
Genome-Wide Survey and Development of the First Microsatellite Markers Database (AnCorDB) in Anemone coronaria L.

 ,
, 
 ,
, 
 and
and
Abstract
:1. Introduction
2. Results and Discussion
2.1. Draft Genome Assembly and Annotation
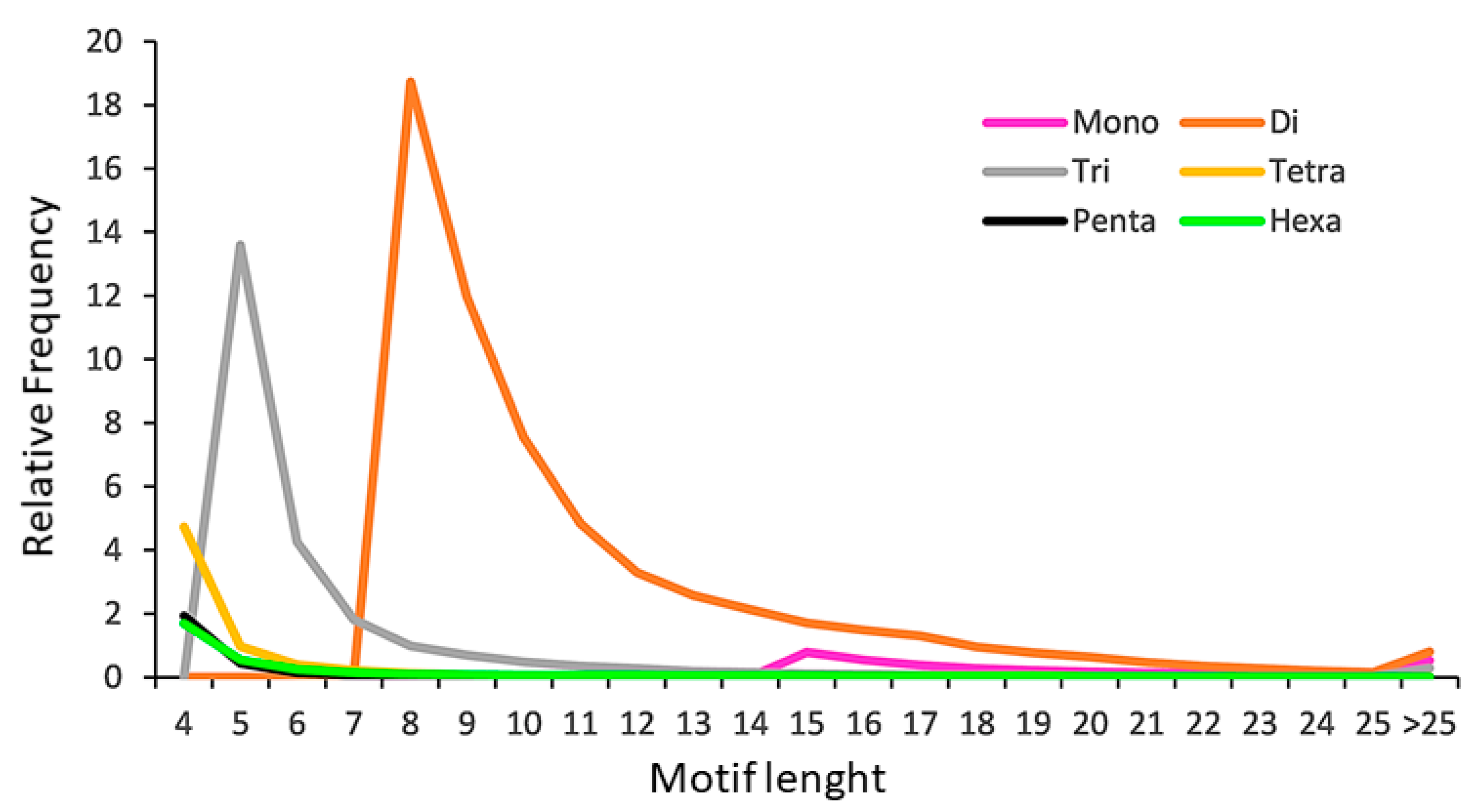
2.2. The SSR Content of the Poppy Anemone Draft Genome
2.3. Gene Context of SSRs
2.4. AnCorDB Construction, System Architecture, Features and Utility
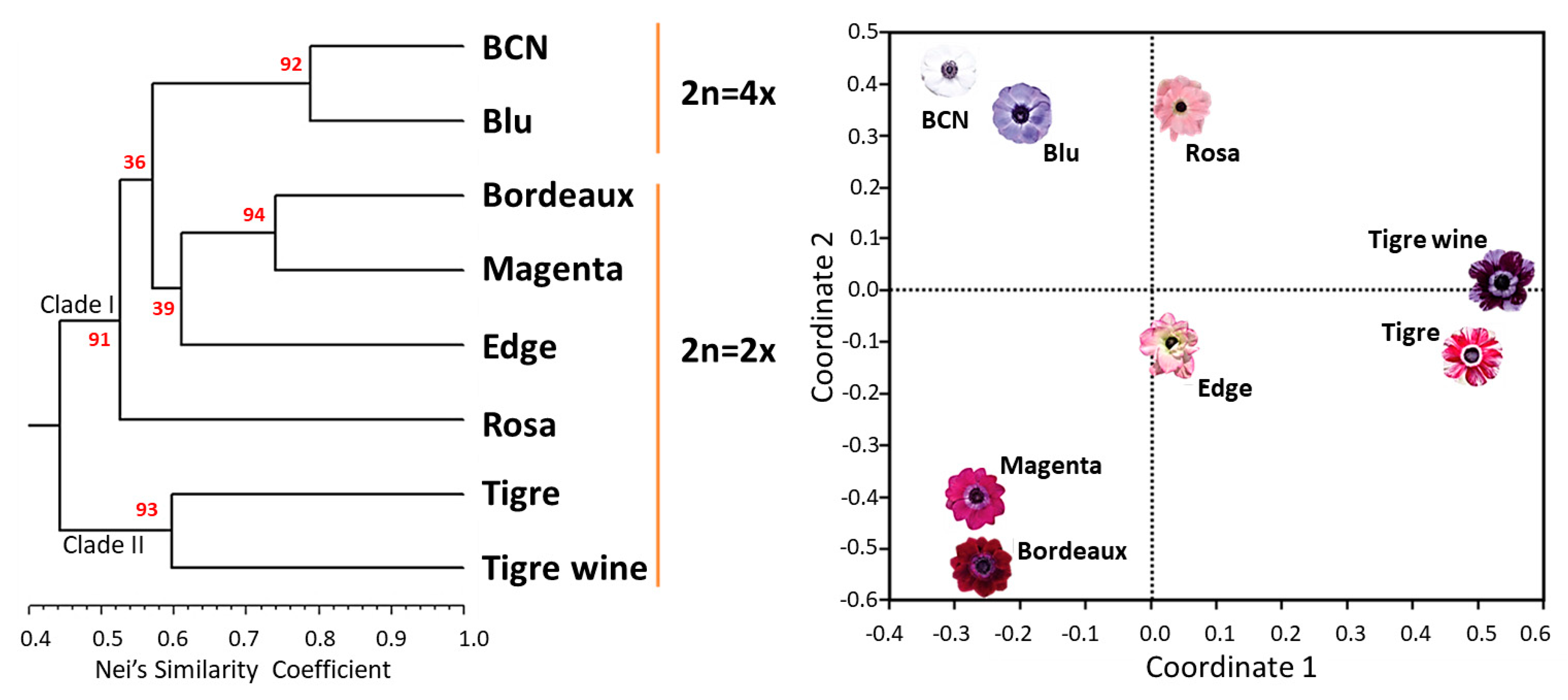
2.5. SSR Validation and Varietal Fingerprinting
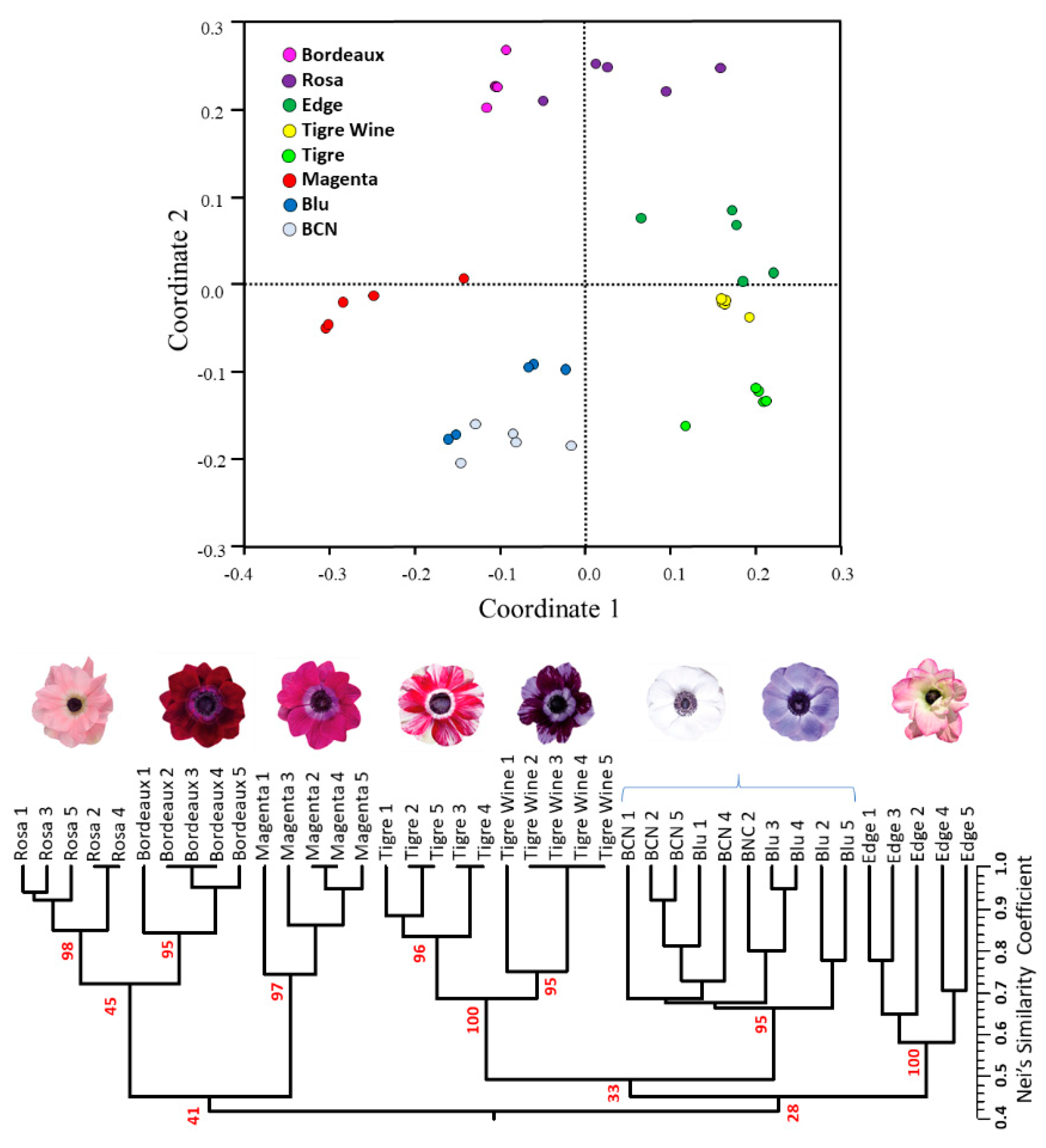
2.6. Intra-Cultivar Variability Assessment
3. Materials and Methods
3.1. Draft Genome Sequencing, Assembly, and Annotation
3.2. SSR-Mining
3.3. AnCorDB, an SSR Database for Poppy Anemone
3.4. Marker Validation
3.5. SSR Fingerprinting and Intra-Cultivar Variation Assessment
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Laura, M.; Borghi, C.; Bobbio, V.; Allavena, A. The Effect on the Transcriptome of Anemone coronaria following Infection with Rust (Tranzschelia discolor). PLoS ONE 2015, 10, e0118565. [Google Scholar] [CrossRef] [Green Version]
- Laura, M.; Allavena, A. Anemone coronaria Breeding: Current Status and Perspectives. Eur. J. Hortic. Sci. 2007, 72, 241–247. [Google Scholar]
- Horovitz, A. The pollination syndrome of Anemone coronaria L.; an insect-biased mutualism. Acta Hortic. 1991, 283–287. [Google Scholar] [CrossRef]
- Horovitz, A.; Galil, J.; Zohary, D. Biological Flora of Israel. 6. Anemone coronaria L. Isr. J. Bot. 1975, 126, 239–242. [Google Scholar]
- Laura, M.; Safaverdi, G.; Allavena, A. Androgenetic Plants of Anemone coronaria Derived through Anther Culture. Plant Breed. 2006, 125, 629–634. [Google Scholar] [CrossRef]
- Nissim, Y.; Jinggui, F.; Arik, S.; Neta, P.; Uri, L.; Avner, C. Phenotypic and Genotypic Analysis of a Commercial Cultivar and Wild Populations of Anemone coronaria. Euphytica 2004, 136, 51–62. [Google Scholar] [CrossRef]
- Shamay, A.; Fang, J.; Pollak, N.; Cohen, A.; Yonash, N.; Lavi, U. Discovery of C-SNPs in Anemone coronaria L. and Assessment of Genetic Variation. Genet. Resour. Crop Evol. 2006, 53, 821–829. [Google Scholar] [CrossRef]
- Wenzel, W.; Hemleben, V. A Comparative Study of Genomes in Angiosperms. Plant Syst. Evol. 1982, 139, 209–227. [Google Scholar] [CrossRef]
- Veselý, P.; Bureš, P.; Šmarda, P.; Pavlíček, T. Genome Size and DNA Base Composition of Geophytes: The Mirror of Phenology and Ecology? Ann. Bot. 2012, 109, 65–75. [Google Scholar] [CrossRef] [Green Version]
- Acquadro, A.; Magurno, F.; Portis, E.; Lanteri, S. DbEST-Derived Microsatellite Markers in Celery (Apium graveolens L. Var. Dulce). Mol. Ecol. Notes 2006, 6, 1080–1082. [Google Scholar] [CrossRef]
- Barchi, L.; Lanteri, S.; Portis, E.; Acquadro, A.; Valè, G.; Toppino, L.; Rotino, G.L. Identification of SNP and SSR Markers in Eggplant Using RAD Tag Sequencing. BMC Genom. 2011, 12, 304. [Google Scholar] [CrossRef] [Green Version]
- Lanteri, S.; Portis, E.; Acquadro, A.; Mauro, R.; Mauromicale, G. Morphology and SSR Fingerprinting of Newly Developed Cynara cardunculus Genotypes Exploitable as Ornamentals. Euphytica 2012, 184, 311–321. [Google Scholar] [CrossRef]
