
Second Cancer Onset in Myeloproliferative Neoplasms: What, When, Why?

 ,
, 

 ,
,  , and
, and 
Abstract
:1. Introduction
2. Risk of SC Onset in MPNs: An Overview
3. Possible Links between MPNs and SC
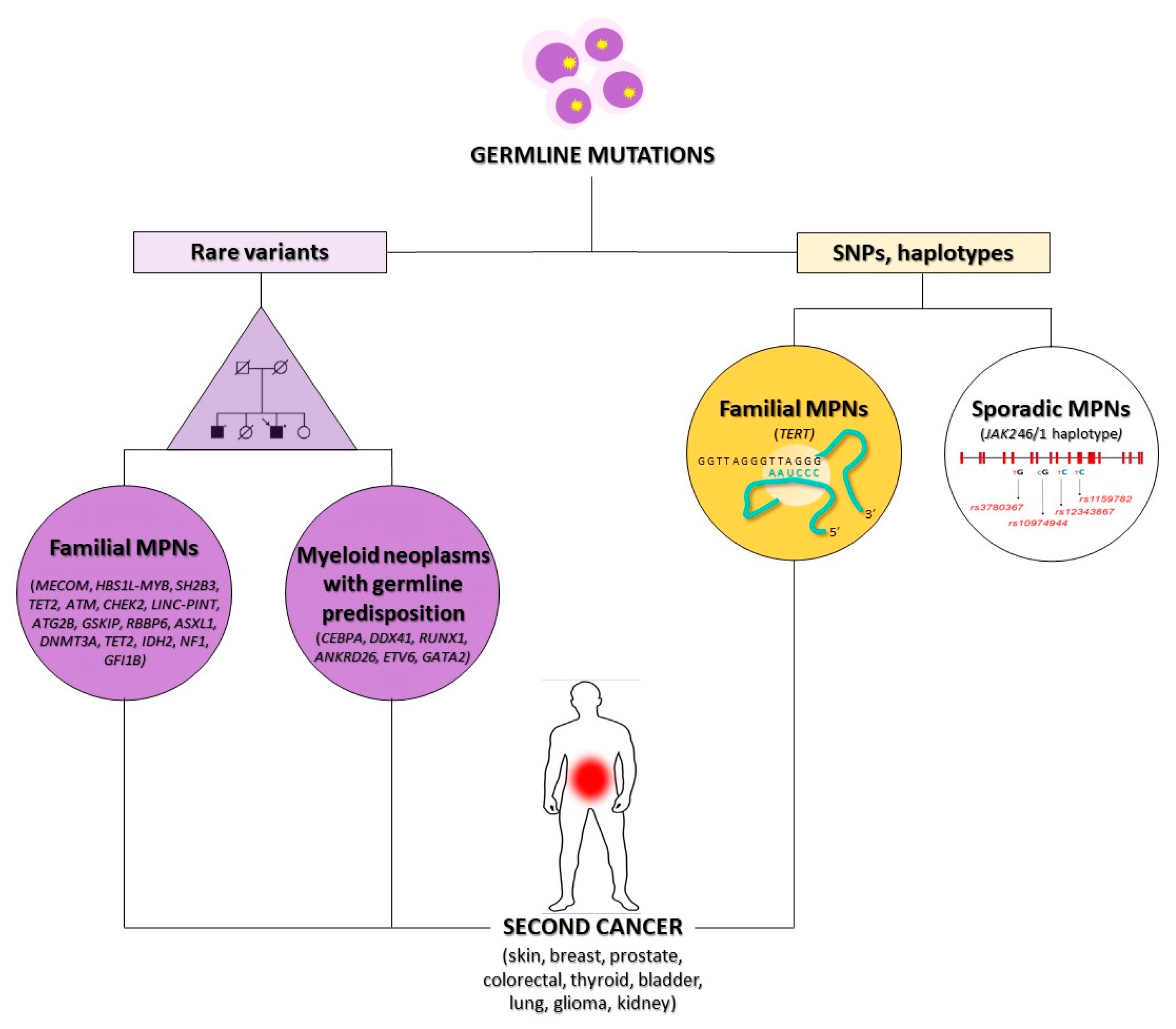
3.1. Genetic Susceptibility
3.2. Effect of Cytotoxic Drugs
3.3. Influence of Chronic Inflammation
3.4. Immune Deregulation
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| MPNs | myeloproliferative neoplasms |
| PV | polycythemia vera |
| ET | essential thrombocythemia |
| PMF | primary myelofibrosis |
| CML | chronic myeloid leukemia |
| SC | second cancer |
| GWA | genome-wide association |
| WES | whole exome sequencing |
| AML | acute myeloid leukemia |
| MDS | myelodysplastic syndromes |
| aCML | atypical CML |
| CMML | chronic myelomonocytic leukemia |
| HSCs | hematopoietic stem cells |
| HPV | human papillomavirus |
| IFN-alpha | interferon-alpha |
| 32P | phosphorus-32 |
| CI | chronic inflammation |
| ROS | reactive oxygen species |
| APCs | antigen presenting cells |
| DCs | dendritic cells |
| T-reg | regulatory T cells |
| NK | natural killer |
References
- Grinfeld, J.; Nangalia, J.; Baxter, E.J.; Wedge, D.C.; Angelopoulos, N.; Cantrill, R.; Godfrey, A.L.; Papaemmanuil, E.; Gundem, G.; MacLean, C.; et al. Classification and Personalized Prognosis in Myeloproliferative Neoplasms. N. Engl. J. Med. 2018, 379, 1416–1430. [Google Scholar] [CrossRef] [PubMed]
- Hultcrantz, M.; Kristinsson, S.Y.; Andersson, T.M.L.; Landgren, O.; Eloranta, S.; Derolf, Å.R.; Dickman, P.W.; Björkholm, M. Patterns of survival among patients with myeloproliferative neoplasms diagnosed in Sweden from 1973 to 2008: A population-based study. J. Clin. Oncol. 2012, 30, 2995–3001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landtblom, A.R.; Bower, H.; Andersson, T.M.L.; Dickman, P.W.; Samuelsson, J.; Björkholm, M.; Kristinsson, S.Y.; Hultcrantz, M. Second malignancies in patients with myeloproliferative neoplasms: A population-based cohort study of 9379 patients. Leukemia 2018, 32, 2203–2210. [Google Scholar] [CrossRef] [PubMed]
- Barbui, T.; Ghirardi, A.; Masciulli, A.; Carobbio, A.; Palandri, F.; Vianelli, N.; De Stefano, V.; Betti, S.; Di Veroli, A.; Iurlo, A.; et al. Second cancer in Philadelphia negative myeloproliferative neoplasms (MPN-K). A nested case-control study. Leukemia 2019, 33, 1996–2005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chattopadhyay, S.; Zheng, G.; Sud, A.; Yu, H.; Sundquist, K.; Sundquist, J.; Försti, A.; Hemminki, A.; Houlston, R.; Hemminki, K. Risk of second primary cancer following myeloid neoplasia and risk of myeloid neoplasia as second primary cancer: A nationwide, observational follow up study in Sweden. Lancet Haematol. 2018, 5, e368–e377. [Google Scholar] [CrossRef] [Green Version]
- Frederiksen, H.; Farkas, D.K.; Christiansen, C.F.; Hasselbalch, H.C.; Sørensen, H.T. Chronic myeloproliferative neoplasms and subsequent cancer risk: A Danish population-based cohort study. Blood 2011, 118, 6515–6520. [Google Scholar] [CrossRef] [Green Version]
