, B.Y.; Kobayashi, Y.; Takaoka, K.; et al. Osteoprotegerin regulates bone formation through a coupling mechanism with bone resorption. Endocrinology 2003, 144, 5441–5449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lotinun, S.; Kiviranta, R.; Matsubara, T.; Alzate, J.A.; Neff, L.; Luth, A.; Koskivirta, I.; Kleuser, B.; Vacher, J.; Vuorio, E.; et al. Osteoclast-specific cathepsin K deletion stim.mulates S1P-dependent bone formation. J. Clin. Investig. 2013, 123, 666–681. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Irie, N.; Takada, Y.; Shimoda, K.; Miyamoto, T.; Nishiwaki, T.; Suda, T.; Matsuo, K. Bidirectional ephrinB2-EphB4 signaling controls bone homeostasis. Cell Metab. 2006, 4, 111–121. [Google Scholar] [CrossRef] [Green Version]
- Takeshita, S.; Fumoto, T.; Matsuoka, K.; Park, K.A.; Aburatani, H.; Kato, S.; Ito, M.; Ikeda, K. Osteoclast-secreted CTHRC1 in the coupling of bone resorption to formation. J. Clin. Investig. 2013, 123, 3914–3924. [Google Scholar] [CrossRef] [Green Version]
- Purdue, P.E.; Crotti, T.N.; Shen, Z.; Swantek, J.; Li, J.; Hill, J.; Hanidu, A.; Dimock, J.; Nabozny, G.; Goldring, S.R.; et al. Comprehensive profiling analysis of actively resorbing osteoclasts identifies critical signaling pathways regulated by bone substrate. Sci. Rep. 2014, 4, 7595. [Google Scholar] [CrossRef] [Green Version]
- Matsuoka, K.; Park, K.A.; Ito, M.; Ikeda, K.; Takeshita, S. Osteoclast-derived complement component 3a stimulates osteoblast differentiation. J. Bone Miner. Res. 2014, 29, 1522–1530. [Google Scholar] [CrossRef]
- Weivoda, M.M.; Ruan, M.; Pederson, L.; Hachfeld, C.; Davey, R.A.; Zajac, J.D.; Westendorf, J.J.; Khosla, S.; Oursler, M.J. Osteoclast TGF-beta Receptor Signaling Induces Wnt1 Secretion and Couples Bone Resorption to Bone Formation. J. Bone Miner. Res. 2016, 31, 76–85. [Google Scholar] [CrossRef] [Green Version]
- Ota, K.; Quint, P.; Ruan, M.; Pederson, L.; Westendorf, J.J.; Khosla, S.; Oursler, M.J. TGF-beta induces Wnt10b in osteoclasts from female mice to enhance coupling to osteoblasts. Endocrinology 2013, 154, 3745–3752. [Google Scholar] [CrossRef] [Green Version]
- Pederson, L.; Ruan, M.; Westendorf, J.J.; Khosla, S.; Oursler, M.J. Regulation of bone formation by osteoclasts involves Wnt/BMP signaling and the chemokine sphingosine-1-phosphate. Proc. Natl. Acad. Sci. USA 2008, 105, 20764–20769. [Google Scholar] [CrossRef] [Green Version]
- Walker, E.C.; McGregor, N.E.; Poulton, I.J.; Pompolo, S.; Allan, E.H.; Quinn, J.M.; Gillespie, M.T.; Martin, T.J.; Sims, N.A. Cardiotrophin-1 is an osteoclast-derived stimulus of bone formation required for normal bone remodeling. J. Bone Miner. Res. 2008, 23, 2025–2032. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.; Cui, Z.; Wang, L.; Xia, Z.; Hu, Y.; Xian, L.; Li, C.; Xie, L.; Crane, J.; Wan, M.; et al. PDGF-BB secreted by preosteoclasts induces angiogenesis during coupling with osteogenesis. Nat. Med. 2014, 20, 1270–1278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikebuchi, Y.; Aoki, S.; Honma, M.; Hayashi, M.; Sugamori, Y.; Khan, M.; Kariya, Y.; Kato, G.; Tabata, Y.; Penninger, J.M.; et al. Coupling of bone resorption and formation by RANKL reverse signalling. Nature 2018, 561, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Redlich, K.; Smolen, J.S. Inflammatory bone loss: Pathogenesis and therapeutic intervention. Nat. Rev. Drug Discov. 2012, 11, 234–250. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y. Managing Osteoporosis and Joint Damage in Patients with Rheumatoid Arthritis: An Overview. J. Clin. Med. 2021, 10, 1241. [Google Scholar] [CrossRef]
- Tsukasaki, M.; Takayanagi, H. Osteoimmunology: Evolving concepts in bone-immune interactions in health and disease. Nat. Rev. Immunol. 2019, 19, 626–642. [Google Scholar] [CrossRef]
- Chotiyarnwong, P.; McCloskey, E.V. Pathogenesis of glucocorticoid-induced osteoporosis and options for treatment. Nat. Rev. Endocrinol. 2020, 16, 437–447. [Google Scholar] [CrossRef]
- Charoenngam, N. Vitamin D and Rheumatic Diseases: A Review of Clinical Evidence. Int. J. Mol. Sci. 2021, 22, 10659. [Google Scholar] [CrossRef] [PubMed]
- Adami, G.; Saag, K.G. Osteoporosis Pathophysiology, Epidemiology, and Screening in Rheumatoid Arthritis. Curr. Rheumatol. Rep. 2019, 21, 34. [Google Scholar] [CrossRef]
- Jimi, E.; Takakura, N.; Hiura, F.; Nakamura, I.; Hirata-Tsuchiya, S. The Role of NF-κB in Physiological Bone Development and Inflammatory Bone Diseases: Is NF-κB Inhibition “Killing Two Birds with One Stone”? Cells 2019, 8, 1636. [Google Scholar] [CrossRef] [Green Version]
- Berardi, S.; Corrado, A.; Maruotti, N.; Cici, D.; Cantatore, F.P. Osteoblast role in the pathogenesis of rheumatoid arthritis. Mol. Biol. Rep. 2021, 48, 2843–2852. [Google Scholar] [CrossRef] [PubMed]
- Diarra, D.; Stolina, M.; Polzer, K.; Zwerina, J.; Ominsky, M.S.; Dwyer, D.; Korb, A.; Smolen, J.; Hoffmann, M.; Scheinecker, C.; et al. Dickkopf-1 is a master regulator of joint remodeling. Nat. Med. 2007, 13, 156–163. [Google Scholar] [CrossRef] [PubMed]
- Tsukasaki, M.; Yamada, A.; Suzuki, D.; Aizawa, R.; Miyazono, A.; Miyamoto, Y.; Suzawa, T.; Takami, M.; Yoshimura, K.; Morimura, N.; et al. Expression of POEM, a positive regulator of osteoblast differentiation, is suppressed by TNF-α. BioChem. Biophys. Res. Commun. 2011, 410, 766–770. [Google Scholar] [CrossRef] [PubMed]
- Qin, Z.; Fang, Z.; Zhao, L.; Chen, J.; Li, Y.; Liu, G. High dose of TNF-α suppressed osteogenic differentiation of human dental pulp stem cells by activating the Wnt/β-catenin signaling. J. Mol. Histol. 2015, 46, 409–420. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Yan, K.; Liu, S.; Yu, X.; Xu, J.; Liu, J.; Li, S. Semaphorin 3A promotes the osteogenic differentiation of rat bone marrow-derived mesenchymal stem cells in inflammatory environments by suppressing the Wnt/β-catenin signaling pathway. J. Mol. Histol. 2021, 52, 1245–1255. [Google Scholar] [CrossRef]