- Gharsallah, C.; Ben Abdelkrim, A.; Fakhfakh, H.; Salhi-Hannachi, A.; Gorsane, F. SSR Marker-Assisted Screening of Commercial Tomato Genotypes under Salt Stress. Breed. Sci. 2016, 66, 823–830. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; He, R.; Zheng, J.; Hu, Z.; Wu, J.; Leng, P. Development of EST-SSR Markers and Association Mapping with Floral Traits in Syringa oblata. BMC Plant Biol. 2020, 20, 436. [Google Scholar] [CrossRef]
- Huang, C.-W.; Chu, P.-Y.; Wu, Y.-F.; Chan, W.-R.; Wang, Y.-H. Identification of Functional SSR Markers in Freshwater Ornamental Shrimps Neocaridina denticulata Using Transcriptome Sequencing. Mar. Biotechnol. 2020, 22, 772–785. [Google Scholar] [CrossRef]
- Li, Q.; Su, X.; Ma, H.; Du, K.; Yang, M.; Chen, B.; Fu, S.; Fu, T.; Xiang, C.; Zhao, Q.; et al. Development of Genic SSR Marker Resources from RNA-Seq Data in Camellia japonica and Their Application in the Genus Camellia. Sci. Rep. 2021, 11, 9919. [Google Scholar] [CrossRef]
- Acquadro, A.; Portis, E.; Lee, D.; Donini, P.; Lanteri, S. Development and Characterization of Microsatellite Markers in Cynara cardunculus L. Genome 2005, 48, 217–225. [Google Scholar] [CrossRef]
- Portis, E.; Scaglione, D.; Acquadro, A.; Mauromicale, G.; Mauro, R.; Knapp, S.J.; Lanteri, S. Genetic Mapping and Identification of QTL for Earliness in the Globe Artichoke/Cultivated Cardoon Complex. BMC Res. Notes 2012, 5, 252. [Google Scholar] [CrossRef] [Green Version]
- Feng, S.; He, R.; Lu, J.; Jiang, M.; Shen, X.; Jiang, Y.; Wang, Z.; Wang, H. Development of SSR Markers and Assessment of Genetic Diversity in Medicinal Chrysanthemum morifolium Cultivars. Front. Genet. 2016, 7, 113. [Google Scholar] [CrossRef]
- Aiello, D.; Ferradini, N.; Torelli, L.; Volpi, C.; Lambalk, J.; Russi, L.; Albertini, E. Evaluation of Cross-Species Transferability of SSR Markers in Foeniculum vulgare. Plants 2020, 9, 175. [Google Scholar] [CrossRef] [Green Version]
- Jaillon, O.; Aury, J.-M.; Noel, B.; Policriti, A.; Clepet, C.; Casagrande, A.; Choisne, N.; Aubourg, S.; Vitulo, N.; Jubin, C.; et al. The Grapevine Genome Sequence Suggests Ancestral Hexaploidization in Major Angiosperm Phyla. Nature 2007, 449, 463–467. [Google Scholar] [CrossRef] [PubMed]
- Scaglione, D.; Reyes-Chin-Wo, S.; Acquadro, A.; Froenicke, L.; Portis, E.; Beitel, C.; Tirone, M.; Mauro, R.; Lo Monaco, A.; Mauromicale, G.; et al. The Genome Sequence of the Outbreeding Globe Artichoke Constructed de Novo Incorporating a Phase-Aware Low-Pass Sequencing Strategy of F1 Progeny. Sci. Rep. 2016, 6, 19427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barchi, L.; Pietrella, M.; Venturini, L.; Minio, A.; Toppino, L.; Acquadro, A.; Andolfo, G.; Aprea, G.; Avanzato, C.; Bassolino, L.; et al. A Chromosome-Anchored Eggplant Genome Sequence Reveals Key Events in Solanaceae Evolution. Sci. Rep. 2019, 9, 11769. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Itgen, M.W.; Wang, H.; Gong, Y.; Jiang, J.; Li, J.; Sun, C.; Sessions, S.K.; Mueller, R.L. Gigantic Genomes Provide Empirical Tests of Transposable Element Dynamics Models. Genom. Proteom. Bioinform. 2021, 19, 123–139. [Google Scholar] [CrossRef] [PubMed]
- Scheeff, E.D.; Bourne, P.E. Structural Evolution of the Protein Kinase-like Superfamily. PLoS Comput. Biol. 2005, 1, e49. [Google Scholar] [CrossRef] [PubMed]