- Mora, B.; Rumi, E.; Guglielmelli, P.; Barraco, D.; Maffioli, M.; Rambaldi, A.; Caramella, M.; Komrokji, R.; Gotlib, J.; Kiladjian, J.J.; et al. Second primary malignancies in postpolycythemia vera and postessential thrombocythemia myelofibrosis: A study on 2233 patients. Cancer Med. 2019, 8, 4089. [Google Scholar] [CrossRef]
- Brabrand, M.; Frederiksen, H. Risks of solid and lymphoid malignancies in patients with myeloproliferative neoplasms: Clinical implications. Cancers 2020, 12, 3061. [Google Scholar] [CrossRef]
- Blasco, M.A. Telomeres and human disease: Ageing, cancer and beyond. Nat. Rev. Genet. 2005, 6, 611–622. [Google Scholar] [CrossRef]
- Oddsson, A.; Kristinsson, S.Y.; Helgason, H.; Gudbjartsson, D.F.; Masson, G.; Sigurdsson, A.; Jonasdottir, A.; Jonasdottir, A.; Steingrimsdottir, H.; Vidarsson, B.; et al. The germline sequence variant rs2736100_C in TERT associates with myeloproliferative neoplasms. Leukemia 2014, 28, 1371–1374. [Google Scholar] [CrossRef] [Green Version]
- Hinds, D.A.; Barnholt, K.E.; Mesa, R.A.; Kiefer, A.K.; Do, C.B.; Eriksson, N.; Mountain, J.L.; Francke, U.; Tung, J.Y.; Nguyen, H.; et al. Germ line variants predispose to both JAK2 V617F clonal hematopoiesis and myeloproliferative neoplasms. Blood 2016, 128, 1121–1128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tapper, W.; Jones, A.V.; Kralovics, R.; Harutyunyan, A.S.; Zoi, K.; Leung, W.; Godfrey, A.L.; Guglielmelli, P.; Callaway, A.; Ward, D.; et al. Genetic variation at MECOM, TERT, JAK2 and HBS1L-MYB predisposes to myeloproliferative neoplasms. Nat. Commun. 2015, 6, 6691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malato, A.; Rossi, E.; Palumbo, G.A.; Guglielmelli, P.; Pugliese, N. Drug-related cutaneous adverse events in philadelphia chromosome-negative myeloproliferative neoplasms: A literature review. Int. J. Mol. Sci. 2020, 21, 3900. [Google Scholar] [CrossRef] [PubMed]
- Erixon, K.; Ahnström, G. Single-strand breaks in DNA during repair of UV-induced damage in normal human and xeroderma pigmentosum cells as determined by alkaline DNA unwinding and hydroxylapatite chromatography: Effects of hydroxyurea, 5-fluorodeoxyuridine and 1-β-d-arabinofuranosylcytosine on the kinetics of repair. Mutat. Res. Mol. Mech. Mutagen. 1979, 59, 257–271. [Google Scholar]
- Gavini, D.R.; Salvi, D.J.; Shah, P.H.; Uma, D.; Lee, J.H.; Hamid, P. Non-melanoma Skin Cancers in Patients on Hydroxyurea for Philadelphia Chromosome-Negative Myeloproliferative Neoplasms: A Systematic Review. Cureus 2021, 13, 1–8. [Google Scholar] [CrossRef]
- Zhang, W.; Remenyik, E.; Zelterman, D.; Brash, D.E.; Wikonkal, N.M. Escaping the stem cell compartment: Sustained UVB exposure allows p53-mutant keratinocytes to colonize adjacent epidermal proliferating units without incurring additional mutations. Proc. Natl. Acad. Sci. USA 2001, 98, 13948–13953. [Google Scholar] [CrossRef] [Green Version]