- Glass, D.A., 2nd; Bialek, P.; Ahn, J.D.; Starbuck, M.; Patel, M.S.; Clevers, H.; Taketo, M.M.; Long, F.; McMahon, A.P.; Lang, R.A.; et al. Canonical Wnt signaling in differentiated osteoblasts controls osteoclast differentiation. Dev. Cell 2005, 8, 751–764. [Google Scholar] [CrossRef] [Green Version]
- Colditz, J.; Thiele, S.; Baschant, U.; Niehrs, C.; Bonewald, L.F.; Hofbauer, L.C.; Rauner, M. Postnatal Skeletal Deletion of Dickkopf-1 Increases Bone Formation and Bone Volume in Male and Female Mice, Despite Increased Sclerostin Expression. J. Bone Miner. Res. 2018, 33, 1698–1707. [Google Scholar] [CrossRef] [Green Version]
- Stach, C.M.; Bäuerle, M.; Englbrecht, M.; Kronke, G.; Engelke, K.; Manger, B.; Schett, G. Periarticular bone structure in rheumatoid arthritis patients and healthy individuals assessed by high-resolution computed tomography. Arthritis Rheum. 2010, 62, 330–339. [Google Scholar] [CrossRef]
- Zhu, T.Y.; Griffith, J.F.; Qin, L.; Hung, V.W.; Fong, T.N.; Au, S.K.; Li, M.; Lam, Y.Y.; Wong, C.K.; Kwok, A.W.; et al. Alterations of bone density, microstructure, and strength of the distal radius in male patients with rheumatoid arthritis: A case-control study with HR-pQCT. J. Bone Miner. Res. 2014, 29, 2118–2129. [Google Scholar] [CrossRef]
- Yamanaka, H.; Sugiyama, N.; Inoue, E.; Taniguchi, A.; Momohara, S. Estimates of the prevalence of and current treatment practices for rheumatoid arthritis in Japan using reimbursement data from health insurance societies and the IORRA cohort (I). Mod. Rheumatol. 2014, 24, 33–40. [Google Scholar] [CrossRef]
- Komori, T. Glucocorticoid Signaling and Bone Biology. Horm. Metab. Res. Horm.—Stoffwechs. Horm. Metab. 2016, 48, 755–763. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, F.; Hayashi, M.; Mouri, Y.; Nakamura, S.; Adachi, T.; Nakashima, T. Mechanotransduction via the Piezo1-Akt pathway underlies Sost suppression in osteocytes. Biochem. Biophys. Res. Commun. 2020, 521, 806–813. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Gao, B.; Fan, Y.; Liu, Y.; Feng, S.; Cong, Q.; Zhang, X.; Zhou, Y.; Yadav, P.S.; Lin, J.; et al. Piezo1/2 mediate mechanotransduction essential for bone formation through concerted activation of NFAT-YAP1-β-catenin. eLife 2020, 9, e52779. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, F.; Hayashi, M.; Ono, T.; Nakashima, T. The regulation of RANKL by mechanical force. J. Bone Miner. Metab. 2021, 39, 34–44. [Google Scholar] [CrossRef]
- Lin, J.; Liu, J.; Davies, M.L.; Chen, W. Serum Vitamin D Level and Rheumatoid Arthritis Disease Activity: Review and Meta-Analysis. PLoS ONE 2016, 11, e0146351. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.H.; Bae, S.C. Vitamin D level in rheumatoid arthritis and its correlation with the disease activity: A meta-analysis. Clin. Exp. Rheumatol. 2016, 34, 827–833. [Google Scholar] [PubMed]
- Hasegawa, T.; Kikuta, J.; Sudo, T.; Matsuura, Y.; Matsui, T.; Simmons, S.; Ebina, K.; Hirao, M.; Okuzaki, D.; Yoshida, Y.; et al. Identification of a novel arthritis-associated osteoclast precursor macrophage regulated by FoxM1. Nat. Immunol. 2019, 20, 1631–1643. [Google Scholar] [CrossRef] [PubMed]
- Narisawa, M.; Kubo, S.; Okada, Y.; Yamagata, K.; Nakayamada, S.; Sakata, K.; Yamaoka, K.; Tanaka, Y. Human dendritic cell-derived osteoclasts with high bone resorption capacity and T cell stimulation ability. Bone 2021, 142, 115616. [Google Scholar] [CrossRef]
- Ranges, G.E.; Sriram, S.; Cooper, S.M. Prevention of type II collagen-induced arthritis by in vivo treatment with anti-L3T4. J. Exp. Med. 1985, 162, 1105–1110. [Google Scholar] [CrossRef] [Green Version]
- Okada, Y.; Wu, D.; Trynka, G.; Raj, T.; Terao, C.; Ikari, K.; Kochi, Y.; Ohmura, K.; Suzuki, A.; Yoshida, S.; et al. Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature 2014, 506, 376–381. [Google Scholar] [CrossRef]
- Kochi, Y.; Okada, Y.; Suzuki, A.; Ikari, K.; Terao, C.; Takahashi, A.; Yamazaki, K.; Hosono, N.; Myouzen, K.; Tsunoda, T.; et al. A regulatory variant in CCR6 is associated with rheumatoid arthritis susceptibility. Nat. Genet. 2010, 42, 515–519. [Google Scholar] [CrossRef] [PubMed]
- Mueller, A.L.; Payandeh, Z.; Mohammadkhani, N.; Mubarak, S.M.H.; Zakeri, A.; Alagheband Bahrami, A.; Brockmueller, A.; Shakibaei, M. Recent Advances in Understanding the Pathogenesis of Rheumatoid Arthritis: New Treatment Strategies. Cells 2021, 10, 3017. [Google Scholar] [CrossRef] [PubMed]
- Kremer, J.M.; Dougados, M.; Emery, P.; Durez, P.; Sibilia, J.; Shergy, W.; Steinfeld, S.; Tindall, E.; Becker, J.C.; Li, T.; et al. Treatment of rheumatoid arthritis with the selective costimulation modulator abatacept: Twelve-month results of a phase iib, double-blind, randomized, placebo-controlled trial. Arthritis Rheum. 2005, 52, 2263–2271. [Google Scholar] [CrossRef] [PubMed]
- Takayanagi, H.; Ogasawara, K.; Hida, S.; Chiba, T.; Murata, S.; Sato, K.; Takaoka, A.; Yokochi, T.; Oda, H.; Tanaka, K.; et al. T-cell-mediated regulation of osteoclastogenesis by signalling cross-talk between RANKL and IFN-gamma. Nature 2000, 408, 600–605. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Suematsu, A.; Okamoto, K.; Yamaguchi, A.; Morishita, Y.; Kadono, Y.; Tanaka, S.; Kodama, T.; Akira, S.; Iwakura, Y.; et al. Th17 functions as an osteoclastogenic helper T cell subset that links T cell activation and bone destruction. J. Exp. Med. 2006, 203, 2673–2682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jovanovic, D.V.; Di Battista, J.A.; Martel-Pelletier, J.; Jolicoeur, F.C.; He, Y.; Zhang, M.; Mineau, F.; Pelletier, J.P. IL-17 stimulates the production and expression of proinflammatory cytokines, IL-beta and TNF-alpha, by human macrophages. J. Immunol. 1998, 160, 3513–3521. [Google Scholar] [PubMed]