- Shalaeva, D.N.; Cherepanov, D.A.; Galperin, M.Y.; Golovin, A.V.; Mulkidjanian, A.Y. Evolution of Cation Binding in the Active Sites of P-Loop Nucleoside Triphosphatases in Relation to the Basic Catalytic Mechanism. eLife 2018, 7, e37373. [Google Scholar] [CrossRef]
- Di Nardo, G.; Gilardi, G. Natural Compounds as Pharmaceuticals: The Key Role of Cytochromes P450 Reactivity. Trends Biochem. Sci. 2020, 45, 511–525. [Google Scholar] [CrossRef]
- Acquadro, A.; Barchi, L.; Portis, E.; Nourdine, M.; Carli, C.; Monge, S.; Valentino, D.; Lanteri, S. Whole Genome Resequencing of Four Italian Sweet Pepper Landraces Provides Insights on Sequence Variation in Genes of Agronomic Value. Sci. Rep. 2020, 10, 9189. [Google Scholar] [CrossRef]
- Pavese, V.; Cavalet-Giorsa, E.; Barchi, L.; Acquadro, A.; Torello Marinoni, D.; Portis, E.; Lucas, S.J.; Botta, R. Whole-Genome Assembly of Corylus avellana cv. “Tonda Gentile Delle Langhe” Using Linked-Reads (10× Genomics). G3 Genes|Genomes|Genetics 2021, 11, jkab152. [Google Scholar] [CrossRef]
- Hamarsheh, O.; Amro, A. Characterization of Simple Sequence Repeats (SSRs) from Phlebotomus papatasi (Diptera: Psychodidae) Expressed Sequence Tags (ESTs). Parasit Vectors 2011, 4, 189. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.; Xie, Y.; Zhang, D.; Chen, H. Analysis of SSR Loci and Development of SSR Primers in Eucalyptus. J. For. Res. 2018, 29, 273–282. [Google Scholar] [CrossRef]
- Manee, M.M.; Al-Shomrani, B.M.; Al-Fageeh, M.B. Genome-Wide Characterization of Simple Sequence Repeats in Palmae Genomes. Genes Genom. 2020, 42, 597–608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, S.; Wang, S.; He, K.; Jiang, M.; Li, F. Large-Scale Analysis Reveals That the Genome Features of Simple Sequence Repeats Are Generally Conserved at the Family Level in Insects. BMC Genom. 2017, 18, 848. [Google Scholar] [CrossRef] [PubMed]
- Chadha, S.; Gopalakrishna, T. Informativeness of Dinucleotide Repeat-Based Primers in Fungal Pathogen of Rice Magnaporthe grisea. Microbiol. Res. 2009, 164, 276–281. [Google Scholar] [CrossRef] [PubMed]
- Patil, P.G.; Singh, N.V.; Bohra, A.; Raghavendra, K.P.; Mane, R.; Mundewadikar, D.M.; Babu, K.D.; Sharma, J. Comprehensive Characterization and Validation of Chromosome-Specific Highly Polymorphic SSR Markers From Pomegranate (Punica granatum L.) cv. Tunisia Genome. Front. Plant Sci. 2021, 12, 337. [Google Scholar] [CrossRef]
- Sahu, K.K.; Chattopadhyay, D. Genome-Wide Sequence Variations between Wild and Cultivated Tomato Species Revisited by Whole Genome Sequence Mapping. BMC Genom. 2017, 18, 430. [Google Scholar] [CrossRef] [Green Version]
- Portis, E.; Portis, F.; Valente, L.; Moglia, A.; Barchi, L.; Lanteri, S.; Acquadro, A. A Genome-Wide Survey of the Microsatellite Content of the Globe Artichoke Genome and the Development of a Web-Based Database. PLoS ONE 2016, 11, e0162841. [Google Scholar] [CrossRef]
- Portis, E.; Lanteri, S.; Barchi, L.; Portis, F.; Valente, L.; Toppino, L.; Rotino, G.L.; Acquadro, A. Comprehensive Characterization of Simple Sequence Repeats in Eggplant (Solanum melongena L.) Genome and Construction of a Web Resource. Front. Plant Sci. 2018, 9, 401. [Google Scholar] [CrossRef] [Green Version]