- Coombs, C.C.; Zehir, A.; Devlin, S.M.; Kishtagari, A.; Syed, A.; Jonsson, P.; Hyman, D.M.; Solit, D.B.; Robson, M.E.; Baselga, J.; et al. Therapy-Related Clonal Hematopoiesis in Patients with Non-hematologic Cancers Is Common and Associated with Adverse Clinical Outcomes. Cell Stem Cell 2017, 21, 374–382.e4. [Google Scholar] [CrossRef] [Green Version]
- Kahn, J.D.; Miller, P.G.; Silver, A.J.; Sellar, R.S.; Bhatt, S.; Gibson, C.; McConkey, M.; Adams, D.; Mar, B.; Mertins, P.; et al. PPM1D-truncating mutations confer resistance to chemotherapy and sensitivity to PPM1D inhibition in hematopoietic cells. Blood 2018, 132, 1095–1105. [Google Scholar] [CrossRef]
- Hasselbalch, H.C. Chronic inflammation as a promotor of mutagenesis in essential thrombocythemia, polycythemia vera and myelofibrosis. A human inflammation model for cancer development? Leuk. Res. 2013, 37, 214–220. [Google Scholar] [CrossRef]
- Ferguson, L.R. Chronic inflammation and mutagenesis. Mutat. Res. 2010, 690, 3–11. [Google Scholar] [CrossRef]
- Cooke, M.S.; Evans, M.D.; Dizdaroglu, M.; Lunec, J. Oxidative DNA damage: Mechanisms, mutation, and disease. FASEB J. 2003, 17, 1195–1214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grivennikov, S.I.; Karin, M. Dangerous liaisons: STAT3 and NF-kappaB collaboration and crosstalk in cancer. Cytokine Growth Factor Rev. 2010, 21, 11–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hahn, M.A.; Hahn, T.; Lee, D.H.; Esworthy, R.S.; Kim, B.W.; Riggs, A.D.; Chu, F.F.; Pfeifer, G.P. Methylation of polycomb target genes in intestinal cancer is mediated by inflammation. Cancer Res. 2008, 68, 10280–10289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foran, E.; Garrity-Park, M.M.; Mureau, C.; Newell, J.; Smyrk, T.C.; Limburg, P.J.; Egan, L.J. Upregulation of DNA methyltransferase-mediated gene silencing, anchorage-independent growth, and migration of colon cancer cells by interleukin-6. Mol. Cancer Res. 2010, 8, 471–481. [Google Scholar] [CrossRef] [Green Version]
- O’Hagan, H.M.; Wang, W.; Sen, S.; DeStefano Shields, C.; Lee, S.S.; Zhang, Y.W.; Clements, E.G.; Cai, Y.; Van Neste, L.; Easwaran, H.; et al. Oxidative damage targets complexes containing DNA methyltransferases, SIRT1, and polycomb members to promoter CpG Islands. Cancer Cell 2011, 20, 606–619. [Google Scholar] [CrossRef] [Green Version]
- Cook, E.K.; Luo, M.; Rauh, M.J. Clonal hematopoiesis and inflammation: Partners in leukemogenesis and comorbidity. Exp. Hematol. 2020, 83, 85–94. [Google Scholar] [CrossRef] [Green Version]
- Abegunde, S.O.; Buckstein, R.; Wells, R.A.; Rauh, M.J. An inflammatory environment containing TNFα favors Tet2-mutant clonal hematopoiesis. Exp. Hematol. 2018, 59, 60–65. [Google Scholar] [CrossRef]
- Cai, Z.; Kotzin, J.J.; Ramdas, B.; Chen, S.; Nelanuthala, S.; Palam, L.R.; Pandey, R.; Mali, R.S.; Liu, Y.; Kelley, M.R.; et al. Inhibition of Inflammatory Signaling in Tet2 Mutant Preleukemic Cells Mitigates Stress-Induced Abnormalities and Clonal Hematopoiesis. Cell Stem Cell 2018, 23, 833–849.e5. [Google Scholar] [CrossRef] [Green Version]
- Hasselbalch, H.C. Perspectives on the increased risk of second cancer in patients with essential thrombocythemia, polycythemia vera and myelofibrosis. Eur. J. Haematol. 2015, 94, 96–98. [Google Scholar] [CrossRef]