- Ogura, H.; Murakami, M.; Okuyama, Y.; Tsuruoka, M.; Kitabayashi, C.; Kanamoto, M.; Nishihara, M.; Iwakura, Y.; Hirano, T. Interleukin-17 promotes autoimmunity by triggering a positive-feedback loop via interleukin-6 induction. Immunity 2008, 29, 628–636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakaguchi, S.; Mikami, N.; Wing, J.B.; Tanaka, A.; Ichiyama, K.; Ohkura, N. Regulatory T Cells and Human Disease. Annu. Rev. Immunol. 2020, 38, 541–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komatsu, N.; Okamoto, K.; Sawa, S.; Nakashima, T.; Oh-hora, M.; Kodama, T.; Tanaka, S.; Bluestone, J.A.; Takayanagi, H. Pathogenic conversion of Foxp3+ T cells into TH17 cells in autoimmune arthritis. Nat. Med. 2014, 20, 62–68. [Google Scholar] [CrossRef]
- Lubberts, E.; Koenders, M.I.; Oppers-Walgreen, B.; van den Bersselaar, L.; Coenen-de Roo, C.J.; Joosten, L.A.; van den Berg, W.B. Treatment with a neutralizing anti-murine interleukin-17 antibody after the onset of collagen-induced arthritis reduces joint inflammation, cartilage destruction, and bone erosion. Arthritis Rheum. 2004, 50, 650–659. [Google Scholar] [CrossRef] [PubMed]
- Kotake, S.; Udagawa, N.; Takahashi, N.; Matsuzaki, K.; Itoh, K.; Ishiyama, S.; Saito, S.; Inoue, K.; Kamatani, N.; Gillespie, M.T.; et al. IL-17 in synovial fluids from patients with rheumatoid arthritis is a potent stimulator of osteoclastogenesis. J. Clin. Investig. 1999, 103, 1345–1352. [Google Scholar] [CrossRef] [PubMed]
- Hirota, K.; Yoshitomi, H.; Hashimoto, M.; Maeda, S.; Teradaira, S.; Sugimoto, N.; Yamaguchi, T.; Nomura, T.; Ito, H.; Nakamura, T.; et al. Preferential recruitment of CCR6-expressing Th17 cells to inflamed joints via CCL20 in rheumatoid arthritis and its animal model. J. Exp. Med. 2007, 204, 2803–2812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nistala, K.; Moncrieffe, H.; Newton, K.R.; Varsani, H.; Hunter, P.; Wedderburn, L.R. Interleukin-17-producing T cells are enriched in the joints of children with arthritis, but have a reciprocal relationship to regulatory T cell numbers. Arthritis Rheum. 2008, 58, 875–887. [Google Scholar] [CrossRef] [PubMed]
- Genovese, M.C.; Durez, P.; Richards, H.B.; Supronik, J.; Dokoupilova, E.; Mazurov, V.; Aelion, J.A.; Lee, S.H.; Codding, C.E.; Kellner, H.; et al. Efficacy and safety of secukinumab in patients with rheumatoid arthritis: A phase II, dose-finding, double-blind, randomised, placebo controlled study. Ann. Rheum. Dis. 2013, 72, 863–869. [Google Scholar] [CrossRef]
- Tlustochowicz, W.; Rahman, P.; Seriolo, B.; Krammer, G.; Porter, B.; Widmer, A.; Richards, H.B. Efficacy and Safety of Subcutaneous and Intravenous Loading Dose Regimens of Secukinumab in Patients with Active Rheumatoid Arthritis: Results from a Randomized Phase II Study. J. Rheumatol. 2016, 43, 495–503. [Google Scholar] [CrossRef] [PubMed]
- Mease, P.J.; Jeka, S.; Jaller, J.J.; Kitumnuaypong, T.; Louthrenoo, W.; Mann, H.; Matsievskaia, G.; Soriano, E.R.; Jia, B.; Wang, C.; et al. CNTO6785, a Fully Human Antiinterleukin 17 Monoclonal Antibody, in Patients with Rheumatoid Arthritis with Inadequate Response to Methotrexate: A Randomized, Placebo-controlled, Phase II, Dose-ranging Study. J. Rheumatol. 2018, 45, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Smolen, J.S.; Agarwal, S.K.; Ilivanova, E.; Xu, X.L.; Miao, Y.; Zhuang, Y.; Nnane, I.; Radziszewski, W.; Greenspan, A.; Beutler, A.; et al. A randomised phase II study evaluating the efficacy and safety of subcutaneously administered ustekinumab and guselkumab in patients with active rheumatoid arthritis despite treatment with methotrexate. Ann. Rheum. Dis. 2017, 76, 831–839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yago, T.; Nanke, Y.; Kawamoto, M.; Kobashigawa, T.; Yamanaka, H.; Kotake, S. IL-23 and Th17 Disease in Inflammatory Arthritis. J. Clin. Med. 2017, 6, 81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taams, L.S. Interleukin-17 in rheumatoid arthritis: Trials and tribulations. J. Exp. Med. 2020, 217, e20192048. [Google Scholar] [CrossRef] [PubMed]
- Edwards, J.C.; Cambridge, G. Sustained improvement in rheumatoid arthritis following a protocol designed to deplete B lymphocytes. Rheumatology 2001, 40, 205–211. [Google Scholar] [CrossRef] [Green Version]
- Syversen, S.W.; Gaarder, P.I.; Goll, G.L.; Ødegård, S.; Haavardsholm, E.A.; Mowinckel, P.; van der Heijde, D.; Landewé, R.; Kvien, T.K. High anti-cyclic citrullinated peptide levels and an algorithm of four variables predict radiographic progression in patients with rheumatoid arthritis: Results from a 10-year longitudinal study. Ann. Rheum. Dis. 2008, 67, 212–217. [Google Scholar] [CrossRef] [PubMed]
- Harre, U.; Georgess, D.; Bang, H.; Bozec, A.; Axmann, R.; Ossipova, E.; Jakobsson, P.J.; Baum, W.; Nimmerjahn, F.; Szarka, E.; et al. Induction of osteoclastogenesis and bone loss by human autoantibodies against citrullinated vimentin. J. Clin. Investig. 2012, 122, 1791–1802. [Google Scholar] [CrossRef] [PubMed]
- Harre, U.; Lang, S.C.; Pfeifle, R.; Rombouts, Y.; Frühbeißer, S.; Amara, K.; Bang, H.; Lux, A.; Koeleman, C.A.; Baum, W.; et al. Glycosylation of immunoglobulin G determines osteoclast differentiation and bone loss. Nat. Commun. 2015, 6, 6651. [Google Scholar] [CrossRef] [PubMed]