- An, J.; Yin, M.; Zhang, Q.; Gong, D.; Jia, X.; Guan, Y.; Hu, J. Genome Survey Sequencing of Luffa cylindrica L. and Microsatellite High Resolution Melting (SSR-HRM) Analysis for Genetic Relationship of Luffa Genotypes. Int. J. Mol. Sci. 2017, 18, 1942. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.; Huang, S.; Fu, D.; Yu, J.; Wang, X.; Hua, W.; Liu, S.; Liu, G.; Wang, H. Evolutionary Dynamics of Microsatellite Distribution in Plants: Insight from the Comparison of Sequenced Brassica, Arabidopsis and Other Angiosperm Species. PLoS ONE 2013, 8, e59988. [Google Scholar] [CrossRef] [Green Version]
- Cheng, J.; Zhao, Z.; Li, B.; Qin, C.; Wu, Z.; Trejo-Saavedra, D.L.; Luo, X.; Cui, J.; Rivera-Bustamante, R.F.; Li, S.; et al. A Comprehensive Characterization of Simple Sequence Repeats in Pepper Genomes Provides Valuable Resources for Marker Development in Capsicum. Sci. Rep. 2016, 6, 18919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tóth, G.; Gáspári, Z.; Jurka, J. Microsatellites in Different Eukaryotic Genomes: Survey and Analysis. Genome Res. 2000, 10, 967–981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mun, J.H.; Kim, D.J.; Choi, H.K.; Gish, J.; Debellé, F.; Mudge, J.; Denny, R.; Endré, G.; Saurat, O.; Dudez, A.M.; et al. Distribution of Microsatellites in the Genome of Medicago Truncatula: A Resource of Genetic Markers That Integrate Genetic and Physical Maps. Genetics 2006, 172, 2541–2555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scaglione, D.; Acquadro, A.; Portis, E.; Taylor, C.A.; Lanteri, S.; Knapp, S.J. Ontology and Diversity of Transcript-Associated Microsatellites Mined from a Globe Artichoke EST Database. BMC Genom. 2009, 10, 454. [Google Scholar] [CrossRef] [Green Version]
- Cavagnaro, P.F.; Senalik, D.A.; Yang, L.; Simon, P.W.; Harkins, T.T.; Kodira, C.D.; Huang, S.; Weng, Y. Genome-Wide Characterization of Simple Sequence Repeats in Cucumber (Cucumis sativus L.). BMC Genom. 2010, 11, 569. [Google Scholar] [CrossRef] [Green Version]
- Morgante, M.; Hanafey, M.; Powell, W. Microsatellites Are Preferentially Associated with Nonrepetitive DNA in Plant Genomes. Nat. Genet. 2002, 30, 194–200. [Google Scholar] [CrossRef]
- Subramanian, S.; Mishra, R.K.; Singh, L. Genome-Wide Analysis of Microsatellite Repeats in Humans: Their Abundance and Density in Specific Genomic Regions. Genome Biol. 2003, 4, R13. [Google Scholar] [CrossRef] [Green Version]
- Andersen, J.R.; Lübberstedt, T. Functional Markers in Plants. Trends Plant Sci. 2003, 8, 554–560. [Google Scholar] [CrossRef]
- Li, Y.-C.; Korol, A.B.; Fahima, T.; Nevo, E. Microsatellites within Genes: Structure, Function, and Evolution. Mol. Biol. Evol. 2004, 21, 991–1007. [Google Scholar] [CrossRef]
- Brouwer, J.R.; Willemsen, R.; Oostra, B.A. Microsatellite Repeat Instability and Neurological Disease. Bioessays 2009, 31, 71–83. [Google Scholar] [CrossRef]
