
Vasopressin and Its Analogues: From Natural Hormones to Multitasking Peptides

 , , , and
, , , and
Abstract
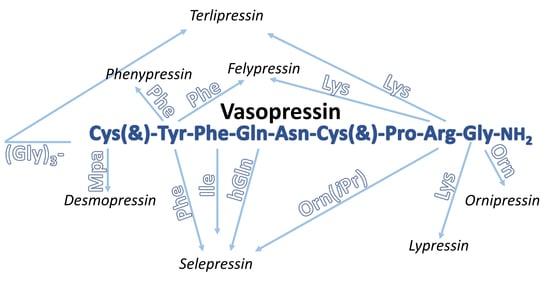
:
1. Introduction
2. Vasopressin
2.1. Vasopressin Analogues
2.1.1. Non-Peptide Synthetic Analogues of AVP
2.1.2. Natural Peptide Analogues of Vasopressin
Lypressin

| Analogue | Sequence | Source | Main Application | Refs. |
|---|---|---|---|---|
| Arginine vasopressin, argipressin, AVP | Cys(&)-Tyr-Phe-Gln-Asn-Cys(&)-Pro-Arg-Gly-NH2 | human and other mammals | antidiuretic effect, maintenances cardiovascular homeostasis, increases blood pressure in septic shock | [38,44,45,46,49,65] |
| Lysine vasopressin, lypressin, LVP | Cys(&)-Tyr-Phe-Gln-Asn-Cys(&)-Pro-Lys-Gly-NH2 | pigs | antidiuretic agent, hemostatic, vasoconstrictor agent | [64,86,87] |
| Phenypressin | Cys(&)-Phe-Phe-Gln-Asn-Cys(&)-Pro-Arg-Gly-NH2 | marsupials (gray and red kangaroo, tammar and quokka wallaby) | increases the reabsorption of water in the kidneys and blood pressure | [90,91] |
| Peptide | Antidiuretic Activity Units/mg | Vasopressor Activity Units/mg | Oxytocic Activity Units/mg | Other Activities and Comments |
|---|---|---|---|---|
| LVP | 203 ± 7 [94] 240 ± 13 [95,96] | 243 ± 3 [94] 266 ± 18 [96] | 7.3 ± 0.2 [96] 4.8 ± 0.3 [94] | AVD a = 48 ± 2 units/mg [94] |
| dLVP (12) b | 301 ± 11 [96] 550 ± 1.7 [97] | 126 ± 2 [96] 145 ± 7 [97] | 12 ± 0.5 [96,97] | - |
| [Dbt2]dLVP (13) c | nr d | nr | nr | AVD = pA2 = 6.97 e; M = 1.1 × 10−7; no measurable antagonistic, oxytocic and pressor activities [98] |
| [Tyr(OMe)2] LVP (14) | 1.5–3 [99] | 79 [99] | antioxytocic and antipressor properties, antagonistic character of these analogues results from the bulky, lipophilic substituents on the aromatic ring rather than from the blocking or elimination of the phenolic group [99] | |
| [Tyr(OEt)2] LVP (15) | nr | 5 [99] | ||
| [Tyr(OX)2] dLVP (16–19) f | 0.5–2.0 units/μmol [100] | 0.5–3.0 units/μmol [100] | weak agonistic properties; in the rat, none of the analogues inhibited the antidiuretic action of LVP when the two substances were administered together in a single injection; completed inhibition was obtained when X = Et; antagonistic potency decrease with increasing size of alkyl substitution [100] | |
| [Thi3]LVP (20) | 332 ± 32 [101,102] | 243 ± 5 [101,102] | 19 ± 0.5 [101,102] | AVD = 87 ± 4 units/mg; steric size in position 3 plays significant role in the manifestation of vasopressin-like activities [101,102] |
| [Ile3]LVP (21) | 24 ± 3 [101,102] | 130 ± 13 [101,102] | 78 ± 10 [101,102] | AVD = 210 ± 3 units/mg [101,102] |
| [Ser3]LVP (22) | ~0.08 [102] | <0.01 [102] | nr | uterotonic activity ≤ 0.01 units/mg [102] |
| [Tyr3]LVP (23) | 0.18 [102] | 1.6 [102] | ||
| [diHPhe3] LVP (24) | 125–130 [102] | 129–132 [102] | uterotonic activity = 6 units/mg; effective agonist; position 3 is not very restrictive for vasopressor receptors and antidiuretic potency [102] | |
| [Leu4]LVP (25) | 1–2 [95] | 1.33 [95] | negligible [95] | AVD = 1 unit/mg [95] |
| [Leu4]dLVP (28) | 5–6 [95] 10.5 ± 0.9 [97] | 0.55 [95] 0.9 ± 0.1 [97] | 0.054 ± 0.002 [97] | AVD = 4.60 units/mg [95] high affinity for the rat V1b receptor, very low affinities for the rat V1a and V2 receptor, potent agonists for the rat V1b receptor, weak agonists for the rat antidiuretic activity [97], Gln is not essential for biological activity [95] |
| [Abu4]LVP (26) | 707 [95] | nr | nr | Gln is not essential for biological activity [95] |
| [Abu4]dLVP (29) | 729 [95] | 3.5 [95] | ||
| [Ala4]LVP (27) | 707 ± 107 [96] | 10.2 ± 0.6 [96] | 1.54 ± 0.1 [96] | - |
| [Ala4]dLVP (30) | 729 ± 26 [96] | 3.5 ± 0.2 [96] | 1.51 ± 0.05 [96] | |
| [Cha4]dLVP (31) | 0.82 ± 0.01 [97] | 0.043 ± 0.008 [97] | pA2 = 6.48 ± 0.03 M = 3.41 × 10−7 ± 0.2 [97] | high affinity for the rat V1b receptor, very low affinities for the rat V1a and V2 receptor, potent agonists for the rat V1b receptor, weak agonists for the rat antidiuretic activity [97] |
| [Orn4]dLVP (32) | 7.8 ± 0.4 [97] | 0.23 ± 0.02 [97] | 3.1 ± 0.1 [97] | - |
| [Arg4]dLVP (33) | 784 ± 54 [97] | 83 ± 4 [97] | 0.15 ± 0.02 [97] | |
| [diMeGln4] LVP (34) | 1.88 ± 0.04 [94] | 1.27 ± 0.03 [94] | <0.05 [94] | AVD ≤ 0.1 units/mg [94] |
| [Ala5]LVP (35) | ~0.2 [96] | 0.15 ± 0.01 [96] | <0.001 [96] | carboxamide group is essential for activity [96] |
| [Ala5]dLVP (36) | ~0.05 [96] | ~0.015 [96] | <0.002 [96] | |
| [diMeAsn5] LVP (37) | 5.5 ± 0.3 [103] | 2.55 ± 0.05 [103] | <0.05 [103] | AVD = 0.39 ± 0.03 units/mg; hydrogen atoms of carboxamide group are not essential for antidiuretic activity [103] |
| [Lys(N-Gly)38]dLVP (38) | nr | nr | nr | more powerful and prolonged analgesia compared to LVP [104] |
| [Eda9]LVP (39) | <0.05; 0.002 [105] | - | ||
| [Eda9]dLVP (40) | 126 [105] |
Phenypressin
2.1.3. Synthetic Peptide Analogues of Vasopressin
| Peptide | Antidiuretic Activity Units/mg | Vasopressor Activity Units/mg (for Agonist) pA2 (for Antagonist) | Oxytocic Activity Units/mg (for Agonist) pA2 (for Antagonist) |
|---|---|---|---|
| AVP (Figure 2a) | 323 [118] 450 (t1/2 = 60) 450 (t1/2 = 200) [119] | 369 [118] 412 [119] | 14 [118] |
| [Tyr(OMe)2]AVP (41) | 386 ± 36 [109] | pA2= 9.7 ± 0.5 a [109] | pA2 = 7.44 ± 0.12 (no Mg2+) and 6.34 ± 0.19 (in 0.5 mM Mg2+) [109] |
| [Cys1(N-Ac), Tyr(OMe)2]AVP (42) | 0.026 ± 0.002 [110] | pA2= 7.18 ± 0.08 [110] | pA2 = 7.29 ± 0.08 (no Mg2+) and 6.73 ± 0.14 (in 0.5 mM Mg2+) [110] |
| [diPhe2]AVP (43) | 450 (t1/2 = 60) 9000 (t1/2 = 200) [112] | 0 [112] | pA2 = 7.00 ± 0.20 [112] |
| [d-diPhe2]AVP (44) | 1000 (t1/2 = 60) 45,000 (t1/2 = 200) [111,112] | 0 [111] | pA2 = 7.82 ± 0.39 [111] |
| [Aic2]AVP (45) | 450 (t1/2 = 60) 45,000 (t1/2 = 200) [77] | 9.4 ± 2.8 [77] | pA2 = 7.27 ± 0.22 (no Mg2+) [77] |
| [Apc2]AVP (46) | 1800 (t1/2 = 60) 1800 (t1/2 = 200) [56] | 13.4 ± 3.8 [56] | 0.2 units/mg pA2 = 6.0 (no Mg2+) [56] |
| [l-1-Nal3, d-Arg8]dAVP (47) | 2.2 ± 0.83 [113] | nr b | nr |
| [l-2-Nal3, d-Arg8]dAVP (48) | 3.79 ± 1.31 [113] | nr | weak [113] |
| [Sar7]AVP (49) | 188 ± 19 [114] | 3.6 ± 0.2 [114] | nr |
| [NMeAla7]AVP (50) | 343 ± 54 [114] | 10.6 ± 0.4 [114] | nr |
| [HNle8]AVP (51) | 10 [116] | 21.4 ± 1.0 [116] | nr |
| [His8]AVP (52) | nr | 1.5 [117] | nr |
| [HyLeu8]AVP (53) | 70 [116] | 30 [116] | nr |
Desmopressin
Selepressin
Felypressin
Ornipressin
Terlipressin
2.2. AVP and Its Analogues in Treatment of SARS-CoV-2
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Koopmanschap, G.; Ruijter, E.; Orru, R.V.A. Isocyanide-based multicomponent reactions towards cyclic constrained peptidomimetics. Beilstein J. Org. Chem. 2014, 10, 544–598. [Google Scholar] [CrossRef] [PubMed]
- Grauer, A.; König, B. Peptidomimetics—A versatile route to biologically active compounds. Eur. J. Org. Chem. 2009, 30, 5099–5111. [Google Scholar] [CrossRef]
- Vaisman, A.; Lushchak, O. Geroscience. In Encyclopedia of Biomedical Gerontology, Reference Modul in Biomedical Science, 1st ed.; Elsevier Inc. Academic Press: Aalborg, Denmark, 2020; pp. 154–159. [Google Scholar] [CrossRef]
- Dawgul, M.A.; Greber, K.E.; Sawicki, W.; Kamysz, W. Human host defense peptides—Role in maintaining human homeostasis and pathological processes. Curr. Med. Chem. 2017, 24, 654–672. [Google Scholar] [CrossRef]
- Lenci, E.; Trabocchi, A. Peptidomimetic toolbox for drug discovery. Chem. Soc. Rev. 2020, 49, 3262–3277. [Google Scholar] [CrossRef] [PubMed]
- Galicia-Garcia, U.; Benito-Vicente, A.; Jebari, S.; Larrea-Sebal, A.; Siddiqi, H.; Uribe, B.K.; Ostolaza, H.; Martín, C. Pathophysiology of type 2 diabetes mellitus. Int. J. Mol. Sci. 2020, 21, 6275. [Google Scholar] [CrossRef] [PubMed]