- Negishi-Koga, T.; Gober, H.J.; Sumiya, E.; Komatsu, N.; Okamoto, K.; Sawa, S.; Suematsu, A.; Suda, T.; Sato, K.; Takai, T.; et al. Immune complexes regulate bone metabolism through FcRγ signalling. Nat. Commun. 2015, 6, 6637. [Google Scholar] [CrossRef] [PubMed]
- Rao, D.A.; Gurish, M.F.; Marshall, J.L.; Slowikowski, K.; Fonseka, C.Y.; Liu, Y.; Donlin, L.T.; Henderson, L.A.; Wei, K.; Mizoguchi, F.; et al. Pathologically expanded peripheral T helper cell subset drives B cells in rheumatoid arthritis. Nature 2017, 542, 110–114. [Google Scholar] [CrossRef]
- Yeo, L.; Toellner, K.M.; Salmon, M.; Filer, A.; Buckley, C.D.; Raza, K.; Scheel-Toellner, D. Cytokine mRNA profiling identifies B cells as a major source of RANKL in rheumatoid arthritis. Ann. Rheum. Dis. 2011, 70, 2022–2028. [Google Scholar] [CrossRef] [Green Version]
- Ota, Y.; Niiro, H.; Ota, S.; Ueki, N.; Tsuzuki, H.; Nakayama, T.; Mishima, K.; Higashioka, K.; Jabbarzadeh-Tabrizi, S.; Mitoma, H.; et al. Generation mechanism of RANKL(+) effector memory B cells: Relevance to the pathogenesis of rheumatoid arthritis. Arthritis Res. Ther. 2016, 18, 67. [Google Scholar] [CrossRef] [Green Version]
- Meednu, N.; Zhang, H.; Owen, T.; Sun, W.; Wang, V.; Cistrone, C.; Rangel-Moreno, J.; Xing, L.; Anolik, J.H. Production of RANKL by Memory B Cells: A Link Between B Cells and Bone Erosion in Rheumatoid Arthritis. Arthritis Rheumatol. 2016, 68, 805–816. [Google Scholar] [CrossRef]
- Xu, S.; Zhang, Y.; Liu, B.; Li, K.; Huang, B.; Yan, B.; Zhang, Z.; Liang, K.; Jia, C.; Lin, J.; et al. Activation of mTORC1 in B Lymphocytes Promotes Osteoclast Formation via Regulation of β-Catenin and RANKL/OPG. J. Bone Miner. Res. 2016, 31, 1320–1333. [Google Scholar] [CrossRef] [Green Version]
- Komatsu, N.; Win, S.; Yan, M.; Huynh, N.C.; Sawa, S.; Tsukasaki, M.; Terashima, A.; Pluemsakunthai, W.; Kollias, G.; Nakashima, T.; et al. Plasma cells promote osteoclastogenesis and periarticular bone loss in autoimmune arthritis. J. Clin. Investig. 2021, 131, e143060. [Google Scholar] [CrossRef]
- Maeda, K.; Kobayashi, Y.; Udagawa, N.; Uehara, S.; Ishihara, A.; Mizoguchi, T.; Kikuchi, Y.; Takada, I.; Kato, S.; Kani, S.; et al. Wnt5a-Ror2 signaling between osteoblast-lineage cells and osteoclast precursors enhances osteoclastogenesis. Nat. Med. 2012, 18, 405–412. [Google Scholar] [CrossRef] [PubMed]
- MacLauchlan, S.; Zuriaga, M.A.; Fuster, J.J.; Cuda, C.M.; Jonason, J.; Behzadi, F.; Duffen, J.P.; Haines, G.K., 3rd; Aprahamian, T.; Perlman, H.; et al. Genetic deficiency of Wnt5a diminishes disease severity in a murine model of rheumatoid arthritis. Arthritis Res. Ther. 2017, 19, 166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sen, M.; Lauterbach, K.; El-Gabalawy, H.; Firestein, G.S.; Corr, M.; Carson, D.A. Expression and function of wingless and frizzled homologs in rheumatoid arthritis. Proc. Natl. Acad. Sci. USA 2000, 97, 2791–2796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rauner, M.; Stein, N.; Winzer, M.; Goettsch, C.; Zwerina, J.; Schett, G.; Distler, J.H.; Albers, J.; Schulze, J.; Schinke, T.; et al. WNT5A is induced by inflammatory mediators in bone marrow stromal cells and regulates cytokine and chemokine production. J. Bone Miner. Res. 2012, 27, 575–585. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Trillo, A.; Mosquera, N.; Pena, C.; Rivas-Tobío, F.; Mera-Varela, A.; Gonzalez, A.; Conde, C. Non-Canonical WNT5A Signaling Through RYK Contributes to Aggressive Phenotype of the Rheumatoid Fibroblast-Like Synoviocytes. Front. Immunol. 2020, 11, 555245. [Google Scholar] [CrossRef] [PubMed]
- Mahmoud, D.E.; Kaabachi, W.; Sassi, N.; Mokhtar, A.; Moalla, M.; Ammar, L.B.; Jemmali, S.; Rekik, S.; Tarhouni, L.; Kallel-Sellami, M.; et al. SFRP5 Enhances Wnt5a Induced-Inflammation in Rheumatoid Arthritis Fibroblast-Like Synoviocytes. Front. Immunol. 2021, 12, 663683. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Wei, K.; Slowikowski, K.; Fonseka, C.Y.; Rao, D.A.; Kelly, S.; Goodman, S.M.; Tabechian, D.; Hughes, L.B.; Salomon-Escoto, K.; et al. Defining inflammatory cell states in rheumatoid arthritis joint synovial tissues by integrating single-cell transcriptomics and mass cytometry. Nat. Immunol. 2019, 20, 928–942. [Google Scholar] [CrossRef] [PubMed]
- Mizoguchi, F.; Slowikowski, K.; Wei, K.; Marshall, J.L.; Rao, D.A.; Chang, S.K.; Nguyen, H.N.; Noss, E.H.; Turner, J.D.; Earp, B.E.; et al. Functionally distinct disease-associated fibroblast subsets in rheumatoid arthritis. Nat. Commun. 2018, 9, 789. [Google Scholar] [CrossRef] [Green Version]
- Croft, A.P.; Campos, J.; Jansen, K.; Turner, J.D.; Marshall, J.; Attar, M.; Savary, L.; Wehmeyer, C.; Naylor, A.J.; Kemble, S.; et al. Distinct fibroblast subsets drive inflammation and damage in arthritis. Nature 2019, 570, 246–251. [Google Scholar] [CrossRef]
- Wei, K.; Korsunsky, I.; Marshall, J.L.; Gao, A.; Watts, G.F.M.; Major, T.; Croft, A.P.; Watts, J.; Blazar, P.E.; Lange, J.K.; et al. Notch signalling drives synovial fibroblast identity and arthritis pathology. Nature 2020, 582, 259–264. [Google Scholar] [CrossRef]
- Wei, K.; Nguyen, H.N.; Brenner, M.B. Fibroblast pathology in inflammatory diseases. J. Clin. Investig. 2021, 131, e149538. [Google Scholar] [CrossRef] [PubMed]