- Golubov, A.; Yao, Y.; Maheshwari, P.; Bilichak, A.; Boyko, A.; Belzile, F.; Kovalchuk, I. Microsatellite Instability in Arabidopsis Increases with Plant Development1[W][OA]. Plant Physiol. 2010, 154, 1415–1427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelson, D.L.; Orr, H.T.; Warren, S.T. The Unstable Repeats—Three Evolving Faces of Neurological Disease. Neuron 2013, 77, 825–843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vieira, D.D.S.S.; Emiliani, G.; Michelozzi, M.; Centritto, M.; Luro, F.; Morillon, R.; Loreto, F.; Gesteira, A.; Maserti, B. Polyploidization Alters Constitutive Content of Volatile Organic Compounds (VOC) and Improves Membrane Stability under Water Deficit in Volkamer Lemon (Citrus limonia Osb.) Leaves. Environ. Exp. Bot. 2016, 126, 1–9. [Google Scholar] [CrossRef]
- Varshney, R.K.; Graner, A.; Sorrells, M.E. Genic Microsatellite Markers in Plants: Features and Applications. Trends Biotechnol. 2005, 23, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.-K.; Paik, H.; Choi, J.P.; Han, J.-H.; Choe, J.-K.; Hur, C.-G. Functional Domain Marker (FDM): An In Silico Demonstration in Solanaceae Using Simple Sequence Repeats (SSRs). Plant Mol. Biol. Rep. 2010, 28, 352–356. [Google Scholar] [CrossRef]
- Kujur, A.; Bajaj, D.; Saxena, M.S.; Tripathi, S.; Upadhyaya, H.D.; Gowda, C.L.L.; Singh, S.; Jain, M.; Tyagi, A.K.; Parida, S.K. Functionally Relevant Microsatellite Markers from Chickpea Transcription Factor Genes for Efficient Genotyping Applications and Trait Association Mapping. DNA Res. 2013, 20, 355–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.; Jia, X.; Liu, Z.; Zhang, Z.; Wang, Y.; Liu, Z.; Xie, W. Development and Characterization of Transcription Factor Gene-Derived Microsatellite (TFGM) Markers in Medicago truncatula and Their Transferability in Leguminous and Non-Leguminous Species. Molecules 2015, 20, 8759–8771. [Google Scholar] [CrossRef] [Green Version]
- Treangen, T.J.; Salzberg, S.L. Repetitive DNA and Next-Generation Sequencing: Computational Challenges and Solutions. Nat. Rev. Genet. 2011, 13, 36–46. [Google Scholar] [CrossRef]
- Wang, H.; Yang, B.; Wang, H.; Xiao, H. Impact of Different Numbers of Microsatellite Markers on Population Genetic Results Using SLAF-Seq Data for Rhododendron Species. Sci. Rep. 2021, 11, 8597. [Google Scholar] [CrossRef]
- Stoll, A.; Harpke, D.; Schütte, C.; Stefanczyk, N.; Brandt, R.; Blattner, F.R.; Quandt, D. Development of Microsatellite Markers and Assembly of the Plastid Genome in Cistanthe longiscapa (Montiaceae) Based on Low-Coverage Whole Genome Sequencing. PLoS ONE 2017, 12, e0178402. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Yin, Q.; Do, V.T.; Meng, K.; Chen, S.; Liao, B.; Fan, Q. Development and Characterization of Genomic Microsatellite Markers in the Tree Species, Rhodoleia championii, R. parvipetala, and R. forrestii (Hamamelidaceae). Mol. Biol. Rep. 2019, 46, 6547–6556. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Long, C.; Pang, X.; Ning, D.; Wu, T.; Dong, M.; Han, X.; Guo, H. The Newly Developed Genomic-SSR Markers Uncover the Genetic Characteristics and Relationships of Olive Accessions. PeerJ 2020, 8, e8573. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Liu, C.-M.; Luo, R.; Sadakane, K.; Lam, T.-W. MEGAHIT: An Ultra-Fast Single-Node Solution for Large and Complex Metagenomics Assembly via Succinct de Bruijn Graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef] [Green Version]