- Cantley, J.; Ashcroft, F.M. Q&A: Insulin secretion and type 2 diabetes: Why do β-cells fail? BMC Biol. 2015, 13, 33. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Chen, W.D.; Wang, Y.-D. β-Amyloid: The key peptide in the pathogenesis of Alzheimer’s disease. Front. Pharmacol. 2015, 6, 221. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.-F.; Xu, T.-H.; Zhou, Y.-R.; Jiang, Y.; Melcher, K.; Xu, H.E. Amyloid beta: Structure, biology and structure-based therapeutic development. Acta Pharmacol. Sin. 2017, 38, 1205–1235. [Google Scholar] [CrossRef]
- Martin, V.V.; Egelund, P.G.H.; Johansson, H.; Thordal Le Quement, S.; Wojcik, F.; Sejer Pedersen, D. Greening the synthesis of peptide therapeutics: An industrial perspective. RSC Adv. 2020, 10, 42457–42492. [Google Scholar] [CrossRef]
- Lee, A.C.-L.; Harris, J.L.; Khanna, K.K.; Hong, J.H. A comprehensive review on current advances in peptide drug development and design. Int. J. Mol. Sci. 2019, 20, 2383. [Google Scholar] [CrossRef] [Green Version]
- Lau, J.L.; Dunn, M.K. Therapeutic peptides: Historical perspectives, current development trends, and future directions. Bioorg. Med. Chem. 2018, 26, 2700–2707. [Google Scholar] [CrossRef] [PubMed]
- Adessi, C.; Soto, C. Converting a Peptide into a Drug: Strategies to Improve Stability and Bioavailability. Curr. Med. Chem. 2005, 9, 963–978. [Google Scholar] [CrossRef] [PubMed]
- Albericio, F.; Kruger, H.G. Therapeutic peptides. Future Med. Chem. 2012, 4, 1527–1531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henninot, A.; Collins, J.C.; Nuss, J.M. The Current State of Peptide Drug Discovery: Back to the Future? J. Med. Chem. 2018, 61, 1382–1414. [Google Scholar] [CrossRef] [PubMed]
- Räder, A.F.B.; Weinmüller, M.; Reichart, F.; Schumacher-Klinger, A.; Merzbach, S.; Gilon, C.; Hoffmann, A.; Kessler, H. Orally Active Peptides: Is There a Magic Bullet? Angew. Chemie-Int. Ed. 2018, 57, 14414–14438. [Google Scholar] [CrossRef]
- Chia, C.S.B. A Review on the Metabolism of 25 Peptide Drugs. Int. J. Pept. Res. Ther. 2021, 27, 1397–1418. [Google Scholar] [CrossRef]
- Hamman, J.H.; Enslin, G.M.; Kotzé, A.F. Oral delivery of peptide drugs: Barriers and developments. BioDrugs 2005, 19, 165–177. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.; Patel, M.; Yang, X.; Mitra, A. Recent advances in protein and Peptide drug delivery: A special emphasis on polymeric nanoparticles. Protein Pept. Lett. 2014, 21, 1102–1120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huttunen, K.M.; Raunio, H.; Rautio, J. Prodrugs-from serendipity to rational design. Pharmacol. Rev. 2011, 63, 750–771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshimura, M.; Conway-Campbell, B.; Ueta, Y. Arginine vasopressin: Direct and indirect action on metabolism. Peptides 2021, 142, 170555. [Google Scholar] [CrossRef]
- Garrahy, A.; Thompson, C.J. Vasopressin. In Encyclopedia of Endocrine Diseases, 2nd ed.; Academic Press: Oxford, UK, 2018; pp. 29–35. [Google Scholar] [CrossRef]
- Russell, J.A. Vasopressor therapy in critically ill patients with shock. Intensive Care Med. 2019, 45, 1503–1517. [Google Scholar] [CrossRef]
- Bankir, L.; Bichet, D.G.; Morgenthaler, N.G. Vasopressin: Physiology, assessment and osmosentation. J. Int. Med. 2017, 282, 284–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, H.-H.; Yuan, X.S.; Chen, Z.-K.; Chen, P.-P.; Xiang, Z.; Qu, W.-M.; Li, R.-X.; Zhou, G.-M.; Huang, Z.-H. Presynaptic inputs to vasopressin neurons in the hypothalamic supraoptic nucleus and paraventricular nucleus in mice. Exp. Neurol. 2021, 343, 113784. [Google Scholar] [CrossRef] [PubMed]
- Russell, J.A.; Gordon, A.C.; Williams, M.D.; Oyd, J.H.; Walley, K.R.; Kissoon, N. Vasopressor Therapy in the Intensive Care Unit. Semin. Respir. Crit. Care Med. 2021, 42, 59–77. [Google Scholar] [CrossRef] [PubMed]
- Mitra, A.K. Oxytocin and vasopressin: The social networking buttons of the body. AIMS Mol. Sci. 2021, 8, 32–50. [Google Scholar] [CrossRef]
- Oliver, H.; Shafer, E. On the physiological action of extracts of the pituitary body and certain other glandular organs. J. Physiol. 1895, 18, 277–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- du Vigneaud, V.; Popenoe, E.A.; Lawler, H. Partial purification and amino acid content of vasopressin from hog posterior. pituitary glands. J. Am. Chem. Soc. 1952, 74, 3713. [Google Scholar] [CrossRef]
- Berde, B.; Weidmann, H.; Cerletti, A. On phenylalanine-2-lysine-vasopressin. Helv. Physiol. Pharmacol. Acta 1961, 19, 285. [Google Scholar] [PubMed]
- Huguenin, R.L.; Boissonnas, R.A. Syntheses de la Phe2-arginine-vasopressine et de la Phe2-arginine vasotocineet nouvelles synthese de l’argininevasopressine et de l’arginine-vasotocine. Helv. Chim. Acta 1962, 45, 1629. [Google Scholar] [CrossRef]
- Berde, B.; Boissonnas, R.A.; Hueuenin, R.L.; Strümer, E. Vasopressin analogues with selective pressor activity. Experientia 1964, 20, 42–43. [Google Scholar] [CrossRef] [PubMed]
- Kasafire, E.; Rabek, V.; Rudinger, J.; Šorm, F. Amino acids and peptides. Synthesis of ten extended-chain analogues of lysine vasopressin. Coll. Czech. Chem. Commun. 1966, 31, 4581. [Google Scholar] [CrossRef]
- Chauvet, M.T.; Hurpet, D.; Chauvet, J.; Acher, R. Phenypressin (Phe2-Arg8-vasopressin), a new neurohypophysial peptide found in marsupials. Nature 1980, 287, 640–642. [Google Scholar] [CrossRef] [PubMed]
- Wisniewski, K.; Galyean, R.; Croston, G.; Laporte, R.; Taki, H.; Schteingart, C.D.; Rivière, P.-J.M. Synthesis and in vitro. pharmacological profile of potent and selective peptidic V1a receptor agonists. In Pept. Youth; Springer: New York, NY, USA, 2009; pp. 507–508. [Google Scholar] [CrossRef]
- Callahan, J.F.; Ashton-Shue, D.; Bryan, H.G.; Bryan, W.M.; Heckman, G.D.; Kinter, L.B.; McDonald, J.E.; Moore, M.L.; Schmidt, D.B.; Silvestri, J.S.; et al. Structure-Activity Relationships of Novel Vasopressin Antagonists Containing C-Terminal Diaminoalkanes and (Aminoalkyl)guanidines. J. Med. Chem. 1989, 32, 391–396. [Google Scholar] [CrossRef] [PubMed]
- Rubini, C.; Osler, A.; Calderan, A.; Guiotto, A.; Ruzza, P. Mechanistic studies of amide bond scission during acidolytic deprotection of Pip containing peptide. J. Pept. Sci. 2008, 14, 989–997. [Google Scholar] [CrossRef] [PubMed]
- Dekan, Z.; Kremsmayr, T.; Keov, P.; Godin, M.; Teakle, N.; Dürrauer, L.; Xiang, H.; Gharib, D.; Bergmayr, C.; Hellinger, R.; et al. Nature-inspired dimerization as a strategy to modulate neuropeptide pharmacology exemplified with vasopressin and oxytocin. Chem. Sci. 2021, 12, 4057–4062. [Google Scholar] [CrossRef] [PubMed]
- Wilds, A.L.; Ralls, W.; Tyner, A.; Daniel, R.; Kraychy, S.; Harnik, M. The synthesis of an octapeptide amide with the hormonal activity of oxytocin. J. Am. Chem. Soc. 1953, 75, 4879–4880. [Google Scholar]
- Alexander, R.; Aragón, O.R.; Bookwala, J.; Cherbuin, N.; Gatt, J.M.; Kahrilas, I.J.; Kästern, N.; Lawrence, A.; Lowe, L.; Morrison, R.G.; et al. The neuroscience of positive emotions and affect: Implications for cultivating happiness and wellbeing. Neurosci. Biobehav. Rev. 2021, 121, 220–249. [Google Scholar] [CrossRef] [PubMed]
- Goh, K.K.; Chen, C.-H.; Lane, H.-Y. Oxytocin in schizophrenia: Pathophysiology and implications for future treatment. Int. J. Mol. Sci. 2021, 22, 2146. [Google Scholar] [CrossRef] [PubMed]
- Gimpl, G.; Fahrenholz, F. The oxytocin receptor system: Structure, function, and regulation. Physiol. Rev. 2001, 81, 629–683. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Huang, X.; Ebstein, R.P.; Yu, R. Intranasal oxytocin in the treatment of autism spectrum disorders: A multilevel meta-analysis. Neurosci. Biobehav. Rev. 2021, 122, 18–27. [Google Scholar] [CrossRef]