- Uehara, S.; Udagawa, N.; Mukai, H.; Ishihara, A.; Maeda, K.; Yamashita, T.; Murakami, K.; Nishita, M.; Nakamura, T.; Kato, S.; et al. Protein kinase N3 promotes bone resorption by osteoclasts in response to Wnt5a-Ror2 signaling. Sci. Signal. 2017, 10, eaan0023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maeda, K.; Kobayashi, Y.; Koide, M.; Uehara, S.; Okamoto, M.; Ishihara, A.; Kayama, T.; Saito, M.; Marumo, K. The Regulation of Bone Metabolism and Disorders by Wnt Signaling. Int. J. Mol. Sci. 2019, 20, 5525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yokota, K.; Sato, K.; Miyazaki, T.; Aizaki, Y.; Tanaka, S.; Sekikawa, M.; Kozu, N.; Kadono, Y.; Oda, H.; Mimura, T. Characterization and Function of Tumor Necrosis Factor and Interleukin-6-Induced Osteoclasts in Rheumatoid Arthritis. Arthritis Rheumatol. 2021, 73, 1145–1154. [Google Scholar] [CrossRef] [PubMed]
- Rossini, M.; Viapiana, O.; Adami, S.; Fracassi, E.; Idolazzi, L.; Dartizio, C.; Povino, M.R.; Orsolini, G.; Gatti, D. In patients with rheumatoid arthritis, Dickkopf-1 serum levels are correlated with parathyroid hormone, bone erosions and bone mineral density. Clin. Exp. Rheumatol. 2015, 33, 77–83. [Google Scholar]
- Murray, E.; Ellis, A.; Butylkova, Y.; Skup, M.; Kalabic, J.; Garg, V. Systematic review and network meta-analysis: Effect of biologics on radiographic progression in rheumatoid arthritis. J. Comp. Eff. Res. 2018, 7, 959–974. [Google Scholar] [CrossRef] [Green Version]
- Segawa, Y.; Yamaura, M.; Aota, S.; Omata, T.; Tuzuike, N.; Itokazu, Y.; Oka, H.; Tamaki, H.; Nakamura, T. Methotrexate maintains bone mass by preventing both a decrease in bone formation and an increase in bone resorption in adjuvant-induced arthritic rats. Bone 1997, 20, 457–464. [Google Scholar] [CrossRef]
- Kanagawa, H.; Masuyama, R.; Morita, M.; Sato, Y.; Niki, Y.; Kobayashi, T.; Katsuyama, E.; Fujie, A.; Hao, W.; Tando, T.; et al. Methotrexate inhibits osteoclastogenesis by decreasing RANKL-induced calcium influx into osteoclast progenitors. J. Bone Miner. Metab. 2016, 34, 526–531. [Google Scholar] [CrossRef]
- Poutoglidou, F.; Pourzitaki, C.; Manthou, M.E.; Samoladas, E.; Saitis, A.; Malliou, F.; Kouvelas, D. Infliximab prevents systemic bone loss and suppresses tendon inflammation in a collagen-induced arthritis rat model. Inflammopharmacology 2021, 29, 661–672. [Google Scholar] [CrossRef]
- Kaste, S.C.; Jones-Wallace, D.; Rose, S.R.; Boyett, J.M.; Lustig, R.H.; Rivera, G.K.; Pui, C.H.; Hudson, M.M. Bone mineral decrements in survivors of childhood acute lymphoblastic leukemia: Frequency of occurrence and risk factors for their development. Leukemia 2001, 15, 728–734. [Google Scholar] [CrossRef] [Green Version]
- di Munno, O.; Mazzantini, M.; Sinigaglia, L.; Bianchi, G.; Minisola, G.; Muratore, M.; la Corte, R.; di Matteo, L.; Canesi, B.; Caminiti, M.; et al. Effect of low dose methotrexate on bone density in women with rheumatoid arthritis: Results from a multicenter cross-sectional study. J. Rheumatol. 2004, 31, 1305–1309. [Google Scholar] [PubMed]
- Vis, M.; Voskuyl, A.E.; Wolbink, G.J.; Dijkmans, B.A.; Lems, W.F. Bone mineral density in patients with rheumatoid arthritis treated with infliximab. Ann. Rheum. Dis. 2005, 64, 336–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vis, M.; Havaardsholm, E.A.; Haugeberg, G.; Uhlig, T.; Voskuyl, A.E.; van de Stadt, R.J.; Dijkmans, B.A.; Woolf, A.D.; Kvien, T.K.; Lems, W.F. Evaluation of bone mineral density, bone metabolism, osteoprotegerin and receptor activator of the NFkappaB ligand serum levels during treatment with infliximab in patients with rheumatoid arthritis. Ann. Rheum. Dis. 2006, 65, 1495–1499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marotte, H.; Pallot-Prades, B.; Grange, L.; Gaudin, P.; Alexandre, C.; Miossec, P. A 1-year case-control study in patients with rheumatoid arthritis indicates prevention of loss of bone mineral density in both responders and nonresponders to infliximab. Arthritis Res. Ther. 2007, 9, R61. [Google Scholar] [CrossRef] [Green Version]
- Chopin, F.; Garnero, P.; le Henanff, A.; Debiais, F.; Daragon, A.; Roux, C.; Sany, J.; Wendling, D.; Zarnitsky, C.; Ravaud, P.; et al. Long-term effects of infliximab on bone and cartilage turnover markers in patients with rheumatoid arthritis. Ann. Rheum. Dis. 2008, 67, 353–357. [Google Scholar] [CrossRef] [PubMed]
- Eekman, D.A.; Vis, M.; Bultink, I.E.; Kuik, D.J.; Voskuyl, A.E.; Dijkmans, B.A.; Lems, W.F. Stable bone mineral density in lumbar spine and hip in contrast to bone loss in the hands during long-term treatment with infliximab in patients with rheumatoid arthritis. Ann. Rheum. Dis. 2011, 70, 389–390. [Google Scholar] [CrossRef]
- Wijbrandts, C.A.; Klaasen, R.; Dijkgraaf, M.G.; Gerlag, D.M.; van Eck-Smit, B.L.; Tak, P.P. Bone mineral density in rheumatoid arthritis patients 1 year after adalimumab therapy: Arrest of bone loss. Ann. Rheum. Dis. 2009, 68, 373–376. [Google Scholar] [CrossRef] [PubMed]
- Krieckaert, C.L.; Nurmohamed, M.T.; Wolbink, G.; Lems, W.F. Changes in bone mineral density during long-term treatment with adalimumab in patients with rheumatoid arthritis: A cohort study. Rheumatology 2013, 52, 547–553. [Google Scholar] [CrossRef] [Green Version]
- Gulyás, K.; Horváth, Á.; Végh, E.; Pusztai, A.; Szentpétery, Á.; Pethö, Z.; Váncsa, A.; Bodnár, N.; Csomor, P.; Hamar, A.; et al. Effects of 1-year anti-TNF-α therapies on bone mineral density and bone biomarkers in rheumatoid arthritis and ankylosing spondylitis. Clin. Rheumatol. 2020, 39, 167–175. [Google Scholar] [CrossRef] [Green Version]
- Orsolini, G.; Adami, G.; Adami, S.; Viapiana, O.; Idolazzi, L.; Gatti, D.; Rossini, M. Short-Term Effects of TNF Inhibitors on Bone Turnover Markers and Bone Mineral Density in Rheumatoid Arthritis. Calcif. Tissue Int. 2016, 98, 580–585. [Google Scholar] [CrossRef]
- Abu-Shakra, M.; Zisman, D.; Balbir-Gurman, A.; Amital, H.; Levy, Y.; Langevitz, P.; Tishler, M.; Molad, Y.; Aamar, S.; Roser, I.; et al. Effect of Tocilizumab on Fatigue and Bone Mineral Density in Patients with Rheumatoid Arthritis. Isr. Med. Assoc. J. IMAJ 2018, 20, 239–244. [Google Scholar] [PubMed]