- Hasselbalch, H.C. Perspectives on chronic inflammation in essential thrombocythemia, polycythemia vera, and myelofibrosis: Is chronic inflammation a trigger and driver of clonal evolution and development of accelerated atherosclerosis and second cancer? Blood 2012, 119, 3219–3225. [Google Scholar] [CrossRef] [Green Version]
- Hasselbalch, H.C. The role of cytokines in the initiation and progression of myelofibrosis. Cytokine Growth Factor Rev. 2013, 24, 133–145. [Google Scholar] [CrossRef] [PubMed]
- Wada, J.; Suauki, H.; Fuchino, R.; Yamasaki, A.; Nagai, S.; Yanai, K.; Koga, K.; Nakamura, M.; Tanaka, M.; Morisaki, T.; et al. The Contribution of Vascular Endothelial Growth Factor to the Induction of Regulatory T-Cells in Malignant Effusions. Anticancer Res. 2009, 29, 881–888. [Google Scholar] [PubMed]
- Dong, M.; Blobe, G.C. Role of transforming growth factor-β in hematologic malignancies. Blood 2006, 107, 4589–4596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorelik, L.; Flavell, R.A. Transforming growth factor-beta in T-cell biology. Nat. Rev. Immunol. 2002, 2, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Riedl, E.; Strobl, H.; Majdic, O.; Knapp, W. TGF-beta 1 promotes in vitro generation of dendritic cells by protecting progenitor cells from apoptosis. J. Immunol. 1997, 158, 1591–1597. [Google Scholar] [PubMed]
- Bogdans, C.; Paik, J.; Vodovotz, Y.; Nathan, C. Contrasting Mechanisms for Suppression of Macrophage Cytokine Release by Transforming Growth Factor-@ and Interleukin-10. J. Biol. Chem. 1992, 267, 23301–23308. [Google Scholar] [CrossRef]
- Skov, V.; Larsen, T.S.; Thomassen, M.; Riley, C.H.; Jensen, M.K.; Bjerrum, O.W.; Kruse, T.A.; Hasselbalch, H.C. Molecular profiling of peripheral blood cells from patients with polycythemia vera and related neoplasms: Identification of deregulated genes of significance for inflammation and immune surveillance. Leuk. Res. 2012, 36, 1387–1392. [Google Scholar] [CrossRef]
- Frederiksen, H.; Farkas, D.K.; Christiansen, C.F.; Larsen, T.S.; Hasselbalch, H.C.; Stentoft, J.; Sørensen, H.T. Survival of patients with chronic myeloproliferative neoplasms and new primary cancers: A population-based cohort study. Lancet Haematol. 2015, 2, e289–e296. [Google Scholar] [CrossRef] [Green Version]
- Bao, E.L.; Nandakumar, S.K.; Liao, X.; Bick, A.G.; Karjalainen, J.; Tabaka, M.; Gan, O.I.; Havulinna, A.S.; Kiiskinen, T.T.J.; Lareau, C.A.; et al. Inherited myeloproliferative neoplasm risk affects haematopoietic stem cells. Nature 2020, 586, 769–775. [Google Scholar] [CrossRef]
- Rumi, E.; Cazzola, M. Advances in understanding the pathogenesis of familial myeloproliferative neoplasms. Br. J. Haematol. 2017, 178, 689–698. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Xu, Y.; Mei, H.; Peng, L.; Li, X.; Tang, J. The TERT rs2736100 polymorphism increases cancer risk: A meta-analysis. Oncotarget 2017, 8, 38693–38705. [Google Scholar] [CrossRef] [PubMed]
