
Drug Metabolism for the Identification of Clinical Biomarkers in Breast Cancer



Abstract
:1. Breast Cancer Therapeutic Options
2. Immunometabolism
2.1. Limitations of Breast Cancer Subtyping
2.2. Metabolism Regulates Immune Cell Activation
2.3. Drug-Induced Alterations
3. Finding Biomarkers
3.1. Pharmacogenomics, Defining a Metabolic Background of Tumors: Genome Take on Drug Metabolism
3.2. Pharmacomicrobiomics, Using the Microbiome to Predict Resistance and Enhance Immunotherapies
3.2.1. Factors Influencing Gut Microbiota Composition
3.2.2. The Microbiome Intervein with Neurophysiological Function
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Weigelt, B.; Geyer, F.C.; Reis-Filho, J.S. Histological types of breast cancer: How special are they? Mol. Oncol. 2010, 4, 192–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hon, J.D.C.; Singh, B.; Sahin, A.; Du, G.; Wang, J.; Wang, V.Y.; Deng, F.-M.; Zhang, D.Y.; Monaco, M.E.; Lee, P. Breast cancer molecular subtypes: From TNBC to QNBC. Am. J. Cancer Res. 2016, 6, 1864–1872. [Google Scholar]
- Lamb, C.A.; Vanzulli, S.I.; Lanari, C. Hormone receptors in breast cancer: More than estrogen receptors. Medicina 2019, 79, 540–545. [Google Scholar] [PubMed]
- Early Breast Cancer Trialists’ Collaborative Group (EBCTCG). Aromatase inhibitors versus tamoxifen in early breast cancer: Patient-level meta-analysis of the randomised trials. Lancet 2015, 386, 1341–1352. [Google Scholar] [CrossRef]
- Pistelli, M.; Della Mora, A.; Ballatore, Z.; Berardi, R. Aromatase Inhibitors in Premenopausal Women with Breast Cancer: The State of the Art and Future Prospects. Curr. Oncol. 2018, 25, e168–e175. [Google Scholar] [CrossRef] [Green Version]
- Oh, D.-Y.; Bang, Y.-J. HER2-targeted therapies—A role beyond breast cancer. Nat. Rev. Clin. Oncol. 2020, 17, 33–48. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Yost, S.E.; Yuan, Y. Neoadjuvant Treatment for Triple Negative Breast Cancer: Recent Progresses and Challenges. Cancers 2020, 12, 1404. [Google Scholar] [CrossRef] [PubMed]
- Ahmadian, E.; Samiei, M.; Hasanzadeh, A.; Kavetskyy, T.; Jafari, S.; Alipour, M.; Salatin, S.; Rameshrad, M.; Sharifi, S.; Eftekhari, A.; et al. Monitoring of drug resistance towards reducing the toxicity of pharmaceutical compounds: Past, present and future. J. Pharm. Biomed. Anal. 2020, 186, 113265. [Google Scholar] [CrossRef]
- Henriques, B.; Mendes, F.; Martins, D. Immunotherapy in Breast Cancer: When, How, and What Challenges? Biomedicines 2021, 9, 1687. [Google Scholar] [CrossRef]
- Kreutzfeldt, J.; Rozeboom, B.; Dey, N.; De, P. The trastuzumab era: Current and upcoming targeted HER2+ breast cancer therapies. Am. J. Cancer Res. 2020, 10, 1045–1067. [Google Scholar] [PubMed]
- Makhoul, I.; Atiq, M.; Alwbari, A.; Kieber-Emmons, T. Breast Cancer Immunotherapy: An Update. Breast Cancer Basic Clin. Res. 2018, 12, 1178223418774802. [Google Scholar] [CrossRef] [PubMed]
- Gjoerup, O.; Brown, C.A.; Ross, J.S.; Huang, R.S.P.; Schrock, A.; Creeden, J.; Fabrizio, D.; Tolba, K. Identification and Utilization of Biomarkers to Predict Response to Immune Checkpoint Inhibitors. AAPS J. 2020, 22, 132. [Google Scholar] [CrossRef] [PubMed]
- Sivapiragasam, A.; Kumar, P.A.; Sokol, E.S.; Albacker, L.A.; Killian, J.K.; Ramkissoon, S.H.; Huang, R.S.P.; Severson, E.A.; Brown, C.A.; Danziger, N.; et al. Predictive Biomarkers for Immune Checkpoint Inhibitors in Metastatic Breast Cancer. Cancer Med. 2021, 10, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Tower, H.; Ruppert, M.; Britt, K. The Immune Microenvironment of Breast Cancer Progression. Cancers 2019, 11, 1375. [Google Scholar] [CrossRef] [Green Version]