- Kume, K.; Amano, K.; Yamada, S.; Kanazawa, T.; Ohta, H.; Hatta, K.; Amano, K.; Kuwaba, N. The effect of tocilizumab on bone mineral density in patients with methotrexate-resistant active rheumatoid arthritis. Rheumatology 2014, 53, 900–903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Briot, K.; Rouanet, S.; Schaeverbeke, T.; Etchepare, F.; Gaudin, P.; Perdriger, A.; Vray, M.; Steinberg, G.; Roux, C. The effect of tocilizumab on bone mineral density, serum levels of Dickkopf-1 and bone remodeling markers in patients with rheumatoid arthritis. Jt. Int. Bone Spine 2015, 82, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Boyapati, A.; Msihid, J.; Fiore, S.; van Adelsberg, J.; Graham, N.M.; Hamilton, J.D. Sarilumab plus methotrexate suppresses circulating biomarkers of bone resorption and synovial damage in patients with rheumatoid arthritis and inadequate response to methotrexate: A biomarker study of MOBILITY. Arthritis Res. Ther. 2016, 18, 225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabay, C.; Msihid, J.; Zilberstein, M.; Paccard, C.; Lin, Y.; Graham, N.M.H.; Boyapati, A. Identification of sarilumab pharmacodynamic and predictive markers in patients with inadequate response to TNF inhibition: A biomarker substudy of the phase 3 TARGET study. RMD Open 2018, 4, e000607. [Google Scholar] [CrossRef] [PubMed]
- Gabay, C.; Burmester, G.R.; Strand, V.; Msihid, J.; Zilberstein, M.; Kimura, T.; van Hoogstraten, H.; Boklage, S.H.; Sadeh, J.; Graham, N.M.H.; et al. Sarilumab and adalimumab differential effects on bone remodelling and cardiovascular risk biomarkers, and predictions of treatment outcomes. Arthritis Res. Ther. 2020, 22, 70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tada, M.; Inui, K.; Sugioka, Y.; Mamoto, K.; Okano, T.; Koike, T. Abatacept might increase bone mineral density at femoral neck for patients with rheumatoid arthritis in clinical practice: AIRTIGHT study. Rheumatol. Int. 2018, 38, 777–784. [Google Scholar] [CrossRef] [Green Version]
- Hamar, A.; Szekanecz, Z.; Pusztai, A.; Czókolyová, M.; Végh, E.; Pethő, Z.; Bodnár, N.; Gulyás, K.; Horváth, Á.; Soós, B.; et al. Effects of one-year tofacitinib therapy on bone metabolism in rheumatoid arthritis. Osteoporos. Int. 2021, 32, 1621–1629. [Google Scholar] [CrossRef] [PubMed]
- Thudium, C.S.; Bay-Jensen, A.C.; Cahya, S.; Dow, E.R.; Karsdal, M.A.; Koch, A.E.; Zhang, W.; Benschop, R.J. The Janus kinase 1/2 inhibitor baricitinib reduces biomarkers of joint destruction in moderate to severe rheumatoid arthritis. Arthritis Res. Ther. 2020, 22, 235. [Google Scholar] [CrossRef]
- Mageed, R.A.; Adams, G.; Woodrow, D.; Podhajcer, O.L.; Chernajovsky, Y. Prevention of collagen-induced arthritis by gene delivery of soluble p75 tumour necrosis factor receptor. Gene Ther. 1998, 5, 1584–1592. [Google Scholar] [CrossRef] [Green Version]
- Saidenberg-Kermanac’h, N.; Corrado, A.; Lemeiter, D.; deVernejoul, M.C.; Boissier, M.C.; Cohen-Solal, M.E. TNF-alpha antibodies and osteoprotegerin decrease systemic bone loss associated with inflammation through distinct mechanisms in collagen-induced arthritis. Bone 2004, 35, 1200–1207. [Google Scholar] [CrossRef] [PubMed]
- De Benedetti, F.; Rucci, N.; Del Fattore, A.; Peruzzi, B.; Paro, R.; Longo, M.; Vivarelli, M.; Muratori, F.; Berni, S.; Ballanti, P.; et al. Impaired skeletal development in interleukin-6-transgenic mice: A model for the impact of chronic inflammation on the growing skeletal system. Arthritis Rheum. 2006, 54, 3551–3563. [Google Scholar] [CrossRef] [PubMed]
- Peruzzi, B.; Cappariello, A.; Del Fattore, A.; Rucci, N.; De Benedetti, F.; Teti, A. c-Src and IL-6 inhibit osteoblast differentiation and integrate IGFBP5 signalling. Nat. Commun. 2012, 3, 630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeuchi, T.; Yoshida, H.; Tanaka, S. Role of interleukin-6 in bone destruction and bone repair in rheumatoid arthritis. Autoimmun. Rev. 2021, 20, 102884. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Feng, W.; Yimin; Cui, J.; Lv, S.; Hasegawa, T.; Sun, B.; Li, J.; Oda, K.; Amizuka, N.; et al. Histological Evidence of Increased Osteoclast Cell Number and Asymmetric Bone Resorption Activity in the Tibiae of Interleukin-6-Deficient Mice. J. Histochem. Cytochem. Off. J. Histochem. Soc. 2014, 62, 556–564. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, H.; Suzuki, M.; Tanaka, K.; Takeda, S.; Yogo, K.; Matsumoto, Y. Anti-interleukin-6 receptor antibody prevents loss of bone structure and bone strength in collagen-induced arthritis mice. Scand. J. Rheumatol. 2018, 47, 384–391. [Google Scholar] [CrossRef] [PubMed]
- Finzel, S.; Kraus, S.; Figueiredo, C.P.; Regensburger, A.; Kocijan, R.; Rech, J.; Schett, G. Comparison of the effects of tocilizumab monotherapy and adalimumab in combination with methotrexate on bone erosion repair in rheumatoid arthritis. Ann. Rheum. Dis. 2019, 78, 1186–1191. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.Y.; Liu, Y.Y.; Ye, H.; Guo, J.P.; Li, R.; Liu, X.; Li, Z.G. Circulating Dickkopf-1 is correlated with bone erosion and inflammation in rheumatoid arthritis. J. Rheumatol. 2011, 38, 821–827. [Google Scholar] [CrossRef] [PubMed]
- Seror, R.; Boudaoud, S.; Pavy, S.; Nocturne, G.; Schaeverbeke, T.; Saraux, A.; Chanson, P.; Gottenberg, J.E.; Devauchelle-Pensec, V.; Tobón, G.J.; et al. Increased Dickkopf-1 in Recent-onset Rheumatoid Arthritis is a New Biomarker of Structural Severity. Data from the ESPOIR Cohort. Sci. Rep. 2016, 6, 18421. [Google Scholar] [CrossRef]