- Anelli, L.; Orsini, P.; Zagaria, A.; Minervini, A.; Coccaro, N.; Parciante, E.; Minervini, C.F.; Cumbo, C.; Tota, G.; Impera, L.; et al. Erythrocytosis with JAK2 GGCC_46/1 haplotype and without JAK2 V617F mutation is associated with CALR rs1049481_G allele. Leukemia 2020, 35, 619–622. [Google Scholar] [CrossRef] [PubMed]
- Anelli, L.; Zagaria, A.; Specchia, G.; Albano, F. The JAK2 GGCC (46/1) Haplotype in Myeloproliferative Neoplasms: Causal or Random? Int. J. Mol. Sci. 2018, 19, 1152. [Google Scholar] [CrossRef] [Green Version]
- Jones, A.V.; Chase, A.; Silver, R.T.; Oscier, D.; Zoi, K.; Wang, Y.L.; Cario, H.; Pahl, H.L.; Collins, A.; Reiter, A.; et al. JAK2 haplotype is a major risk factor for the development of myeloproliferative neoplasms. Nat. Genet. 2009, 41, 446–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trifa, A.P.; Bănescu, C.; Tevet, M.; Bojan, A.; Dima, D.; Urian, L.; Török-Vistai, T.; Popov, V.M.; Zdrenghea, M.; Petrov, L.; et al. TERT rs2736100 A>C SNP and JAK2 46/1 haplotype significantly contribute to the occurrence of JAK2 V617F and CALR mutated myeloproliferative neoplasms—A multicentric study on 529 patients. Br. J. Haematol. 2016, 174, 218–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Swierczek, S.I.; Drummond, J.; Hickman, K.; Kim, S.; Walker, K.; Doddapaneni, H.; Muzny, D.M.; Gibbs, R.A.; Wheeler, D.A.; et al. Whole-exome sequencing of polycythemia vera revealed novel driver genes and somatic mutation shared by T cells and granulocytes. Leukemia 2014, 28, 935–938. [Google Scholar] [CrossRef] [Green Version]
- Mbita, Z.; Meyer, M.; Skepu, A.; Hosie, M.; Rees, J.; Dlamini, Z. De-regulation of the RBBP6 isoform 3/DWNN in human cancers. Mol. Cell. Biochem. 2012, 362, 249–262. [Google Scholar] [CrossRef]
- Stolarova, L.; Kleiblova, P.; Janatova, M.; Soukupova, J.; Zemankova, P.; Macurek, L.; Kleibl, Z. CHEK2 Germline Variants in Cancer Predisposition: Stalemate Rather than Checkmate. Cells 2020, 9, 2675. [Google Scholar] [CrossRef]
- Shen, L.; Yin, Z.H.; Wan, Y.; Zhang, Y.; Li, K.; Zhou, B. Sen Association between ATM polymorphisms and cancer risk:a meta-analysis. Mol. Biol. Rep. 2012, 39, 5719–5725. [Google Scholar] [CrossRef]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef]
- Crysandt, M.; Brings, K.; Beier, F.; Thiede, C.; Brümmendorf, T.H.; Jost, E. Germ line predisposition to myeloid malignancies appearing in adulthood. Expert Rev. Hematol. 2018, 11, 625–636. [Google Scholar] [CrossRef] [PubMed]
- Quesada, A.E.; Routbort, M.J.; DiNardo, C.D.; Bueso-Ramos, C.E.; Kanagal-Shamanna, R.; Khoury, J.D.; Thakral, B.; Zuo, Z.; Yin, C.C.; Loghavi, S.; et al. DDX41 mutations in myeloid neoplasms are associated with male gender, TP53 mutations and high-risk disease. Am. J. Hematol. 2019, 94, 757–766. [Google Scholar] [CrossRef] [PubMed]
- Goyal, T.; Tu, Z.J.; Wang, Z.; Cook, J.R. Clinical and Pathologic Spectrum of DDX41-Mutated Hematolymphoid Neoplasms. Am. J. Clin. Pathol. 2021, 156, 829–838. [Google Scholar] [CrossRef] [PubMed]