- Land, H.; Schütz, G.; Schmale, H.; Richter, R. Nucleotide sequence of cloned cDNA encoding bovine arginine vasopressin-neurophysin II precursor. Nature 1982, 295, 299–303. [Google Scholar] [CrossRef] [PubMed]
- Küchler, S.; Perwitz, N.; Schick, R.R.; Klein, J.; Westphal, S. Arginine-vasopressin directly promotes a thermogenic and pro-inflammatory adipokine expression profile in brown adipocytes. Regul. Pept. 2010, 164, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Mohan, S.; Moffett, R.C.; Thomas, K.G.; Irwin, N.; Flatt, P.R. Vasopressin receptors in islets enhance glucose tolerance, pancreatic beta-cell secretory function, proliferation and survival. Biochimie 2019, 158, 191–198. [Google Scholar] [CrossRef]
- Cotte, N.; Balestre, M.-N.; Phalipou, S.; Hibert, M.; Manning, M.; Barberis, C.; Mouillac, B. Identification of residues responsible for the selective binding of peptide antagonists and agonists in the V2 vasopressin receptor. J. Biol. Chem. 1998, 273, 29462–29468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimada, Y.; Taniguchi, N.; Matsuhisa, A.; Akane, H.; Kawano, N.; Suzuki, T.; Tobe, T.; Kakefuda, A.; Yatsu, T.; Tahara, A.; et al. Synthesis and biological activity of novel 4,4-difluorobenzazepine derivatives as non-peptide antagonists of the arginine vasopressin V 1A receptor. Bioorg. Med. Chem. 2006, 14, 1827–1837. [Google Scholar] [CrossRef] [PubMed]
- Zeynalov, E.; Jones, S.M.; Elliott, J.P. Vasopressin and vasopressin receptors in brain edema. Vitam. Horm. 2020, 113, 291–312. [Google Scholar] [CrossRef] [PubMed]
- Tsukamoto, I.; Koshio, H.; Akamatsu, S.; Kuramochi, T.; Saitoh, C.; Yatsu, T.; Yanai-Inamura, H.; Kitada, C.; Yamamoto, E.; Sakamoto, S.; et al. Preparation of (4,4-difluoro-1,2,3,4-tetrahydro-5H-1-benzazepin-5-ylidene)acetamide derivatives as novel arginine vasopressin V2 receptor agonists. Bioorg. Med. Chem. 2008, 16, 9524–9535. [Google Scholar] [CrossRef]
- Sobolewski, D.; Prahl, A.; Derdowska, I.; Kwiatkowska, A.; Slaninova, J.; Lammek, B. Influence of Conformationally Constrained Amino Acids Replacing Positions 2 and 3 of Arginine Vasopressin (AVP) and Its Analogues on Their Pharmacological Properties. Protein Pept. Lett. 2017, 14, 213–217. [Google Scholar] [CrossRef] [PubMed]
- Demiselle, J.; Fage, N.; Radermacher, P.; Asfar, P. Vasopressin and its analogues in shock states: A review. Ann. Intensive Care 2020, 10, 9. [Google Scholar] [CrossRef] [PubMed]
- Corbani, M.; Trueba, M.; Stoev, S.; Murat, B.; Mion, J.; Boulay, V.; Guillon, G.; Manning, M. Design, synthesis, and pharmacological characterization of fluorescent peptides for imaging human V1b vasopressin or oxytocin receptors. J. Med. Chem. 2011, 54, 2864–2877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Estrada, E.F.; Barra, V.; Caorsi, C.E.; Troncoso, S.; González, C.B.; Ruiz-Opazo, N. Identification of the V1 Vasopressin Receptor by Chemical Cross-Linking and Ligand Affinity Blotting. Biochemistry 1991, 30, 8611–8616. [Google Scholar] [CrossRef] [PubMed]
- Łempicka, E.; Derdowska, I.; Kowalczyk, W.; Dawidowska, O.; Prahl, A.; Janecki, M.; Jasiński, T.; Trzeciak, H.I.; Lammek, B. Analogues of arginine vasopressin modified in position 2 and 3 with conformationally constrained dipeptide fragments. J. Pept. Sci. 2005, 11, 91–96. [Google Scholar] [CrossRef] [PubMed]
- Kowalczyk, W.; Prahl, A.; Dawidowska, O.; Sobolewski, D.; Hardtrodt, B.; Lammek, B. The influence of 1-aminocyclopentane-1-carboxylic acid at position 2 or 3 of AVP and its analogues on their pharmacological properties. J. Pept. Sci. 2005, 11, 584–588. [Google Scholar] [CrossRef] [PubMed]
- Cheetham, T.; Baylis, P.H. Diabetes insipidus in children: Pathophysiology, diagnosis and management. Pediatr. Drugs 2002, 4, 785–796. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, N.; Fohiaer, J.; Marples, D.; Kwon, T.-H.; Agre, P.; Knepper, M.A. Aquaporins in the kidney: From molecules to medicine. Physiol. Rev. 2002, 1, 205–244. [Google Scholar] [CrossRef] [PubMed]
- Bockenhauer, D.; Bichet, D. Pathophysiology, diagnosis and management of nephrogenic diabetes insipidus. Nat. Rev. Nephrol. 2015, 11, 576–588. [Google Scholar] [CrossRef] [PubMed]
- Yea, C.M.; Allan, C.E.; Ashworth, D.M.; Barnett, J.; Baxter, A.J.; Broadbridge, J.D.; Franklin, R.J.; Hampton, S.L.; Hudson, P.; Horton, J.A.; et al. New benzylureas as a novel series of potent, nonpeptidic vasopressin V2 receptor agonists. J. Med. Chem. 2008, 51, 8124–8134. [Google Scholar] [CrossRef] [PubMed]
- Tsukamoto, I.; Koshio, H.; Kuramochi, T.; Saitoh, C.; Yanai-Inamura, H.; Kitada-Nozawa, C.; Yamamoto, E.; Yatsu, T.; Shimada, Y.; Sakamoto, S.; et al. Synthesis and structure-activity relationships of amide derivatives of (4,4-difluoro-1,2,3,4-tetrahydro-5H-1-benzazepin-5-ylidene)acetic acid as selective arginine vasopressin V2 receptor agonists. Bioorg. Med. Chem. 2009, 17, 3130–3141. [Google Scholar] [CrossRef]
- Holz, G.G.; Kang, G.; Harbeck, M.; Roe, M.W.; Chepurny, O.G. Cell physiology of cAMP sensor. Epac. J. Physiol. 2006, 577, 5–15. [Google Scholar] [CrossRef] [PubMed]
- Chou, C.-L.; Christense, B.M.; Frische, S.; Vorum, H.; Desai, R.A.; Hoffert, J.D.; de Lanerolle, P.; Nielsen, S.; Knepper, M.A. Non-muscle myosin II and myosin light chain kinase are downstream targets for vasopressin signaling in the renal collecting duct. J. Biol. Chem. 2004, 279, 49026–49035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Favory, R.; Salgado, D.R.; Vincent, J.-L. Investigational vasopressin receptor modulators in the pipeline. Expert Opin. Investig. Drugs 2009, 18, 1119–1131. [Google Scholar] [CrossRef] [PubMed]
- Felmet, K.; Carcillo, J.A. Neuroendocrine-Immune Mediator Coordination and Disarray in Critical Illness. In Pediatric Critical Care; Mosby Inc.: Maryland Heights, MO, USA, 2006; pp. 1462–1473. [Google Scholar] [CrossRef]
- Narayen, G.; Mandal, S. Vasopressin receptor antagonists and their role in clinical medicine. Indian J. Endocrinol. Metab. 2012, 16, 183–191. [Google Scholar] [CrossRef]
- Masuda, S.; Hattori, A.; Matsumoto, H.; Miyazawa, S.; Natori, Y.; Mizutani, S.; Tsujimoto, M. Involvement of the V2 receptor in vasopressin-stimulated translocation of placental leucine aminopeptidase/oxytocinase in renal cells. Eur. J. Biochem. 2003, 270, 1988–1994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tahara, A.; Tsukada, J.; Tomura, Y.; Wada, K.; Kusayama, T.; Ishii, N.; Tanaka, A. Effect of YM471, a nonpeptide AVP receptor antagonist, on human coronary artery smooth muscle cells. Peptides 2002, 23, 1809–1816. [Google Scholar] [CrossRef]
- Kim, J.W.; Kim, G.; Kim, T.W.; Han, W.; Kim, M.S.; Jeong, C.Y.; Park, D.H. Hemodynamic changes following accidental infiltration of a high dose of vasopressin. J. Int. Med. Res. 2020, 48, 0300060520959494. [Google Scholar] [CrossRef] [PubMed]
- Antoni, F.A. Vasopressin as a Stress Hormone. In Stress: Neuroendocrinology and Neurobiology: Handbook of Stressseries; Academic Press: Cambridge, MA, USA, 2017; Volume 2, pp. 97–108. [Google Scholar] [CrossRef]
- de Franchis, R. Somatostatin, somatostatin analogues and other vasoactive drugs in the treatment of bleeding oesophageal varices. Dig. Liver Dis. 2004, 36, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Vasostrict Información Española De la Droga. Available online: https://www.drugs.com/mtm_esp/vasostrict.html (accessed on 11 December 2021).
- Vasopressin Price of 9 Brands/Trade Names|Medindia. Available online: https://www.medindia.net/drug-price/vasopressin.htm (accessed on 11 December 2021).
- Empressin®—Amomed. Available online: https://www.amomed.com/product/empressin-3/?lang=en&cn-reloaded=1 (accessed on 11 December 2021).
- Reverpleg®—Amomed. Available online: https://www.amomed.com/product/empressin-3-2/?lang=fr (accessed on 11 December 2021).