- Bradnam, K.R.; Fass, J.N.; Alexandrov, A.; Baranay, P.; Bechner, M.; Birol, I.; Boisvert, S.; Chapman, J.A.; Chapuis, G.; Chikhi, R.; et al. Assemblathon 2: Evaluating de Novo Methods of Genome Assembly in Three Vertebrate Species. GigaScience 2013, 2, 2047-217X. [Google Scholar] [CrossRef]
- Smit, S.A.F.; Hubley, R.; Green, P. RepeatMasker Open-4.0. 2013. [Google Scholar]
- Campbell, M.S.; Law, M.; Holt, C.; Stein, J.C.; Moghe, G.D.; Hufnagel, D.E.; Lei, J.; Achawanantakun, R.; Jiao, D.; Lawrence, C.J.; et al. MAKER-P: A Tool Kit for the Rapid Creation, Management, and Quality Control of Plant Genome Annotations. Plant Physiol. 2014, 164, 513–524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanke, M.; Keller, O.; Gunduz, I.; Hayes, A.; Waack, S.; Morgenstern, B. AUGUSTUS: Ab Initio Prediction of Alternative Transcripts. Nucleic Acids Res. 2006, 34, W435–W439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bromberg, Y.; Rost, B. SNAP: Predict Effect of Non-Synonymous Polymorphisms on Function. Nucleic Acids Res. 2007, 35, 3823–3835. [Google Scholar] [CrossRef] [Green Version]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing Genome Assembly and Annotation Completeness with Single-Copy Orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [Green Version]
- Jones, P.; Binns, D.; Chang, H.-Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-Scale Protein Function Classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef] [Green Version]
- Sigrist, C.J.A.; de Castro, E.; Cerutti, L.; Cuche, B.A.; Hulo, N.; Bridge, A.; Bougueleret, L.; Xenarios, I. New and Continuing Developments at PROSITE. Nucleic Acids Res. 2013, 41, D344–D347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mi, H.; Muruganujan, A.; Thomas, P.D. PANTHER in 2013: Modeling the Evolution of Gene Function, and Other Gene Attributes, in the Context of Phylogenetic Trees. Nucleic Acids Res. 2013, 41, D377–D386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lupas, A.; Van Dyke, M.; Stock, J. Predicting Coiled Coils from Protein Sequences. Science 1991, 252, 1162–1164. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.H.; Nikolskaya, A.; Huang, H.; Yeh, L.L.; Natale, D.A.; Vinayaka, C.R.; Hu, Z.; Mazumder, R.; Kumar, S.; Kourtesis, P.; et al. PIRSF: Family Classification System at the Protein Information Resource. Nucleic Acids Res. 2004, 32, D112–D114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lima, T.; Auchincloss, A.H.; Coudert, E.; Keller, G.; Michoud, K.; Rivoire, C.; Bulliard, V.; de Castro, E.; Lachaize, C.; Baratin, D.; et al. HAMAP: A Database of Completely Sequenced Microbial Proteome Sets and Manually Curated Microbial Protein Families in UniProtKB/Swiss-Prot. Nucleic Acids Res. 2009, 37, D471–D478. [Google Scholar] [CrossRef] [PubMed]
- Punta, M.; Coggill, P.C.; Eberhardt, R.Y.; Mistry, J.; Tate, J.; Boursnell, C.; Pang, N.; Forslund, K.; Ceric, G.; Clements, J.; et al. The Pfam Protein Families Database. Nucleic Acids Res. 2012, 40, D290–D301. [Google Scholar] [CrossRef]
- de Lima Morais, D.A.; Fang, H.; Rackham, O.J.L.; Wilson, D.; Pethica, R.; Chothia, C.; Gough, J. SUPERFAMILY 1.75 including a Domain-Centric Gene Ontology Method. Nucleic Acids Res. 2011, 39, D427–D434. [Google Scholar] [CrossRef] [Green Version]