- Geyer, J.T. Myeloid Neoplasms with Germline Predisposition. Pathobiology 2019, 86, 5–6. [Google Scholar] [CrossRef]
- Wlodarski, M.W.; Collin, M.; Horwitz, M.S. GATA2 deficiency and related myeloid neoplasms. Semin. Hematol. 2017, 54, 81–86. [Google Scholar] [CrossRef]
- Collin, M.; Dickinson, R.; Bigley, V. Haematopoietic and immune defects associated with GATA2 mutation. Br. J. Haematol. 2015, 169, 173–187. [Google Scholar] [CrossRef] [Green Version]
- Rütsche, C.V.; Haralambieva, E.; Lysenko, V.; Balabanov, S.; Theocharides, A.P.A. A patient with a germline GATA2 mutation and primary myelofibrosis. Blood Adv. 2021, 5, 791–795. [Google Scholar] [CrossRef]
- Gunnarsson, N.; Höglund, M.; Stenke, L.; Wallberg-Jonsson, S.; Sandin, F.; Björkholm, M.; Dreimane, A.; Lambe, M.; Markevärn, B.; Olsson-Strömberg, U.; et al. Increased prevalence of prior malignancies and autoimmune diseases in patients diagnosed with chronic myeloid leukemia. Leukemia 2016, 30, 1562–1567. [Google Scholar] [CrossRef]
- Thomas, J.W.; Jamy, O.; Shah, M.V.; Vachhani, P.; Go, R.S.; Goyal, G. Risk of mortality and second malignancies in primary myelofibrosis before and after ruxolitinib approval. Leuk. Res. 2022, 112, 106770. [Google Scholar] [CrossRef]
- Sekhri, R.; Sadjadian, P.; Becker, T.; Kolatzki, V.; Huenerbein, K.; Meixner, R.; Marchi, H.; Wallmann, R.; Fuchs, C.; Griesshammer, M.; et al. Ruxolitinib-treated polycythemia vera patients and their risk of secondary malignancies. Ann. Hematol. 2021, 100, 2707–2716. [Google Scholar] [CrossRef]
- Polverelli, N.; Elli, E.M.; Abruzzese, E.; Palumbo, G.A.; Benevolo, G.; Tiribelli, M.; Bonifacio, M.; Tieghi, A.; Caocci, G.; D’Adda, M.; et al. Second primary malignancy in myelofibrosis patients treated with ruxolitinib. Br. J. Haematol. 2021, 193, 356–368. [Google Scholar] [CrossRef] [PubMed]
- Cuthbert, D.; Stein, B.L. Therapy-associated leukemic transformation in myeloproliferative neoplasms—What do we know? Best Pract. Res. Clin. Haematol. 2019, 32, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Cantisani, C.; Kiss, N.; Naqeshbandi, A.F.; Tosti, G.; Tofani, S.; Cartoni, C.; Carmosino, I.; Cantoresi, F. Nonmelanoma skin cancer associated with Hydroxyurea treatment: Overview of the literature and our own experience. Dermatol. Ther. 2019, 32, e13043. [Google Scholar] [CrossRef]
- Bulte, C.A.; Hoegler, K.M.; Kutlu, Ö.; Khachemoune, A. Hydroxyurea: A reappraisal of its cutaneous side effects and their management. Int. J. Dermatol. 2021, 60, 810–817. [Google Scholar] [CrossRef] [PubMed]
- Gunnarsson, N.; Stenke, L.; Höglund, M.; Sandin, F.; Björkholm, M.; Dreimane, A.; Lambe, M.; Markevärn, B.; Olsson-Strömberg, U.; Richter, J.; et al. Second malignancies following treatment of chronic myeloid leukaemia in the tyrosine kinase inhibitor era. Br. J. Haematol. 2015, 169, 683–688. [Google Scholar] [CrossRef]
- Verma, D.; Kantarjian, H.; Strom, S.S.; Rios, M.B.; Jabbour, E.; Quintas-Cardama, A.; Verstovsek, S.; Ravandi, F.; O’Brien, S.; Cortes, J. Malignancies occurring during therapy with tyrosine kinase inhibitors (TKIs) for chronic myeloid leukemia (CML) and other hematologic malignancies. Blood 2011, 118, 4353–4358. [Google Scholar] [CrossRef]