- Wegiel, B.; Vuerich, M.; Daneshmandi, S.; Seth, P. Metabolic Switch in the Tumor Microenvironment Determines Immune Responses to Anti-cancer Therapy. Front. Oncol. 2018, 8, 284. [Google Scholar] [CrossRef] [Green Version]
- Segovia-Mendoza, M.; Morales-Montor, J. Immune Tumor Microenvironment in Breast Cancer and the Participation of Estrogen and Its Receptors in Cancer Physiopathology. Front. Immunol. 2019, 10, 348. [Google Scholar] [CrossRef] [Green Version]
- Criscitiello, C.; Vingiani, A.; Maisonneuve, P.; Viale, G.; Curigliano, G. Tumor-infiltrating lymphocytes (TILs) in ER+/HER2− breast cancer. Breast Cancer Res. Treat. 2020, 183, 347–354. [Google Scholar] [CrossRef]
- Li, X.; Li, M.; Lian, Z.; Zhu, H.; Kong, L.; Wang, P.; Yu, J. Prognostic Role of Programmed Death Ligand-1 Expression in Breast Cancer: A Systematic Review and Meta-Analysis. Target. Oncol. 2016, 11, 753–761. [Google Scholar] [CrossRef]
- Savas, P.; Salgado, R.; Denkert, C.; Sotiriou, C.; Darcy, P.K.; Smyth, M.J.; Loi, S. Clinical relevance of host immunity in breast cancer: From TILs to the clinic. Nat. Rev. Clin. Oncol. 2016, 13, 228–241. [Google Scholar] [CrossRef]
- Shiga, K.; Hara, M.; Nagasaki, T.; Sato, T.; Takahashi, H.; Takeyama, H. Cancer-Associated Fibroblasts: Their Characteristics and Their Roles in Tumor Growth. Cancers 2015, 7, 2443–2458. [Google Scholar] [CrossRef]
- Sükei, T.; Palma, E.; Urbani, L. Interplay between Cellular and Non-Cellular Components of the Tumour Microenvironment in Hepatocellular Carcinoma. Cancers 2021, 13, 5586. [Google Scholar] [CrossRef] [PubMed]
- Weyand, C.M.; Watanabe, R.; Zhang, H.; Akiyama, M.; Berry, G.J.; Goronzy, J.J. Cytokines, growth factors and proteases in medium and large vessel vasculitis. Clin. Immunol. 2019, 206, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Jabbari, N.; Akbariazar, E.; Feqhhi, M.; Rahbarghazi, R.; Rezaie, J. Breast cancer-derived exosomes: Tumor progression and therapeutic agents. J. Cell. Physiol. 2020, 235, 6345–6356. [Google Scholar] [CrossRef] [PubMed]
- Haibe, Y.; Kreidieh, M.; El Hajj, H.; Khalifeh, I.; Mukherji, D.; Temraz, S.; Shamseddine, A. Resistance Mechanisms to Anti-angiogenic Therapies in Cancer. Front. Oncol. 2020, 10, 221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218, Erratum in Trends Biochem. Sci. 2016, 41, 287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdel-Wahab, A.F.; Mahmoud, W.; Al-Harizy, R.M. Targeting glucose metabolism to suppress cancer progression: Prospective of anti-glycolytic cancer therapy. Pharmacol. Res. 2019, 150, 104511. [Google Scholar] [CrossRef]
- Buck, M.D.; O’Sullivan, D.; Geltink, R.I.K.; Curtis, J.D.; Chang, C.-H.; Sanin, D.E.; Qiu, J.; Kretz, O.; Braas, D.; Van Der Windt, G.J.; et al. Mitochondrial Dynamics Controls T Cell Fate through Metabolic Programming. Cell 2016, 166, 63–76. [Google Scholar] [CrossRef] [Green Version]
- Makowski, L.; Chaib, M.; Rathmell, J.C. Immunometabolism: From basic mechanisms to translation. Immunol. Rev. 2020, 295, 5–14. [Google Scholar] [CrossRef] [Green Version]
- Geltink, R.I.K.; O’Sullivan, D.; Corrado, M.; Bremser, A.; Buck, M.D.; Buescher, J.M.; Firat, E.; Zhu, X.; Niedermann, G.; Caputa, G.; et al. Mitochondrial Priming by CD28. Cell 2017, 171, 385–397.e11. [Google Scholar] [CrossRef] [Green Version]