- Singh, A.; Gupta, M.K.; Mishra, S.P. Study of correlation of level of expression of Wnt signaling pathway inhibitors sclerostin and dickkopf-1 with disease activity and severity in rheumatoid arthritis patients. Drug Discov. Ther. 2019, 13, 22–27. [Google Scholar] [CrossRef] [Green Version]
- Cici, D.; Corrado, A.; Rotondo, C.; Cantatore, F.P. Wnt Signaling and Biological Therapy in Rheumatoid Arthritis and Spondyloarthritis. Int. J. Mol. Sci. 2019, 20, 5552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roser-Page, S.; Vikulina, T.; Zayzafoon, M.; Weitzmann, M.N. CTLA-4Ig-induced T cell anergy promotes Wnt-10b production and bone formation in a mouse model. Arthritis Rheumatol. 2014, 66, 990–999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roser-Page, S.; Vikulina, T.; Weiss, D.; Habib, M.M.; Beck, G.R., Jr.; Pacifici, R.; Lane, T.F.; Weitzmann, M.N. CTLA-4Ig (abatacept) balances bone anabolic effects of T cells and Wnt-10b with antianabolic effects of osteoblastic sclerostin. Ann. N. Y. Acad. Sci. 2018, 1415, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Axmann, R.; Herman, S.; Zaiss, M.; Franz, S.; Polzer, K.; Zwerina, J.; Herrmann, M.; Smolen, J.; Schett, G. CTLA-4 directly inhibits osteoclast formation. Ann. Rheum. Dis. 2008, 67, 1603–1609. [Google Scholar] [CrossRef]
- Bozec, A.; Zaiss, M.M.; Kagwiria, R.; Voll, R.; Rauh, M.; Chen, Z.; Mueller-Schmucker, S.; Kroczek, R.A.; Heinzerling, L.; Moser, M.; et al. T cell costimulation molecules CD80/86 inhibit osteoclast differentiation by inducing the IDO/tryptophan pathway. Sci. Transl. Med. 2014, 6, 235ra260. [Google Scholar] [CrossRef] [PubMed]
- Okada, H.; Kajiya, H.; Omata, Y.; Matsumoto, T.; Sato, Y.; Kobayashi, T.; Nakamura, S.; Kaneko, Y.; Nakamura, S.; Koyama, T.; et al. CTLA4-Ig Directly Inhibits Osteoclastogenesis by Interfering With Intracellular Calcium Oscillations in Bone Marrow Macrophages. J. Bone Miner. Res. 2019, 34, 1744–1752. [Google Scholar] [CrossRef]
- Okada, H.; Okabe, K.; Tanaka, S. Finely-Tuned Calcium Oscillations in Osteoclast Differentiation and Bone Resorption. Int. J. Mol. Sci. 2020, 22, 180. [Google Scholar] [CrossRef]
- Yamaoka, K. Janus kinase inhibitors for rheumatoid arthritis. Curr. Opin. Chem. Biol. 2016, 32, 29–33. [Google Scholar] [CrossRef]
- Sanpaolo, E.R.; Rotondo, C.; Cici, D.; Corrado, A.; Cantatore, F.P. JAK/STAT pathway and molecular mechanism in bone remodeling. Mol. Biol. Rep. 2020, 47, 9087–9096. [Google Scholar] [CrossRef]
- Vidal, B.; Cascão, R.; Finnilä, M.A.J.; Lopes, I.P.; da Glória, V.G.; Saarakkala, S.; Zioupos, P.; Canhão, H.; Fonseca, J.E. Effects of tofacitinib in early arthritis-induced bone loss in an adjuvant-induced arthritis rat model. Rheumatology 2018, 57, 1461–1471. [Google Scholar] [CrossRef] [Green Version]
- LaBranche, T.P.; Jesson, M.I.; Radi, Z.A.; Storer, C.E.; Guzova, J.A.; Bonar, S.L.; Thompson, J.M.; Happa, F.A.; Stewart, Z.S.; Zhan, Y.; et al. JAK inhibition with tofacitinib suppresses arthritic joint structural damage through decreased RANKL production. Arthritis Rheum. 2012, 64, 3531–3542. [Google Scholar] [CrossRef] [PubMed]
- Gaber, T.; Brinkman, A.C.K.; Pienczikowski, J.; Diesing, K.; Damerau, A.; Pfeiffenberger, M.; Lang, A.; Ohrndorf, S.; Burmester, G.R.; Buttgereit, F.; et al. Impact of Janus Kinase Inhibition with Tofacitinib on Fundamental Processes of Bone Healing. Int. J. Mol. Sci. 2020, 21, 865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adam, S.; Simon, N.; Steffen, U.; Andes, F.T.; Scholtysek, C.; Müller, D.I.H.; Weidner, D.; Andreev, D.; Kleyer, A.; Culemann, S.; et al. JAK inhibition increases bone mass in steady-state conditions and ameliorates pathological bone loss by stimulating osteoblast function. Sci. Transl. Med. 2020, 12, eaay4447. [Google Scholar] [CrossRef] [PubMed]
- Fridman, J.S.; Scherle, P.A.; Collins, R.; Burn, T.C.; Li, Y.; Li, J.; Covington, M.B.; Thomas, B.; Collier, P.; Favata, M.F.; et al. Selective inhibition of JAK1 and JAK2 is efficacious in rodent models of arthritis: Preclinical characterization of INCB028050. J. Immunol. 2010, 184, 5298–5307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murakami, K.; Kobayashi, Y.; Uehara, S.; Suzuki, T.; Koide, M.; Yamashita, T.; Nakamura, M.; Takahashi, N.; Kato, H.; Udagawa, N.; et al. A Jak1/2 inhibitor, baricitinib, inhibits osteoclastogenesis by suppressing RANKL expression in osteoblasts in vitro. PLoS ONE 2017, 12, e0181126. [Google Scholar] [CrossRef] [Green Version]
- Ito, M.; Yamazaki, S.; Yamagami, K.; Kuno, M.; Morita, Y.; Okuma, K.; Nakamura, K.; Chida, N.; Inami, M.; Inoue, T.; et al. A novel JAK inhibitor, peficitinib, demonstrates potent efficacy in a rat adjuvant-induced arthritis model. J. Pharmacol. Sci. 2017, 133, 25–33. [Google Scholar] [CrossRef]
- Van Rompaey, L.; Galien, R.; van der Aar, E.M.; Clement-Lacroix, P.; Nelles, L.; Smets, B.; Lepescheux, L.; Christophe, T.; Conrath, K.; Vandeghinste, N.; et al. Preclinical characterization of GLPG0634, a selective inhibitor of JAK1, for the treatment of inflammatory diseases. J. Immunol. 2013, 191, 3568–3577. [Google Scholar] [CrossRef]
- Emery, P.; Durez, P.; Hueber, A.J.; de la Torre, I.; Larsson, E.; Holzkämper, T.; Tanaka, Y. Baricitinib inhibits structural joint damage progression in patients with rheumatoid arthritis-a comprehensive review. Arthritis Res. Ther. 2021, 23, 3. [Google Scholar] [CrossRef]
- Tanaka, Y.; Okumura, H.; Kim, S.; Dorey, J.; Wojciechowski, P.; Chorąży, J.; Kato, D.; Schultz, N.M. Comparative Efficacy and Safety of Peficitinib Versus Tofacitinib and Baricitinib for Treatment of Rheumatoid Arthritis: A Systematic Review and Network Meta-Analysis. Rheumatol. Ther. 2021, 8, 729–750. [Google Scholar] [CrossRef]