- Gugliotta, G.; Castagnetti, F.; Breccia, M.; Albano, F.; Iurlo, A.; Intermesoli, T.; Abruzzese, E.; Levato, L.; D’Adda, M.; Pregno, P.; et al. Incidence of second primary malignancies and related mortality in patients with imatinib-treated chronic myeloid leukemia. Haematologica 2017, 102, 1530. [Google Scholar] [CrossRef] [Green Version]
- Balkwill, F.; Mantovani, A. Inflammation and cancer: Back to Virchow? Lancet 2001, 357, 539–545. [Google Scholar] [CrossRef]
- Hussain, S.P.; Harris, C.C. Inflammation and cancer: An ancient link with novel potentials. Int. J. Cancer 2007, 121, 2373–2380. [Google Scholar] [CrossRef]
- Mantovani, A.; Garlanda, C.; Allavena, P. Molecular pathways and targets in cancer-related inflammation. Ann. Med. 2010, 42, 161–170. [Google Scholar] [CrossRef]
- Barbui, T.; Carobbio, A.; Finazzi, G.; Vannucchi, A.M.; Barosi, G.; Antonioli, E.; Guglielmelli, P.; Pancrazzi, A.; Salmoiraghi, S.; Zilio, P.; et al. Inflammation and thrombosis in essential thrombocythemia and polycythemia vera: Different role of C-reactive protein and pentraxin 3. Haematologica 2011, 96, 315–318. [Google Scholar] [CrossRef] [PubMed]
- Cumbo, C.; Tarantini, F.; Anelli, L.; Zagaria, A.; Redavid, I.; Minervini, C.F.; Coccaro, N.; Tota, G.; Ricco, A.; Parciante, E.; et al. IRF4 expression is low in Philadelphia negative myeloproliferative neoplasms and is associated with a worse prognosis. Exp. Hematol. Oncol. 2021, 10, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Genovese, G.; Kähler, A.K.; Handsaker, R.E.; Lindberg, J.; Rose, S.A.; Bakhoum, S.F.; Chambert, K.; Mick, E.; Neale, B.M.; Fromer, M.; et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N. Engl. J. Med. 2014, 371, 2477–2487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaiswal, S.; Fontanillas, P.; Flannick, J.; Manning, A.; Grauman, P.V.; Mar, B.G.; Lindsley, R.C.; Mermel, C.H.; Burtt, N.; Chavez, A.; et al. Age-Related Clonal Hematopoiesis Associated with Adverse Outcomes. N. Engl. J. Med. 2014, 371, 2488–2498. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, K.; Wang, F.; Kantarjian, H.; Doss, D.; Khanna, K.; Thompson, E.; Zhao, L.; Patel, K.; Neelapu, S.; Gumbs, C.; et al. Preleukaemic clonal haemopoiesis and risk of therapy-related myeloid neoplasms: A case-control study. Lancet Oncol. 2017, 18, 100–111. [Google Scholar] [CrossRef] [Green Version]
- Mayle, A.; Yang, L.; Rodriguez, B.; Zhou, T.; Chang, E.; Curry, C.V.; Challen, G.A.; Li, W.; Wheeler, D.; Rebel, V.I.; et al. Dnmt3a loss predisposes murine hematopoietic stem cells to malignant transformation. Blood 2015, 125, 629–638. [Google Scholar] [CrossRef] [Green Version]
- Moran-Crusio, K.; Reavie, L.; Shih, A.; Abdel-Wahab, O.; Ndiaye-Lobry, D.; Lobry, C.; Figueroa, M.E.; Vasanthakumar, A.; Patel, J.; Zhao, X.; et al. Tet2 Loss Leads to Increased Hematopoietic Stem Cell Self-Renewal and Myeloid Transformation. Cancer Cell 2011, 20, 11–24. [Google Scholar] [CrossRef] [Green Version]