- Heintzman, D.R.; Fisher, E.L.; Rathmell, J.C. Microenvironmental influences on T cell immunity in cancer and inflammation. Cell. Mol. Immunol. 2022, 19, 316–326. [Google Scholar] [CrossRef] [PubMed]
- Crino, P.B. Mechanistic target of rapamycin (mTOR) signaling in status epilepticus. Epilepsy Behav. 2019, 101, 106550. [Google Scholar] [CrossRef] [PubMed]
- Mikó, E.; Kovács, T.; Sebő, É.; Tóth, J.; Csonka, T.; Ujlaki, G.; Sipos, A.; Szabó, J.; Méhes, G.; Bai, P. Microbiome—Microbial Metabolome—Cancer Cell Interactions in Breast Cancer—Familiar, but Unexplored. Cells 2019, 8, 293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, L.; Wick, N.; Germans, S.K.; Peng, Y. The Role of Breast Cancer Stem Cells in Chemoresistance and Metastasis in Triple-Negative Breast Cancer. Cancers 2021, 13, 6209. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Kim, D.H.; Jung, W.-H.; Koo, J.S. Metabolic phenotypes in triple-negative breast cancer. Tumor Biol. 2013, 34, 1699–1712. [Google Scholar] [CrossRef] [PubMed]
- Lanning, N.J.; Castle, J.P.; Singh, S.J.; Leon, A.N.; Tovar, E.A.; Sanghera, A.; MacKeigan, J.P.; Filipp, F.V.; Graveel, C.R. Metabolic profiling of triple-negative breast cancer cells reveals metabolic vulnerabilities. Cancer Metab. 2017, 5, 6. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, N.; Das, G.M. Metabolic Reprogramming in Breast Cancer and Its Therapeutic Implications. Cells 2019, 8, 89. [Google Scholar] [CrossRef] [Green Version]
- Icard, P.; Shulman, S.; Farhat, D.; Steyaert, J.-M.; Alifano, M.; Lincet, H. How the Warburg effect supports aggressiveness and drug resistance of cancer cells? Drug Resist. Updates 2018, 38, 1–11. [Google Scholar] [CrossRef]
- Komurov, K.; Tseng, J.; Muller, M.; Seviour, E.; Moss, T.; Yang, L.; Nagrath, D.; Ram, P. The glucose-deprivation network counteracts lapatinib-induced toxicity in resistant ErbB2-positive breast cancer cells. Mol. Syst. Biol. 2012, 8, 596. [Google Scholar] [CrossRef]
- Zhao, Y.; Liu, H.; Liu, Z.; Ding, Y.; LeDoux, S.P.; Wilson, G.L.; Voellmy, R.; Lin, Y.; Lin, W.; Nahta, R.; et al. Overcoming Trastuzumab Resistance in Breast Cancer by Targeting Dysregulated Glucose Metabolism. Cancer Res. 2011, 71, 4585–4597. [Google Scholar] [CrossRef] [Green Version]
- Zhou, M.; Zhao, Y.; Ding, Y.; Liu, H.; Liu, Z.; Fodstad, O.; Riker, A.I.; Kamarajugadda, S.; Lu, J.; Owen, L.B.; et al. Warburg effect in chemosensitivity: Targeting lactate dehydrogenase-A re-sensitizes Taxol-resistant cancer cells to Taxol. Mol. Cancer 2010, 9, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalezic, A.; Udicki, M.; Galic, B.S.; Aleksic, M.; Korac, A.; Jankovic, A.; Korac, B. Lactate Metabolism in Breast Cancer Microenvironment: Contribution Focused on Associated Adipose Tissue and Obesity. Int. J. Mol. Sci. 2020, 21, 9676. [Google Scholar] [CrossRef] [PubMed]
- Deblois, G.; Smith, H.W.; Tam, I.S.; Gravel, S.-P.; Caron, M.; Savage, P.; Labbé, D.P.; Bégin, L.R.; Tremblay, M.L.; Park, M.; et al. ERRα mediates metabolic adaptations driving lapatinib resistance in breast cancer. Nat. Commun. 2016, 7, 12156. [Google Scholar] [CrossRef] [PubMed]