- Bru, C.; Courcelle, E.; Carrère, S.; Beausse, Y.; Dalmar, S.; Kahn, D. The ProDom Database of Protein Domain Families: More Emphasis on 3D. Nucleic Acids Res. 2005, 33, D212–D215. [Google Scholar] [CrossRef] [Green Version]
- Letunic, I.; Doerks, T.; Bork, P. SMART 7: Recent Updates to the Protein Domain Annotation Resource. Nucleic Acids Res. 2012, 40, D302–D305. [Google Scholar] [CrossRef]
- Lees, J.; Yeats, C.; Perkins, J.; Sillitoe, I.; Rentzsch, R.; Dessailly, B.H.; Orengo, C. Gene3D: A Domain-Based Resource for Comparative Genomics, Functional Annotation and Protein Network Analysis. Nucleic Acids Res. 2012, 40, D465–D471. [Google Scholar] [CrossRef]
- Haft, D.H.; Selengut, J.D.; Richter, R.A.; Harkins, D.; Basu, M.K.; Beck, E. TIGRFAMs and Genome Properties in 2013. Nucleic Acids Res. 2013, 41, D387–D395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Araujo, F.A.; Barh, D.; Silva, A.; Guimarães, L.; Ramos, R.T.J. GO FEAT: A Rapid Web-Based Functional Annotation Tool for Genomic and Transcriptomic Data. Sci. Rep. 2018, 8, 1794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kofler, R.; Schlötterer, C.; Lelley, T. SciRoKo: A New Tool for Whole Genome Microsatellite Search and Investigation. Bioinformatics 2007, 23, 1683–1685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barkley, N.A.; Dean, R.E.; Pittman, R.N.; Wang, M.L.; Holbrook, C.C.; Pederson, G.A. Genetic Diversity of Cultivated and Wild-Type Peanuts Evaluated with M13-Tailed SSR Markers and Sequencing. Genet. Res. 2007, 89, 93–106. [Google Scholar] [CrossRef] [PubMed]
- Rosenthal, A.; Coutelle, O.; Craxton, M. Large-Scale Production of DNA Sequencing Templates by Microtitre Format PCR. Nucleic Acids Res. 1993, 21, 173–174. [Google Scholar] [CrossRef] [Green Version]
- Nei, M.; Li, W.H. Mathematical Model for Studying Genetic Variation in Terms of Restriction Endonucleases. Proc. Natl. Acad. Sci. USA 1979, 76, 5269–5273. [Google Scholar] [CrossRef] [Green Version]
- Sneath, P.H.A.; Sokal, R.R. Numerical Taxonomy: The Principles and Practice of Numerical Classification; W. H. Freeman and Co.: New York, NY, USA, 1973. [Google Scholar]
- Rohlf, F.J. NTSYS-Pc: Numerical Taxonomy and Multivariate Analysis System; Exeter Software: Setauket, NY, USA, 1988; ISBN 978-0-925031-00-6. [Google Scholar]
- Hammer, O.; Harper, D.A.T.; Ryan, P.D. PAST: Paleontological Statistics Software Package for Education and Data Analysis. Palaeontol. Electron. 2001, 4, 9. [Google Scholar]
- Anderson, J.A.; Churchill, G.A.; Autrique, J.E.; Tanksley, S.D.; Sorrells, M.E. Optimizing Parental Selection for Genetic Linkage Maps. Genome 1993, 36, 181–186. [Google Scholar] [CrossRef]
- Mantel, N. The Detection of Disease Clustering and a Generalized Regression Approach. Cancer Res. 1967, 27, 209–220. [Google Scholar]
- Wagner, H.W.; Sefc, K.M. IDENTITY 4.0. Centre for Applied Genetics; References-Scientific Research Publishing; University of Agricultural Sciences: Vienna, Austria, 1999; Available online: https://www.scirp.org/%28S%28vtj3fa45qm1ean45vvffcz55%29%29/reference/referencespapers.aspx?referenceid=564391 (accessed on 8 March 2022).