- Appel, S.; Balabanov, S.; Brümmendorf, T.H.; Brossart, P. Effects of Imatinib on Normal Hematopoiesis and Immune Activation. Stem Cells 2005, 23, 1082–1088. [Google Scholar] [CrossRef]
- Zitvogel, L.; Rusakiewicz, S.; Routy, B.; Ayyoub, M.; Kroemer, G. Immunological off-target effects of imatinib. Nat. Rev. Clin. Oncol. 2016, 13, 431–446. [Google Scholar] [CrossRef]
- Li, Q.; Verma, I.M. NF-kappaB regulation in the immune system. Nat. Rev. Immunol. 2002, 2, 725–734. [Google Scholar] [CrossRef]
- Hasselbalch, H.C. A new era for IFN-α in the treatment of Philadelphia-negative chronic myeloproliferative neoplasms. Expert Rev. Hematol. 2011, 4, 637–655. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Mechanism | Main Pathways | References |
|---|---|---|
| Genetic susceptibility |
| [9,10,11,12] |
| Cytotoxic drugs effect |
| [13,14,15,16,17,18] |
| Chronic inflammation influence |
| [19,20,21,22,23,24,25] [22] [26,27,28] |
| Immune deregulation |
| [29,30,31,32,33,34,35,36] [37] |
| Gene Name | Encoding Protein Name | Function |
|---|---|---|
| JAK2 | Homo sapiens Janus kinase 2 | Protein tyrosine kinase involved in a specific subset of cytokine receptor signaling pathways |
| TERT | Homo sapiens telomerase reverse transcriptase | Ribonucleoprotein polymerase that maintains telomere ends by addition of the telomere repeat TTAGGG |
| DDX41 | Homo sapiens DEAD (Asp-Glu-Ala-Asp) box polypeptide 41 | Putative RNA helicases characterized by the conserved motif Asp-Glu-Ala-Asp (DEAD) and involved in RNA splicing |
| ETV6 | Homo sapiens ETS variant 6 | ETS family transcription factor required for hematopoiesis and maintenance of the developing vascular network |
| GATA2 | Homo sapiens GATA binding protein 2 | Zinc-finger transcription factor regulating genes involved in the development and proliferation of hematopoietic and endocrine cell lineages |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cumbo, C.; Anelli, L.; Zagaria, A.; Coccaro, N.; Tarantini, F.; Specchia, G.; Musto, P.; Albano, F. Second Cancer Onset in Myeloproliferative Neoplasms: What, When, Why? Int. J. Mol. Sci. 2022, 23, 3177. https://doi.org/10.3390/ijms23063177
Cumbo C, Anelli L, Zagaria A, Coccaro N, Tarantini F, Specchia G, Musto P, Albano F. Second Cancer Onset in Myeloproliferative Neoplasms: What, When, Why? International Journal of Molecular Sciences. 2022; 23(6):3177. https://doi.org/10.3390/ijms23063177
Chicago/Turabian StyleCumbo, Cosimo, Luisa Anelli, Antonella Zagaria, Nicoletta Coccaro, Francesco Tarantini, Giorgina Specchia, Pellegrino Musto, and Francesco Albano. 2022. "Second Cancer Onset in Myeloproliferative Neoplasms: What, When, Why?" International Journal of Molecular Sciences 23, no. 6: 3177. https://doi.org/10.3390/ijms23063177
APA StyleCumbo, C., Anelli, L., Zagaria, A., Coccaro, N., Tarantini, F., Specchia, G., Musto, P., & Albano, F. (2022). Second Cancer Onset in Myeloproliferative Neoplasms: What, When, Why? International Journal of Molecular Sciences, 23(6), 3177. https://doi.org/10.3390/ijms23063177