- Tanaka, Y.; Kavanaugh, A.; Wicklund, J.; McInnes, I.B. Filgotinib, a novel JAK1-preferential inhibitor for the treatment of rheumatoid arthritis: An overview from clinical trials. Mod. Rheumatol. 2021, 32, 1–11. [Google Scholar] [CrossRef]
- Van Vollenhoven, R.; Takeuchi, T.; Pangan, A.L.; Friedman, A.; Mohamed, M.F.; Chen, S.; Rischmueller, M.; Blanco, R.; Xavier, R.M.; Strand, V. Efficacy and Safety of Upadacitinib Monotherapy in Methotrexate-Naive Patients With Moderately-to-Severely Active Rheumatoid Arthritis (SELECT-EARLY): A Multicenter, Multi-Country, Randomized, Double-Blind, Active Comparator-Controlled Trial. Arthritis Rheumatol. 2020, 72, 1607–1620. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.W.; Kobayashi, Y.; Nakamichi, Y.; Udagawa, N.; Takahashi, N.; Im, N.K.; Seo, H.J.; Jeon, W.B.; Yonezawa, T.; Cha, B.Y.; et al. Alisol-B, a novel phyto-steroid, suppresses the RANKL-induced osteoclast formation and prevents bone loss in mice. Biochem. Pharm. 2010, 80, 352–361. [Google Scholar] [CrossRef] [PubMed]
- Shakibaei, M.; Buhrmann, C.; Mobasheri, A. Resveratrol-mediated SIRT-1 interactions with p300 modulate receptor activator of NF-kappaB ligand (RANKL) activation of NF-kappaB signaling and inhibit osteoclastogenesis in bone-derived cells. J. Biol. Chem. 2011, 286, 11492–11505. [Google Scholar] [CrossRef] [Green Version]
- Shakibaei, M.; Shayan, P.; Busch, F.; Aldinger, C.; Buhrmann, C.; Lueders, C.; Mobasheri, A. Resveratrol mediated modulation of Sirt-1/Runx2 promotes osteogenic differentiation of mesenchymal stem cells: Potential role of Runx2 deacetylation. PLoS ONE 2012, 7, e35712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamashita, T.; Uehara, S.; Udagawa, N.; Li, F.; Kadota, S.; Esumi, H.; Kobayashi, Y.; Takahashi, N. Arctigenin inhibits osteoclast differentiation and function by suppressing both calcineurin-dependent and osteoblastic cell-dependent NFATc1 pathways. PLoS ONE 2014, 9, e85878. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.Y.; Schneeweiss, S.; Liu, J.; Solomon, D.H. Effects of disease-modifying antirheumatic drugs on nonvertebral fracture risk in rheumatoid arthritis: A population-based cohort study. J. Bone Miner. Res. 2012, 27, 789–796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawai, V.K.; Grijalva, C.G.; Arbogast, P.G.; Curtis, J.R.; Solomon, D.H.; Delzell, E.; Chen, L.; Ouellet-Hellstrom, R.; Herrinton, L.; Liu, L.; et al. Initiation of tumor necrosis factor α antagonists and risk of fractures in patients with selected rheumatic and autoimmune diseases. Arthritis Care Res. 2013, 65, 1085–1094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozen, G.; Pedro, S.; Wolfe, F.; Michaud, K. Medications associated with fracture risk in patients with rheumatoid arthritis. Ann. Rheum. Dis. 2019, 78, 1041–1047. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Agent | Participants (Identifier) | Observation Periods | Effects on Bones | Ref |
|---|---|---|---|---|
| MTX | 731 | 6 months | BMD (LS, FN) →,→ | [151] |
| IFX | 36 | 12 months | BMD (LS, FN) →,→ | [152] |
| 102 | 12 months | BMD (LS, FN, Hand) →,→,↓ CTX, RANKL↓ | [153] | |
| 189 | 12 months | BMD (LS, FN) →,→ CTX→, OCN→ | [154] | |
| 48 | 12 months | BMD (LS, FN) →,→ CTX↓, P1NP→ | [155] | |
| 52 | 3.3 years | BMD (LS, FN, Hand) ↑,→,↓ | [156] | |
| ADA | 46 | 12 months | BMD (LS, FN) →,→ | [157] |
| 184 | 4 years | BMD (LS, FN, Hand) →,↓,↓ | [158] | |
| ETN/CZP | 36 | 12 months | BMD (LS, FN) →,→ | [159] |
| TNFi | 54 | 6 months | BMD (LS, FN) →,↓ CTX↑, P1NP↑ | [160] |
| TCZ | 145 | 96 weeks | BMD (LS, FN) →,→ | [161] |
| 86 | 52 weeks | BMD (LS, FN) →,→ | [162] | |
| 103 | 48 weeks | BMD (LS, FN) →,→ DKK1↓, P1NP↑ | [163] | |
| SAR | 259 (NCT01061736) | 24–52 weeks | BMD not measured sRANKL↓ | [164] |
| 291 (NCT01709578) | 24 weeks | BMD not measured tRANKL↓ | [165] | |
| 207 (NCT02332590) | 24 weeks | BMD not measured tRANKL↓, P1NP↑ | [166] | |
| ABT | 165 (UMIN000005570) | 48 weeks | BMD (LS, FN) →,↑ | [167] |
| TOF | 30 | 6–12 months | BMD (LS, FN) →,→ CTX↓, OCN↑ | [168] |
| BAR | 240 (NCT01721057) | 12 months | BMD not measured CTX↓ | [169] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maeda, K.; Yoshida, K.; Nishizawa, T.; Otani, K.; Yamashita, Y.; Okabe, H.; Hadano, Y.; Kayama, T.; Kurosaka, D.; Saito, M. Inflammation and Bone Metabolism in Rheumatoid Arthritis: Molecular Mechanisms of Joint Destruction and Pharmacological Treatments. Int. J. Mol. Sci. 2022, 23, 2871. https://doi.org/10.3390/ijms23052871
Maeda K, Yoshida K, Nishizawa T, Otani K, Yamashita Y, Okabe H, Hadano Y, Kayama T, Kurosaka D, Saito M. Inflammation and Bone Metabolism in Rheumatoid Arthritis: Molecular Mechanisms of Joint Destruction and Pharmacological Treatments. International Journal of Molecular Sciences. 2022; 23(5):2871. https://doi.org/10.3390/ijms23052871
Chicago/Turabian StyleMaeda, Kazuhiro, Ken Yoshida, Tetsuro Nishizawa, Kazuhiro Otani, Yu Yamashita, Hinako Okabe, Yuka Hadano, Tomohiro Kayama, Daitaro Kurosaka, and Mitsuru Saito. 2022. "Inflammation and Bone Metabolism in Rheumatoid Arthritis: Molecular Mechanisms of Joint Destruction and Pharmacological Treatments" International Journal of Molecular Sciences 23, no. 5: 2871. https://doi.org/10.3390/ijms23052871
APA StyleMaeda, K., Yoshida, K., Nishizawa, T., Otani, K., Yamashita, Y., Okabe, H., Hadano, Y., Kayama, T., Kurosaka, D., & Saito, M. (2022). Inflammation and Bone Metabolism in Rheumatoid Arthritis: Molecular Mechanisms of Joint Destruction and Pharmacological Treatments. International Journal of Molecular Sciences, 23(5), 2871. https://doi.org/10.3390/ijms23052871