- Xiao, X.; Zhu, Y.; Zhen, D.; Chen, X.M.; Yue, W.; Liu, L.; Li, T. Beneficial and side effects of arginine vasopressin and terlipressin for septic shock. J. Surg. Res. 2015, 195, 568–579. [Google Scholar] [CrossRef] [PubMed]
- Kowalczyk, W.; Sobolewski, D.; Prahl, A.; Derdowska, I.; Borovičková, L.; Slaninová, J.; Lammek, B. The effects of N-terminal part modification of arginine vasopressin analogues with 2-aminoindane-2-carboxylic acid: A highly potent V2 agonist. J. Med. Chem. 2007, 50, 2926–2929. [Google Scholar] [CrossRef] [PubMed]
- Kakefuda, A.; Suzuki, T.; Tobe, T.; Tsukada, J.; Tsukamoto, S. Synthesis and pharmacological evaluation of 5-(4-biphenyl)-3-methyl-4-phenyl-1,2,4-triazole derivatives as a novel class of selective antagonists for the human vasopressin V1a receptor. J. Med. Chem. 2002, 45, 2589–2598. [Google Scholar] [CrossRef] [PubMed]
- Guillon, C.D.; Koppel, G.A.; Brownstein, M.J.; Chaney, M.O.; Ferris, C.F.; Lu, S.; Fabio, K.M.; Miller, M.J.; Heindel, N.D.; Hunden, D.C.; et al. Azetidinones as vasopressin V1a antagonists. Bioorganic Med. Chem. 2007, 15, 2054–2080. [Google Scholar] [CrossRef] [Green Version]
- Schnider, P.; Bissantz, C.; Bruns, A.; Dolente, C.; Goetschi, E.; Jakob-Roetne, R.; Künnecke, B.; Mueggler, T.; Muster, W.; Parrott, N.; et al. Discovery of Balovaptan, a Vasopressin 1a Receptor Antagonist for the Treatment of Autism Spectrum Disorder. J. Med. Chem. 2020, 63, 1511–1525. [Google Scholar] [CrossRef] [PubMed]
- Search of: Balovaptan—List Results—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/results?cond=&term=Balovaptan&cntry=&state=&city=&dist= (accessed on 11 December 2021).
- Kondo, K.; Ogawa, H.; Shinohara, T.; Kurimura, M.; Tanda, Y.; Kan, K.; Yamashita, H.; Nakamura, S.; Hirano, T.; Yamamura, Y.; et al. Novel design of nonpeptide AVP V2 receptor agonists: Structural requirements for an agonist having 1-(4-aminobenzoyl)-2,3,4,5-tetrahydro-1H-1-benzazepine as a template. J. Med. Chem. 2000, 43, 4388–4397. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.; Murakami, K.; Nakanishi, H.; Fujisawa, A.; Isoshima, H.; Niwa, M.; Hayakawa, H.; Hase, Y.; Uchida, I.; Watanabe, H.; et al. Synthesis and Structure—Activity Relationships of 5, 6, 7, 8-Tetrahydro-4 H-thieno [3,2-b] azepine Derivatives: Novel Arginine Vasopressin Antagonists. J. Med. Chem. 2004, 47, 101–109. [Google Scholar] [CrossRef]
- Poulani, D. Vasopressin and Oxytocin, 1st ed.; Elsevier Science: Amsterdam, The Netherlands, 2002. [Google Scholar]
- Maxfield, F.R.; Scheraga, H.A. A Raman Spectroscopic Investigation of the Disulfide Conformation in Oxytocin and Lysine Vasopressin. Biochemistry 1977, 16, 4443–4449. [Google Scholar] [CrossRef] [PubMed]
- Lypressin. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Lypressin (accessed on 15 December 2021).
- Sharman, A.; Low, J. Vasopressin and its role in critical care. Contin. Educ. Anaesth. Crit. Care Pain 2008, 8, 134–137. [Google Scholar] [CrossRef] [Green Version]
- Dakters, J.G.; Yashon, D.; Purnell, E.W.; Volk, M.; White, R.J. Ultrasonography for Diagnosis of Orbital Tumors. JAMA J. Am. Med. Assoc. 1968, 203, 803–805. [Google Scholar] [CrossRef]
- Dingman, J.H.; Hauger-Klevene, J.F. Treatment of Diabetes Insipidus: Synthetic Lysine Vasopressin Nasal Solution. J. Clin. Endocr. 1964, 24, 550–553. [Google Scholar] [CrossRef] [PubMed]
- Acher, R. The nonmammalian-mammalian transition through neurohypophysial peptides. Peptides 1985, 6, 309–314. [Google Scholar] [CrossRef]
- Hurpet, D.; Chauvet, M.T.; Chauvet, J.; Acher, R. Marsupial hypothalamo-neurohypophyseal hormones. Int. J. Pept. Protein Res. 1982, 19, 366–371. [Google Scholar] [CrossRef]
- Jarial, K.D.S.; Bhansali, A.; Gupta, V.; Singh, P.; Mukherjee, K.K.; Sharma, A.; Vashishtha, R.K.; Sukumar, S.P.; Sachdeva, N.; Wlia, R. Diagnostic accuracy and comparison of BIPSS in response to lysine vasopressin and hCRH. Endocr. Connect. 2018, 7, 425–432. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Ba, Y.; Xing, Q.; Cai, R.C. Differential diagnostic value of bilateral inferior Petrosal sinus sampling (BIPSS) in ACTH-dependent Cushing syndrome: A systematic review and Meta-analysis. BMC Endocr. Disord. 2020, 20, 143. [Google Scholar] [CrossRef] [PubMed]
- Stahl, G.L.; Smith, C.W.; Walter, R. Oxytocin and Lysine-vasopressin with N5,N5-Dialkylglutamine in the 4 Position: Effect of Introducing Sterically Hindered Groups into the Hydrophilic Cluster of Neurohypophyseal Hormones. J. Med. Chem. 1980, 23, 213–217. [Google Scholar] [CrossRef] [PubMed]
- Dyckes, D.F.; Ferger, M.F.; du Vigneaud, V.; Chan, W.Y. Synthesis and Some of the Pharmacological Properties of [4-Leucine]-8-lysine-vasopressin and [1-Deamino,4-leucine]-8-lysine-vasopressin. J. Med. Chem. 1973, 16, 843–847. [Google Scholar] [CrossRef]
- Gillessen, D.; du Vigneaud, V. The Synthesis and Pharmacological Properties of 4-Decarboxamido-8-lysine-vasopressin, 5-Decarboxamido-8-lysine-vasopressin, and Their 1-Deamino Analogues. J. Biol. Chem. 1967, 242, 4806–4812. [Google Scholar] [CrossRef]
- Pena, A.; Murat, B.; Trueba, M.; Ventura, M.A.; Wo, N.C.; Szeto, H.H.; Cheng, L.L.; Stoev, S.; Guillon, G.; Manning, M. Design and Synthesis of the First Selective Agonists for the Rat Vasopressin V1bReceptor: Based on Modifications of Deamino-[Cys]arginine Vasopressin at Positions 4 and 8. J. Med. Chem. 2007, 50, 835–847. [Google Scholar] [CrossRef] [PubMed]
- Lundell, E.O.; Ferger, M.F. Synthesis and some pharmacological properties of [1-beta-mercaptopropionic acid, 2-3,5-dibromo-L-Tyrosine)]-8-Lysine-Vasopressin. Bioorg. Chem. 1975, 4, 377–384. [Google Scholar] [CrossRef]
- Siedel, W.; Sturm, K.; Geiger, R. Syntheseeines vasopressorisch wirkenden Peptids: 0-Methyl-tyrosin2-Lysin8-Vasopressin (OMTLV). Eur. J. Inorg. Chem. 1963, 96, 1436–1440. [Google Scholar] [CrossRef]
- Larsson, L.E.; Lindeberg, G.; Melin, P.; Pliska, V. Synthesis of O-alkylated lysine-vasopressin, inhibitors of the antidiuretic response to lysine-vasopressin. J. Med. Chem. 1978, 21, 352–356. [Google Scholar] [CrossRef]
- Smith, C.W.; Ferger, M.F.; Chan, W.Y. Synthesis and some pharmacological properties of [3-.beta.-(2-thienyl)-L-alanine]-8-lysine-vasopressin. J. Med. Chem. 1975, 18, 822–825. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.N.; Diamond, L.; Ressler, C.; Sawyer, W.H. [3-(1,4-Cyclohexadienyl)-L-alanine,8-lysine]vasopressin synthesis and some pharmacological properties. J. Med. Chem. 1979, 22, 1487–1492. [Google Scholar] [CrossRef]
- Smith, C.W.; Walter, R.; Stavropoulos, G.; Theodoropoulos, D. [5-(N4,N4-Dimethylasparagine),8-lysine]vasopressin: The first 5-position neurohypophyseal hormone analog to retain significant antidiuretic potency. J. Med. Chem. 1980, 23, 217–219. [Google Scholar] [CrossRef] [PubMed]
- Kordower, J.H.; Sikorszky, V.; Bodnar, R.J. Central antinociceptive effects of lysine-vasopressin and an analogue. Peptides 1982, 3, 613–617. [Google Scholar] [CrossRef]
- Glass, J.D.; Du Vigneaud, V. Solid-phase synthesis and pressor potency of [1-deamino-9-ethylenediamine]-lysine-vasopressin. J. Med. Chem. 1973, 16, 160–161. [Google Scholar] [CrossRef] [PubMed]
- Chauvet, M.T.; Hurpet, D.; Chauvet, J.; Acher, R. Identification of mesotocin, lysine vasopressin, and phenypressin in the eastern gray kangaroo (Macropus giganteus). Gen. Comp. Endocrinol. 1983, 49, 63–72. [Google Scholar] [CrossRef]
- Berde, B.; Boissonnas, R.A. Basic Pharmacological Properties of Synthetic Analogues and Homologues of the Neurohypophysial Hormones. In Neurohypophysial Hormones and Similar Polypeptides; Springer: Berlin/Heidelberg, Germany, 1968; pp. 802–870. [Google Scholar] [CrossRef]
- Kowalczyk, W.; Prahl, A.; Derdowska, I.; Dawidowska, O.; Slaninová, J.; Lammek, B. Highly Potent 1-Aminocyclohexane-1-Carboxylic Acid Substituted V2 Agonists of Arginine Vasopressin. J. Med. Chem. 2004, 47, 6020–6024. [Google Scholar] [CrossRef] [PubMed]
- Bankowski, K.; Manning, M.; Haldar, J.; Sawyer, W.H. Design of potent antagonists of the vasopressor response to arginine-vasopressin. J. Med. Chem. 1978, 21, 850–853. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.A., Jr.; Sawyer, W.H. N-Acetyl-[2-(O-methyl)tyrosine]arginine-vasopressin, an interesting antagonist of the vasopressor response to vasopressin. J. Med. Chem. 1980, 23, 696–698. [Google Scholar] [CrossRef]