- Raymond, M.; Rousset, F. GENEPOP (Version 1.2): Population Genetics Software for Exact Tests and Ecumenicism. J. Hered. 1995, 86, 248–249. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mono- | Di- | Tri- | Tetra- | Penta- | Hexa- | Total/Mean | ||
|---|---|---|---|---|---|---|---|---|
| Perfect SSR | Types | 2 | 4 | 10 | 32 | 91 | 304 | 443 |
| Count | 13,475 | 241,693 | 95,326 | 27,203 | 10,805 | 13,320 | 401,822 | |
| % | 3.4 | 60.2 | 23.7 | 6.8 | 2.7 | 3.3 | 100 | |
| Density (SSR/Mbp) | 2.2 | 39.41 | 15.54 | 4.44 | 1.76 | 2.17 | 65.52 | |
| Cumulative (Mbp) | 0.05 | 1.94 | 1.14 | 0.43 | 0.21 | 0.32 | 4.11 | |
| Cumulative (%) | 0.08% | 47.20% | 27.74% | 10.46% | 5.11% | 7.79% | 100% | |
| Mean Repeat Number | 22.7 | 11.3 | 6.9 | 5.2 | 5.0 | 6.8 | 57.9 | |
| Imperfect SSR | Count | 2823 | 111,281 | 38,183 | 10,719 | 12,920 | 13,061 | 188,987 |
| % | 1.49% | 58.88% | 20.20% | 5.67% | 6.84% | 6.91% | 100% | |
| Density (SSR/Mbp) | 0.46 | 18.14 | 6.23 | 2.15 | 2.11 | 2.13 | 31.22 |
| Alleles (bp) | |||||
|---|---|---|---|---|---|
| SSR | SSR Type | Motif | N° of Repeats | BCN | BLU |
| AnCor49 | Di | AT | 26 | 450; 468 | 435 |
| AnCor74 | Di | GT | 30 | 501; 530 | 529; 539 |
| AnCor87 | Di | TC | 29 | 271 | 262; 275 |
| AnCor115 | Tri | AAC | 28 | 518 | 528 |
| AnCor132 | Tri | AAC | 27 | 533; 589 | 536; 572 |
| AnCor139 | Tri | AAG | 26 | 501 | 504; 512 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martina, M.; Acquadro, A.; Barchi, L.; Gulino, D.; Brusco, F.; Rabaglio, M.; Portis, F.; Portis, E.; Lanteri, S. Genome-Wide Survey and Development of the First Microsatellite Markers Database (AnCorDB) in Anemone coronaria L. Int. J. Mol. Sci. 2022, 23, 3126. https://doi.org/10.3390/ijms23063126
Martina M, Acquadro A, Barchi L, Gulino D, Brusco F, Rabaglio M, Portis F, Portis E, Lanteri S. Genome-Wide Survey and Development of the First Microsatellite Markers Database (AnCorDB) in Anemone coronaria L. International Journal of Molecular Sciences. 2022; 23(6):3126. https://doi.org/10.3390/ijms23063126
Chicago/Turabian StyleMartina, Matteo, Alberto Acquadro, Lorenzo Barchi, Davide Gulino, Fabio Brusco, Mario Rabaglio, Flavio Portis, Ezio Portis, and Sergio Lanteri. 2022. "Genome-Wide Survey and Development of the First Microsatellite Markers Database (AnCorDB) in Anemone coronaria L." International Journal of Molecular Sciences 23, no. 6: 3126. https://doi.org/10.3390/ijms23063126
APA StyleMartina, M., Acquadro, A., Barchi, L., Gulino, D., Brusco, F., Rabaglio, M., Portis, F., Portis, E., & Lanteri, S. (2022). Genome-Wide Survey and Development of the First Microsatellite Markers Database (AnCorDB) in Anemone coronaria L. International Journal of Molecular Sciences, 23(6), 3126. https://doi.org/10.3390/ijms23063126