- Kwiatkowska, A.; Sobolewski, D.; Prahl, A.; Borovicková, L.; Slaninová, J.; Lammek, B. Arginine vasopressin and its analogues--the influence of position 2 modification with 3,3-diphenylalanine enantiomers. Highly potent V2 agonists. Eur. J. Med. Chem. 2009, 44, 2862–2867. [Google Scholar] [CrossRef]
- Lebl, M.; Brtnik, F. Tables of Analogs in: Handbook of Neurohypophyseal Hormone Analogs; Jošt, K., Lebl, M., Brtnik, F., Eds.; CRC Press: Boca Raton, FL, USA, 1987; Part 2; Volume 2, pp. 127–167. [Google Scholar]
- Lammek, B.; Czaja, M.; Derdowska, I.; Rekowski, P.; Trzeciak, H.; Sikora, P.; Szkróbka, W.; Stojko, R.; Kupryszewski, G. Influence of L-naphthylalanine in position 3 of AVP and its analogues on their pharmacological properties. J. Pept. Res. 1997, 49, 261–268. [Google Scholar] [CrossRef]
- Grzonka, Z.; Lammek, B.; Kasprzykowski, F.; Gazis, D.; Schwartz, I.L. Synthesis and some pharmacological properties of oxytocin and vasopressin analogues with sarcosine or N-methyl-L-alanine in position 7. J. Med. Chem. 1983, 26, 555–559. [Google Scholar] [CrossRef]
- Ali, F.E.F.; Bryan, W.; Chang, H.L.; Huffman, W.F.; Moore, M.L.; Heckman, G.; Kinter, L.B.; McDonald, J.; Schmidt, D. Potent vasopressin antagonists lacking the proline residue at position 7. J. Med. Chem. 1986, 29, 984–988. [Google Scholar] [CrossRef]
- Fink, M.L.; Bodanszyky, M. Synthesis and hormonal activities of 8-L-homonorleucine-vasopressin. J. Med. Chem. 1973, 16, 1324–1326. [Google Scholar] [CrossRef]
- Katsoyannis, P.G.; du Vigneaud, V. The synthesis of the histidine analog of the vasopressins. Arch. Biochem. Biophys. 1958, 78, 555–562. [Google Scholar] [CrossRef]
- Manning, M.; Coy, E.J.; Sawyer, W.H.; Acosta, M. Solid-phase synthesis and some pharmacological properties of 4-threonine analogs of vasopressins and vasotocin and of arginine-vasopressin and arginine-vasotocin. J. Med. Chem. 1973, 16, 463–466. [Google Scholar] [CrossRef]
- Paranjape, S.B.; Thibonnier, M. Development and therapeutic indications of orally-active non-peptide vasopressin receptor antagonists. Expert Opin. Investig. Drugs 2001, 10, 825–834. [Google Scholar] [CrossRef]
- Wang, J.; Yadav, V.; Smart, A.L.; Tajiri, S.; Basit, A.W. Toward Oral Delivery of Biopharmaceuticals: An Assessment of the Gastrointestinal Stability of 17 Peptide Drugs. Mol. Pharm. 2015, 12, 966–973. [Google Scholar] [CrossRef] [Green Version]
- Manning, M.; Stoev, S.; Chini, B.; Durroux, T.; Mouillac, B.; Guillon, G. Peptide and non-peptide agonists and antagonists for the vasopressin and oxytocin V1a, V1b, V2 and OT receptors: Research tools and potential therapeutic agents. Prog. Brain Res. 2008, 170, 473–512. [Google Scholar] [CrossRef]
- Zaoral, M.; Kolc, J.; Šorm, F. Amino acids and peptides. LXXI. Synthesis of 1-deamino-8-D-γ-aminobutyrine-vasopressin, 1-deamino-8-D-lysine-vasopressin, and 1-deamino-8-D-arginine-vasopressin. Collect. Czechoslov. Chem. Commun. 1967, 32, 1250–1257. [Google Scholar] [CrossRef]
- Krchnak, V.; Zaoral, M. Synthesis of (1-β-mercaptopropionic acid, 8-D-arginine) vasopressin (DDAVP) in solid phase. Simple optimization. Coll. Czech. Chem. Commun. 1979, 44, 1173–1178. [Google Scholar] [CrossRef]
- Manning, M. Impact of the Merrifield solid phase method on the design and synthesis of selective agonists and antagonists of oxytocin and vasopressin: A historical perspective. Biopolymers 2008, 90, 203–212. [Google Scholar] [CrossRef] [Green Version]
- Lewis, R.J.; Angus, D.C.; Laterre, P.-F.; Kjølbye, A.L.; Van Der Meulen, E.; Blemings, A.; Graves, T.; Russell, J.A.; Carlsen, J.E.; Jacobsen, K.; et al. Rationale and Design of an Adaptive Phase 2b/3 Clinical Trial of Selepressin for Adults in Septic Shock. Selepressin Evaluation Programme for Sepsis-induced Shock—Adaptive Clinical Trial. Ann. Am. Thorac. Soc. 2018, 15, 250–257. [Google Scholar] [CrossRef]
- Barabutis, N.; Marinova, M.; Solopov, P.; Uddin, M.A.; Croston, G.E.; Reinheimer, T.M.; Catravas, J.D. Protective Mechanism of the Selective Vasopressin V1A Receptor Agonist Selepressin against Endothelial Barrier Dysfunction. J. Pharmacol. Exp. Ther. 2020, 375, 286–295. [Google Scholar] [CrossRef]
- Milano, S.P.; Boucheix, O.B.; Reinheimer, T.M. Selepressin, a novel selective V1A receptor agonist: Effect on mesenteric flow and gastric mucosa perfusion in the endotoxemic rabbit. Peptides 2020, 129, 170318. [Google Scholar] [CrossRef]
- Berde, B.; Doepfner, W.; Konzett, H. Some Pharmacological Actions of Four Synthetic Analogues Of Oxytocin. Br. J. Pharmacol. Chemother. 1957, 12, 209–214. [Google Scholar] [CrossRef] [Green Version]
- Figallo, M.A.S.; Cayón, R.T.V.; Lagares, D.T.; Flores, J.R.C.; Portillo, G.M. Use of anesthetics associated to vasoconstrictors for dentistry in patients with cardiopathies. Review of the literature published in the last decade. J. Clin. Exp. Dent. 2012, 4, e107–e111. [Google Scholar] [CrossRef]
- Inagawa, M.; Ichinohe, T.; Kaneko, Y. Felypressin, but Not Epinephrine, Reduces Myocardial Oxygen Tension After an Injection of Dental Local Anesthetic Solution at Routine Doses. J. Oral Maxillofac. Surg. 2010, 68, 1013–1017. [Google Scholar] [CrossRef] [Green Version]
- Agata, H.; Ichinohe, T.; Kaneko, Y. Felypressin-induced reduction in coronary blood flow and myocardial tissue oxygen tension during anesthesia in dogs. Can. J. Anaesth. 1999, 46, 1070–1075. [Google Scholar] [CrossRef] [Green Version]
- Jamil, K.; Pappas, S.C.; Devarakonda, K.R. In vitro binding and receptor-mediated activity of terlipressin at vasopressin receptors V1 and V2. J. Exp. Pharmacol. 2018, 10, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Nilsson, G.; Lindblom, P.; Ohlin, M.; Berling, R.; Vernersson, E. Pharmacokinetics of terlipressin after single i.v. doses to healthy volunteers. Drugs Under Exp. Clin. Res. 1990, 16, 307–314. [Google Scholar]
- Saner, F.H.; Canbay, A.; Gerken, G.; Broelsch, C.E. Pharmacology, clinical efficacy and safety of terlipressin in esophageal varices bleeding, septic shock and hepatorenal syndrome. Expert Rev. Gastroenterol. Hepatol. 2007, 1, 207–217. [Google Scholar] [CrossRef]
- Papaluca, T.; Gow, P. Terlipressin: Current and emerging indications in chronic liver disease. J. Gastroenterol. Hepatol. 2018, 33, 591–598. [Google Scholar] [CrossRef]
- Chang, F.-H.; Soong, Y.-K.; Cheng, P.-J.; Lee, C.-L.; Lai, Y.-M.; Wang, H.-S.; Chou, H.-H. Surgery: Laparoscopic myomectomy of large symptomatic leiomyoma using airlift gasless laparoscopy: A preliminary report. Hum. Reprod. 1996, 11, 1427–1432. [Google Scholar] [CrossRef] [Green Version]
- Assaf, A. Adhesions after laparoscopic myomectomy: Effect of the technique used. Gynaecol. Endosc. 2001, 8, 225–229. [Google Scholar] [CrossRef]
- Samy, A.; Raslan, A.N.; Talaat, B.; El Lithy, A.; El Sharkawy, M.; Sharaf, M.F.; Hussein, A.H.; Amin, A.H.; Ibrahim, A.M.; Elsherbiny, W.S.; et al. Perioperative nonhormonal pharmacological interventions for bleeding reduction during open and minimally invasive myomectomy: A systematic review and network meta-analysis. Fertil. Steril. 2020, 113, 224–233.e6. [Google Scholar] [CrossRef]
- Pastrian, M.B.; Guzmán, F.; Garona, J.; Pifano, M.; Ripoll, G.V.; Cascone, O.; Ciccia, G.N.; Albericio, F.; Gomez, D.E.; Alonso, D.F.; et al. Structure-activity relationship of 1-desamino-8-D-arginine vasopressin as an antiproliferative agent on human vasopressin V2 receptor-expressing cancer cells. Mol. Med. Rep. 2014, 9, 2568–2572. [Google Scholar] [CrossRef]
- Czaplewski, C.; Kaźmierkiewicz, R.; Ciarkowski, J. Molecular modeling of the human vasopressin V2 receptor/agonist complex. J. Comput. Aided. Mol. Des. 1998, 12, 275–287. [Google Scholar] [CrossRef]
- Kowalczyk, W.; Prahl, A.; Derdowska, I.; Sobolewski, D.; Olejnik, J.; Zabrocki, J.; Borovicková, L.; Slaninová, J.; Lammek, B. Analogues of Neurohypophyseal Hormones, Oxytocin and Arginine Vasopressin, Conformationally Restricted in the N-Terminal Part of the Molecule. J. Med. Chem. 2006, 49, 2016–2021. [Google Scholar] [CrossRef]
- Manning, M.; Misicka, A.; Olma, A.; Bankowski, K.; Stoev, S.; Chini, B.; Durroux, T.; Mouillac, B.; Corbani, M.; Guillon, G. Oxytocin and Vasopressin Agonists and Antagonists as Research Tools and Potential Therapeutics. J. Neuroendocrinol. 2012, 24, 609–628. [Google Scholar] [CrossRef] [Green Version]
- Sawyer, W.H.; Acosta, M.; Balaspiri, L.; Judd, J.; Manning, M. Structural Changes in the Arginine Vasopressin Molecule that Enhance Antidiuretic Activity and Specificity1. Endocrinology 1974, 94, 1106–1115. [Google Scholar] [CrossRef]
- Erdélyi, L.S.; Balla, A.; Patócs, A.; Tóth, M.; Várnai, P.; Hunyady, L. Altered Agonist Sensitivity of a Mutant V2 Receptor Suggests a Novel Therapeutic Strategy for Nephrogenic Diabetes Insipidus. Mol. Endocrinol. 2014, 28, 634–643. [Google Scholar] [CrossRef] [Green Version]
- Garona, J.; Pifano, M.; Ripoll, G.; Alonso, D.F.; Eiden, L.E. Development and therapeutic potential of vasopressin synthetic analog [V4Q5]dDAVP as a novel anticancer agent. Vitam. Horm. 2020, 113, 259–289. [Google Scholar] [CrossRef]
- Iannucci, N.B.; Ripoll, G.V.; Garona, J.; Cascone, O.; Ciccia, G.N.; Gomez, D.E.; Alonso, D.F. “Antiproliferative effect of 1-deamino-8-d-arginine vasopressin analogs on human breast cancer cells. Fut. Med. Chem. 2011, 3, 1987–1993. [Google Scholar] [CrossRef]
- Garona, J.; Pifano, M.; Pastrian, M.B.; Gomez, D.E.; Ripoll, G.V.; Alonso, D.F. Addition of vasopressin synthetic analogue [V4Q5]dDAVP to standard chemotherapy enhances tumour growth inhibition and impairs metastatic spread in aggressive breast tumour models. Clin. Exp. Metastasis 2016, 33, 589–600. [Google Scholar] [CrossRef] [Green Version]
- Garona, J.; Pifano, M.; Orlando, U.D.; Pastrian, M.B.; Iannucci, N.B.; Ortega, H.H.; Podesta, E.J.; Gomez, D.E.; Ripoll, G.V.; Alonso, D.F. The novel desmopressin analogue [V4Q5]dDAVP inhibits angiogenesis, tumour growth and metastases in vasopressin type 2 receptor-expressing breast cancer models. Int. J. Oncol. 2015, 46, 2335–2345. [Google Scholar] [CrossRef]
- Pifano, M.; Garona, J.; Capobianco, C.S.; Gonzalez, N.; Alonso, D.F.; Ripoll, G.V. Peptide Agonists of Vasopressin V2 Receptor Reduce Expression of Neuroendocrine Markers and Tumor Growth in Human Lung and Prostate Tumor Cells. Front. Oncol. 2017, 7, 11. [Google Scholar] [CrossRef] [Green Version]
- Garona, J.; Sobol, N.T.; Pifano, M.; Segatori, V.I.; Gomez, D.E.; Ripoll, G.V.; Alonso, D.F. Preclinical Efficacy of [V4 Q5 ]dDAVP, a Second Generation Vasopressin Analog, on Metastatic Spread and Tumor-Associated Angiogenesis in Colorectal Cancer. Cancer Res. Treat. 2019, 51, 438–450. [Google Scholar] [CrossRef] [Green Version]
- Wisniewski, K.; Qi, S.; Kraus, J.; Ly, B.; Srinivasan, K.; Tariga, H.; Croston, G.; La, E.; Wisniewska, H.; Ortiz, C.; et al. Discovery of Potent, Selective, and Short-Acting Peptidic V2 Receptor Agonists. J. Med. Chem. 2019, 62, 4991–5005. [Google Scholar] [CrossRef]
- Gasthuys, E.; Dossche, L.; Michelet, R.; Nørgaard, J.P.; Devreese, M.; Croubels, S.; Vermeulen, A.; Van Bocxlaer, J.; Walle, J.V. Pediatric Pharmacology of Desmopressin in Children with Enuresis: A Comprehensive Review. Pediatr. Drugs 2020, 22, 369–383. [Google Scholar] [CrossRef]
- Robson, W.; Leung, A.; Norgaard, J. The Comparative Safety of Oral Versus Intranasal Desmopressin for the Treatment of Children with Nocturnal Enuresis. J. Urol. 2007, 178, 24–30. [Google Scholar] [CrossRef]
- Chanson, P.; Salenave, S. Treatment of neurogenic diabetes insipidus. Ann. d’Endocrinologie 2011, 72, 496–499. [Google Scholar] [CrossRef]
- Juul, K.V.; Van Herzeele, C.; De Bruyne, P.; Goble, S.; Walle, J.V.; Nørgaard, J.P. Desmopressin melt improves response and compliance compared with tablet in treatment of primary monosymptomatic nocturnal enuresis. Eur. J. Pediatr. 2013, 172, 1235–1242. [Google Scholar] [CrossRef] [Green Version]
- Costantini, E.; Mearini, L.; Lazzeri, M.; Bini, V.; Nunzi, E.; di Biase, M.; Porena, M. Laparoscopic Versus Abdominal Sacrocolpopexy: A Randomized, Controlled Trial. J. Urol. 2016, 196, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Lottmann, H.; Froeling, F.; Alloussi, S.; El-Radhi, A.S.; Rittig, S.; Riis, A.; Persson, B.-E. A randomised comparison of oral desmopressin lyophilisate (MELT) and tablet formulations in children and adolescents with primary nocturnal enuresis. Int. J. Clin. Pract. 2007, 61, 1454–1460. [Google Scholar] [CrossRef] [PubMed]
- Manufactured for: Sanofi-Aventis U.S.; LLC Bridgewater: New York, NY, USA, Patent Nos. 5,498,598; 5,500,413; 5,596,078; 5,674,850; 5,763,407. 2007. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2007/017922s038,018938s027,019955s013lbl.pdf (accessed on 20 October 2021).
- Rittig, S.; Jensen, A.R.; Jensen, K.T.; Pedersen, E.B. Effect of food intake on the pharmacokinetics and antidiuretic activity of oral desmopressin (DDAVP) in hydrated normal subjects. Clin. Endocrinol. 1998, 48, 235. [Google Scholar] [CrossRef]
- van Kerrebroeck, P.; Nørgaard, J.P. Desmopressin for the treatment of primary nocturnal enuresis. Pediatr. Health 2009, 3, 311–327. [Google Scholar] [CrossRef]
- Kottke, D.; Burckhardt, B.B.; Knaab, T.C.; Breitkreutz, J.; Fischer, B. Development and evaluation of a composite dosage form containing desmopressin acetate for buccal administration. Int. J. Pharm. X 2021, 3, 100082. [Google Scholar] [CrossRef] [PubMed]
- Zupančič, O.; Leonaviciute, G.; Lam, H.T.; Partenhauser, A.; Podričnik, S.; Bernkop-Schnürch, A. Development andin vitroevaluation of an oral SEDDS for desmopressin. Drug Deliv. 2016, 23, 2074–2083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Enna, S.; Bylund, D.B. Desmopressin. In xPharm: The Comprehensive Phamcology Reference; Elsevier Inc.: Amsterdam, The Netherlands, 2007; pp. 1–2. [Google Scholar] [CrossRef]
- Suman, S.; Robinson, D.; Bhal, N.; Fraser, S.; MacCormick, A.; Williams, A.; Tadtayev, S. Management of nocturia: Overcoming the challenges of nocturnal polyuria. Br. J. Hosp. Med. 2019, 80, 517–524. [Google Scholar] [CrossRef] [PubMed]
- Aguirre, T.; Teijeiro-Osorio, D.; Rosa, M.; Coulter, I.; Alonso, M.; Brayden, D. Current status of selected oral peptide technologies in advanced preclinical development and in clinical trials. Adv. Drug Deliv. Rev. 2016, 106, 223–241. [Google Scholar] [CrossRef] [Green Version]
- Svensson, P.J.; Bergqvist, P.B.; Juul, K.V.; Berntorp, E. Desmopressin in treatment of haematological disorders and in prevention of surgical bleeding. Blood Rev. 2014, 28, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Loomans, J.I.; Kruip, M.J.; Carcao, M.; Jackson, S.; van Velzen, A.S.; Peters, M.; Santagostino, E.; Platokouki, H.; Beckers, E.; Voorberg, J.; et al. Desmopressin in moderate hemophilia A patients: A treatment worth considering. Haematologica 2018, 103, 550–557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Federici, A.B.; Mazurier, C.; Berntorp, E.; Lee, C.A.; Scharrer, I.; Goudemand, J.; Lethagen, S.; Nitu, I.; Ludwig, G.; Hilbert, L.; et al. Biologic response to desmopressin in patients with severe type 1 and type 2 von Willebrand disease: Results of a multicenter European study. Blood 2004, 103, 2032–2038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldberg, N.; Nisenbaum, R.; Song, H.; Lillicrap, D.; Teitel, J.; James, P.; Sholzberg, M. Desmopressin responsiveness by age in type 1 von Willebrand disease. Res. Pract. Thromb. Haemost. 2020, 4, 1046–1052. [Google Scholar] [CrossRef] [PubMed]
- Pasalic, L.; Favaloro, E.J.; Curnow, J. Treatment of von Willebrand Disease. Semin. Thromb. Hemost. 2016, 42, 133–146. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, J.E.; Oksche, A.; Wollheim, C.B.; Günther, G.; Rosenthal, W.; Vischer, U.M. Vasopressin-induced von Willebrand factor secretion from endothelial cells involves V2 receptors and cAMP. J. Clin. Investig. 2000, 106, 107–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaufmann, J.E.; Vischer, U.M. Cellular mechanisms of the hemostatic effects of desmopressin (DDAVP). J. Thromb. Haemost. 2003, 1, 682–689. [Google Scholar] [CrossRef] [PubMed]
- North, W.G.; Fay, M.J.; Longo, K.A.; Du, J. Expression of all known vasopressin receptor subtypes by small cell tumors implies a multifaceted role for this neuropeptide. Cancer Res. 1998, 58, 1866–1871. [Google Scholar] [PubMed]
- North, W.G. Gene regulation of vasopressin and vasopressin receptors in cancer. Exp. Physiol. 2000, 85, 27–40. [Google Scholar] [CrossRef]
- Petit, T.; Davidson, K.K.; Lawrence, R.A.; Von Hoff, D.D.; Izbicka, E. Neuropeptide receptor status in human tumor cell lines. Anti-Cancer Drugs 2001, 12, 133–136. [Google Scholar] [CrossRef] [PubMed]
- North, W.G.; Fay, M.J.; Du, J. MCF-7 breast cancer cells express normal forms of all vasopressin receptors plus an abnormal V2R. Peptides 1999, 20, 837–842. [Google Scholar] [CrossRef]
- Ripoll, G.V.; Garona, J.; Pifano, M.; Farina, H.G.; Gomez, D.E.; Alonso, D.F. Reduction of tumor angiogenesis induced by desmopressin in a breast cancer model. Breast Cancer Res. Treat. 2013, 142, 9–18. [Google Scholar] [CrossRef] [Green Version]
- Gately, S.; Twardowski, P.; Stack, M.S.; Cundiff, D.L.; Grella, D.; Castellino, F.J.; Enghild, J.J.; Kwaan, H.C.; Lee, F.; Kramer, R.A.; et al. 139 The mechanism of cancer-mediated conversion of plasminogen to the angiogenesis inhibitor angiostatin. Fibrinolysis Proteolysis 1997, 11, 39. [Google Scholar] [CrossRef]
- Alonso, D.F.; Skilton, G.; Farías, E.F.; Joffé, E.B.D.K.; Gomez, D.E. Antimetastatic effect of desmopressin in a mouse mammary tumor model. Breast Cancer Res. Treat. 1999, 57, 271–275. [Google Scholar] [CrossRef] [PubMed]
- Giron, S.; Tejera, A.M.; Ripoll, G.V.; Gomez, D.E. Desmopressin inhibits lung and lymph node metastasis in a mouse mammary carcinoma model of surgical manipulation. J. Surg. Oncol. 2002, 81, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Hermo, G.A.; Torres, P.; Ripoll, G.V.; Scursoni, A.M.; Gomez, D.E.; Alonso, D.F.; Gobello, C. Perioperative desmopressin prolongs survival in surgically treated bitches with mammary gland tumours: A pilot study. Vet. J. 2007, 178, 103–108. [Google Scholar] [CrossRef] [PubMed]
- Hermo, G.A.; Turic, E.; Angelico, D.; Scursoni, A.M.; Gomez, D.E.; Gobello, C.; Alonso, D.F. Effect of Adjuvant Perioperative Desmopressin in Locally Advanced Canine Mammary Carcinoma and its Relation to Histologic Grade. J. Am. Anim. Hosp. Assoc. 2011, 47, 21–27. [Google Scholar] [CrossRef] [Green Version]
- Weinberg, R.S.; Grecco, M.O.; Ferro, G.S.; Seigelshifer, D.J.; Perroni, N.V.; Terrier, F.J.; Sánchez-Luceros, A.; Maronna, E.; Sánchez-Marull, R.; Frahm, I.; et al. A phase II dose-escalation trial of perioperative desmopressin (1-desamino-8-d-arginine vasopressin) in breast cancer patients. SpringerPlus 2015, 4, 428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozgönenel, B.; Rajpurkar, M.; Lusher, J.M. How do you treat bleeding disorders with desmopressin? Postgrad. Med. J. 2007, 83, 159–163. [Google Scholar] [CrossRef]
- Terraube, V.; Marx, I.; Denis, C. Role of von Willebrand factor in tumor metastasis. Thromb. Res. 2007, 120, S64–S70. [Google Scholar] [CrossRef]
- Iseas, S.; Roca, E.L.; O’Connor, J.M.; Eleta, M.; Sanchez-Luceros, A.; Di Leo, D.; Tinelli, M.; Fara, M.L.; Spitzer, E.; Demarco, I.A.; et al. Administration of the vasopressin analog desmopressin for the management of bleeding in rectal cancer patients: Results of a phase I/II trial. Investig. New Drugs 2020, 38, 1580–1587. [Google Scholar] [CrossRef] [Green Version]
- Sobol, N.T.; Solernó, L.M.; Beltrán, B.; Vásquez, L.; Ripoll, G.V.; Garona, J.; Alonso, D.F. Anticancer activity of repurposed hemostatic agent desmopressin on AVPR2expressing human osteosarcoma. Exp. Ther. Med. 2021, 21, 566. [Google Scholar] [CrossRef]
- Hoffman, A.; Sasaki, H.; Roberto, D.; Mayer, M.J.; Klotz, L.; Venkateswaran, V. Effect of Combination therapy of Desmopressin and Docetaxel on prostate cancer cell DU145 proliferation, migration and growth. J. Cancer Biol. Ther. 2016, 2. [Google Scholar] [CrossRef] [Green Version]
- Bass, R.; Roberto, D.; Wang, D.Z.; Cantu, F.P.; Mohamadi, R.M.; Kelley, S.O.; Klotz, L.; Venkateswaran, V. Combining Desmopressin and Docetaxel for the Treatment of Castration-Resistant Prostate Cancer in an Orthotopic Model. Anticancer Res. 2019, 39, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Vignon, P.; Laterre, P.-F.; Daix, T.; François, B. New Agents in Development for Sepsis: Any Reason for Hope? Drugs 2020, 80, 1751–1761. [Google Scholar] [CrossRef] [PubMed]
- Yao, R.-Q.; Xia, D.-M.; Wang, L.-X.; Wu, G.-S.; Zhu, Y.-B.; Zhao, H.-Q.; Liu, Q.; Xia, Z.-F.; Ren, C.; Yao, Y.-M. Clinical Efficiency of Vasopressin or Its Analogs in Comparison with Catecholamines Alone on Patients with Septic Shock: A Systematic Review and Meta-Analysis. Front. Pharmacol. 2020, 11, 563. [Google Scholar] [CrossRef] [PubMed]
- Selepressin Evaluation Programme for Sepsis-Induced Shock—Adaptive Clinical Trial—Full Text View—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT02508649 (accessed on 19 December 2021).
- Russell, J.A.; Vincent, J.-L.; Kjølbye, A.L.; Olsson, H.; Blemings, A.; Spapen, H.; Carl, P.; Laterre, P.-F.; Grundemar, L. Selepressin, a novel selective vasopressin V1A agonist, is an effective substitute for norepinephrine in a phase IIa randomized, placebo-controlled trial in septic shock patients. Crit. Care 2017, 21, 213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribot, S.; Abramowitz, S.; Kelhoffer, W.S.; Green, H.; Small, M.J.; Schwartz, I. CARDIOVASCULAR EFFECTS OF PHENYLALANYL-2-LYSYL-8-VASOPRESSIN (PLV-2). Am. J. Med Sci. 1963, 246, 479–484. [Google Scholar] [CrossRef] [PubMed]
- Cecanho, R.; de Luca, L.A.; Ranali, J. Cardiovascular Effects of Felypressin. Anesth. Prog. 2006, 53, 119–125. [Google Scholar] [CrossRef]
- Brahma, A.K.; Pemberton, C.J.; Ayeko, M.; Morgan, L.H. Single medial injection peribulbar anaesthesia using prilocaine. Anaesthesia 1994, 49, 1003–1005. [Google Scholar] [CrossRef] [PubMed]
- Yagiela, J.A. Vasoconstrictor agents for local anesthesia. Anesth. Prog. 1995, 42, 116–120. [Google Scholar]
- Meechan, J.G.; Cole, B.; Welbury, R.R. The influence of two different dental local anaesthetic solutions on the haemo-dynamic responses of children undergoing restorative dentistry: A randomised, single-blind, split-mouth study. Br. Dent. J. 2001, 190, 502–504. [Google Scholar] [CrossRef] [Green Version]
- Meechan, J.G. The effects of dental local anaesthetics on blood glucose concentration in healthy volunteers and in patients having third molar surgery. Br. Dent. J. 1991, 170, 373–376. [Google Scholar] [CrossRef]
- Volpato, M.C.; Ranali, J.; Amaral, I.M.; Demétrio, C.G.B.; Chalita, L.V. Acute toxicity (LD50 and CD50) of lidocaine and prilocaine in combination with adrenaline and felypressin. Indian J. Dent. Res. 1999, 10, 138–144. [Google Scholar]
- Sunada, K.; Nakamura, K.; Yamashiro, M.; Sumitomo, M.; Furuya, H. Clinically safe dosage of felypressin for patients with essential hypertension. Anesthesia Prog. 1996, 43, 108–115. [Google Scholar]
- Nery, D.T.F.; Ranali, J.; Barbosa, D.Z.; Júnior, H.M.; Pereira, R.M.A.; de Oliviera Afonso Pereira, P. Excisional Biopsy of the Pyogenic Granuloma in Very High-Risk Patient. Case Rep. Dent. 2018, 2018, 5180385. [Google Scholar] [CrossRef]
- Yamashita, K.; Kibe, T.; Shidou, R.; Kohjitani, A.; Nakamura, N.; Sugimura, M. Difference in the Effects of Lidocaine with Epinephrine and Prilocaine with Felypressin on the Autonomic Nervous System During Extraction of the Impacted Mandibular Third Molar: A Randomized Controlled Trial. J. Oral Maxillofac. Surg. 2020, 78, 215.e1–215.e8. [Google Scholar] [CrossRef]
- Kyosaka, Y.; Owatari, T.; Inokoshi, M.; Kubota, K.; Inoue, M.; Minakuchi, S. Cardiovascular Comparison of 2 Types of Local Anesthesia with Vasoconstrictor in Older Adults: A Crossover Study. Anesthesia Prog. 2019, 66, 133–140. [Google Scholar] [CrossRef]
- Ojeda-Yuren, A.S.; Cerda-Reyes, E.; Herrero-Maceda, M.R.; Castro-Narro, G.; Piano, S. An Integrated Review of the Hepatorenal Syndrome. Ann. Hepatol. 2021, 22, 100236. [Google Scholar] [CrossRef]
- Busk, T.M.; Bendtsen, F.; Møller, S. Hepatorenal syndrome in cirrhosis: Diagnostic, pathophysiological, and therapeutic aspects. Expert Rev. Gastroenterol. Hepatol. 2016, 10, 1153–1161. [Google Scholar] [CrossRef]
- Freeman, J.G.; Cobden, I.; Record, C.O. Placebo-Controlled Trial of Terlipressin (Glypressin) in the Management of Acute Variceal Bleeding. J. Clin. Gastroenterol. 1989, 11, 58–60. [Google Scholar] [CrossRef]
- Holmes, C.L.; Patel, B.M.; Russell, J.A.; Walley, K.R. Physiology of Vasopressin Relevant to Management of Septic Shock. Chest 2001, 120, 989–1002. [Google Scholar] [CrossRef] [Green Version]
- Krag, A.; Hobolth, L.; Møller, S.; Bendtsen, F. Hyponatraemia during terlipressin therapy. Gut 2010, 59, 417–418. [Google Scholar] [CrossRef] [Green Version]
- Solà, E.; Lens, S.; Guevara, M.; Martín-Llahí, M.; Fagundes, C.; Pereira, G.; Pavesi, M.; Fernández, J.; González-Abraldes, J.; Escorsell, A.; et al. Hyponatremia in patients treated with terlipressin for severe gastrointestinal bleeding due to portal hypertension. Hepatology 2010, 52, 1783–1790. [Google Scholar] [CrossRef]
- Krag, A.; Bendtsen, F.; Pedersen, E.B.; Holstein-Rathlou, N.-H.; Møller, S. Effects of terlipressin on the aquaretic system: Evidence of antidiuretic effects. Am. J. Physiol. Physiol. 2008, 295, F1295–F1300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leithead, J.A.; Hayes, P.C.; Ferguson, J.W. Review article: Advances in the management of patients with cirrhosis and portal hypertension-related renal dysfunction. Aliment. Pharmacol. Ther. 2014, 39, 699–711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kusano, E.; Tian, S.; Umino, T.; Tetsuka, T.; Ando, Y.; Asano, Y. Arginine vasopressin inhibits interleukin-1β-stimulated nitric oxide and cyclic guanosine monophosphate production via the V1 receptor in cultured rat vascular smooth muscle cells. J. Hypertens. 1997, 15, 627–632. [Google Scholar] [CrossRef] [PubMed]
- Wakatsuki, T.; Nakaya, Y.; Inoue, I. Vasopressin modulates K(+)-channel activities of cultured smooth muscle cells from porcine coronary artery. Am. J. Physiol. Circ. Physiol. 1992, 263, H491–H496. [Google Scholar] [CrossRef]
- Hansen, E.F.; Strandberg, C.; Højgaard, L.; Madsen, J.; Henriksen, J.H.; Schroeder, T.; Becker, U.; Bendtsen, F. Splanchnic haemodynamics after intravenous terlipressin in anaesthetised healthy pigs. J. Hepatol. 1999, 30, 503–510. [Google Scholar] [CrossRef]
- Pliška, V.; Chard, T.; Rudinger, J.; Forsling, M.L. In vivo activation of synthetic hormonogens of lysine vasopressin: Nα-glycyl-glycyl-glycyl-[8-lysine]vasopressin in the cat. Eur. J. Endocrinol. 1976, 81, 474–481. [Google Scholar] [CrossRef] [PubMed]
- Döhler, K.D.; Meyer, M. Vasopressin analogues in the treatment of hepatorenal syndrome and gastrointestinal haemorrhage. Best Pract. Res. Clin. Anaesthesiol. 2008, 22, 335–350. [Google Scholar] [CrossRef] [PubMed]
- Nevens, F.; van Steenbergen, W.; Yap, S.H.; Fevery, J. Assessment of variceal pressureby continous non-invasive endoscopic registration: A placebo controlled evaluation of the effect of terlipressin and octerotide. Gut 1996, 38, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Møller, S.; Hansen, E.F.; Becker, U.; Brinch, K.; Henriksen, J.H.; Bendtsen, F. Central and systemic haemodynamic effects of terlipressin in portal hypertensive patients. Liver Int. 2000, 20, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Ioannou, G.N.; Doust, J.; Rockey, D.C. Terlipressin for acute esophageal variceal hemorrhage. Cochrane Database Syst. Rev. 2003, 2003, CD002147. [Google Scholar] [CrossRef] [PubMed]
- Dell’Era, A.; de Franchis, R.; Iannuzzi, F. Acute variceal bleeding: Pharmacological treatment and primary/secondary prophylaxis. Best Pract. Res. Clin. Gastroenterol. 2008, 22, 279–294. [Google Scholar] [CrossRef] [PubMed]
- Wróblewski, E.; Dabrowski, A. Management of variceal haemorrhage. Gastroenterol. Rev. 2010, 3, 123–144. [Google Scholar] [CrossRef]
- Sridharan, K.; Sivaramakrishnan, G. Vasoactive agents for the management of variceal bleeding: A mixed treatment comparison network meta-analysis and trial sequential analysis of randomized clinical trials. Drug Res. 2019, 69, 487–495. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Lin, S.; Yang, Y.; Chen, Y.; Liu, B.; Li, B.; Wu, Y.; Meng, F.; Zhu, Q.; Li, Y.; et al. Development of hyponatremia after terlipressin in cirrhotic patients with acute gastrointestinal bleeding: A retrospective multicenter observational study. Expert Opin. Drug Saf. 2020, 19, 641–647. [Google Scholar] [CrossRef] [PubMed]
- Meng, Q.; Dang, X.; Li, L.; Liu, Z.; Li, L.; Wang, H. Severe hyponatraemia with neurological manifestations in patients treated with terlipressin: Two case reports. J. Clin. Pharm. Ther. 2019, 44, 981–984. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Zhou, Z.; Jin, X.; Shi, D. Clinical characteristics and risk factors of severe hyponatremia in cirrhotic patients treated with terlipressin. J. Clin. Pharm. Ther. 2020, 45, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Tian, Y.; Lin, H.; Zang, L. Preparation and characterization of PEGylated terlipressin. J. Appl. Polym. Sci. 2010, 116, 3220–3224. [Google Scholar] [CrossRef]
- Gaudino, R.; Orlandi, V.; Cavarzere, P.; Chinello, M.; Antoniazzi, F.; Cesaro, S.; Piacentini, G. Case Report: SARS-CoV-2 Infection in a Child with Suprasellar Tumor and Hypothalamic-Pituitary Failure. Front. Endocrinol. 2021, 12, 596654. [Google Scholar] [CrossRef] [PubMed]
- Al-Kuraishy, H.M.; Al-Gareeb, A.I.; Qusti, S.; Alshammari, E.M.; Atanu, F.O.; Batiha, G.E.-S. Arginine vasopressin and pathophysiology of COVID-19: An innovative perspective. Biomed. Pharmacother. 2021, 143, 112193. [Google Scholar] [CrossRef]
- Sheikh, A.B.; Javed, N.; Sheikh, A.A.E.; Upadhyay, S.; Shekhar, R. Diabetes Insipidus and Concomitant Myocarditis: A Late Sequelae of COVID-19 Infection. J. Investig. Med. High Impact Case Rep. 2021, 9, 1–4. [Google Scholar] [CrossRef]
- Maffucci, I.; Contini, A. In Silico Drug Repurposing for SARS-CoV-2 Main Proteinase and Spike Proteins. J. Proteome Res. 2020, 19, 4637–4648. [Google Scholar] [CrossRef]
- Ahmad, J.; Ikram, S.; Ahmad, F.; Rehman, I.U.; Mushtaq, M. SARS-CoV-2 RNA Dependent RNA polymerase (RdRp)—A drug repurposing study. Heliyon 2020, 6, e04502. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}









| Analogue | Sequence | Functions | Refs. |
|---|---|---|---|
| Desmopressin, dDAVP | dCys(&)-Tyr-Phe-Gln-Asn-Cys(&)-Pro-d-Arg-Gly-NH2 | antidiuretic effect, increases plasma osmolality | [64,120,121,122] |
| Selepressin | Cys(&)-Phe-Ile-hGln-Asn-Cys(&)-Pro-Orn(iPr)-Gly-NH2 | applied in septic shock | [125,126,127] |
| Felypressin | Cys(&)-Phe-Phe-Gln-Asn-Cys(&)-Pro-Lys-Gly-NH2 | vasoconstricting agent, used as an additive in anesthesia during dental procedures | [128,129,130,131] |
| Terlipressin | Gly-Gly-Gly-Cys(&)-Tyr-Phe-Gln-Asn-Cys(&)-Pro-Lys-Gly-NH2 | treats bleeding caused by esophageal varices | [132,133,134,135] |
| Ornipressin | Cys(&)-Tyr-Phe-Gln-Asn-Cys(&)-Pro-Orn-Gly-NH2 | vasoconstricting agent during myomectomy; in cirrhosis, as hepatorenal treatment | [136,137,138] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Glavaš, M.; Gitlin-Domagalska, A.; Dębowski, D.; Ptaszyńska, N.; Łęgowska, A.; Rolka, K. Vasopressin and Its Analogues: From Natural Hormones to Multitasking Peptides. Int. J. Mol. Sci. 2022, 23, 3068. https://doi.org/10.3390/ijms23063068
Glavaš M, Gitlin-Domagalska A, Dębowski D, Ptaszyńska N, Łęgowska A, Rolka K. Vasopressin and Its Analogues: From Natural Hormones to Multitasking Peptides. International Journal of Molecular Sciences. 2022; 23(6):3068. https://doi.org/10.3390/ijms23063068
Chicago/Turabian StyleGlavaš, Mladena, Agata Gitlin-Domagalska, Dawid Dębowski, Natalia Ptaszyńska, Anna Łęgowska, and Krzysztof Rolka. 2022. "Vasopressin and Its Analogues: From Natural Hormones to Multitasking Peptides" International Journal of Molecular Sciences 23, no. 6: 3068. https://doi.org/10.3390/ijms23063068
APA StyleGlavaš, M., Gitlin-Domagalska, A., Dębowski, D., Ptaszyńska, N., Łęgowska, A., & Rolka, K. (2022). Vasopressin and Its Analogues: From Natural Hormones to Multitasking Peptides. International Journal of Molecular Sciences, 23(6), 3068. https://doi.org/10.3390/ijms23063068