- Fiorillo, M.; Sotgia, F.; Sisci, D.; Cappello, A.R.; Lisanti, M.P. Mitochondrial “power” drives tamoxifen resistance: NQO1 and GCLC are new therapeutic targets in breast cancer. Oncotarget 2017, 8, 20309–20327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Liu, Y.; Zhang, J.-T. A new mechanism of drug resistance in breast cancer cells: Fatty acid synthase overexpression-mediated palmitate overproduction. Mol. Cancer Ther. 2008, 7, 263–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krzyszczyk, P.; Acevedo, A.; Davidoff, E.J.; Timmins, L.M.; Marrero-Berrios, I.; Patel, M.; White, C.; Lowe, C.; Sherba, J.J.; Hartmanshenn, C.; et al. The growing role of precision and personalized medicine for cancer treatment. Technology 2018, 6, 79–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Claret, F.X.; Vu, T.T. Trastuzumab: Updated mechanisms of action and resistance in breast cancer. Front. Oncol. 2012, 2, 62. [Google Scholar] [CrossRef] [Green Version]
- Nicolini, A.; Ferrari, P.; Duffy, M.J. Prognostic and predictive biomarkers in breast cancer: Past, present and future. Semin. Cancer Biol. 2018, 52, 56–73. [Google Scholar] [CrossRef]
- Wu, H.-J.; Chu, P.-Y. Recent Discoveries of Macromolecule- and Cell-Based Biomarkers and Therapeutic Implications in Breast Cancer. Int. J. Mol. Sci. 2021, 22, 636. [Google Scholar] [CrossRef] [PubMed]
- Redig, A.J.; McAllister, S.S. Breast cancer as a systemic disease: A view of metastasis. J. Intern. Med. 2013, 274, 113–126. [Google Scholar] [CrossRef] [Green Version]
- Nanda, C.S.; Venkateswaran, S.V.; Patani, N.; Yuneva, M. Defining a metabolic landscape of tumours: Genome meets metabolism. Br. J. Cancer 2020, 122, 136–149. [Google Scholar] [CrossRef] [PubMed]
- Jeibouei, S.; Akbari, M.E.; Kalbasi, A.; Aref, A.R.; Ajoudanian, M.; Rezvani, A.; Zali, H. Personalized medicine in breast cancer: Pharmacogenomics approaches. Pharm. Pers. Med. 2019, 12, 59–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, L.; Whirl-Carrillo, M.; Klein, T.E. PharmGKB, an Integrated Resource of Pharmacogenomic Knowledge. Curr. Protoc. 2021, 1, e226. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, K.; Horie-Inoue, K.; Inoue, S. Identification of estrogen-responsive genes based on the DNA binding properties of estrogen receptors using high-throughput sequencing technology. Acta Pharmacol. Sin. 2015, 36, 24–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruiz-Saenz, A.; Dreyer, C.; Campbell, M.R.; Steri, V.; Gulizia, N.; Moasser, M.M.; Gulizia, N.P. HER2 Amplification in Tumors Activates PI3K/Akt Signaling Independent of HER3. Cancer Res. 2018, 78, 3645–3658. [Google Scholar] [CrossRef] [Green Version]
- Aggarwal, T.; Wadhwa, R.; Gupta, R.; Paudel, K.R.; Collet, T.; Chellappan, D.K.; Gupta, G.; Perumalsamy, H.; Mehta, M.; Satija, S.; et al. MicroRNAs as Biomarker for Breast Cancer. Endocr. Metab. Immune Disord. Drug Targets 2020, 20, 1597–1610. [Google Scholar] [CrossRef]
- Barzaman, K.; Karami, J.; Zarei, Z.; Hosseinzadeh, A.; Kazemi, M.H.; Moradi-Kalbolandi, S.; Safari, E.; Farahmand, L. Breast cancer: Biology, biomarkers, and treatments. Int. Immunopharmacol. 2020, 84, 106535. [Google Scholar] [CrossRef] [PubMed]
- Malone, E.R.; Oliva, M.; Sabatini, P.J.B.; Stockley, T.; Siu, L.L. Molecular profiling for precision cancer therapies. Genome Med. 2020, 12, 8. [Google Scholar] [CrossRef] [Green Version]
- Rosenbaum, J.N.; Weisman, P. The Evolving Role of Companion Diagnostics for Breast Cancer in an Era of Next-Generation Omics. Am. J. Pathol. 2017, 187, 2185–2198. [Google Scholar] [CrossRef] [Green Version]
- Syed, Y.Y. Oncotype DX Breast Recurrence Score®: A Review of its Use in Early-Stage Breast Cancer. Mol. Diagn. Ther. 2020, 24, 621–632. [Google Scholar] [CrossRef]
- Soliman, H.; Shah, V.; Srkalovic, G.; Mahtani, R.; Levine, E.; Mavromatis, B.; Srinivasiah, J.; Kassar, M.; Gabordi, R.; Qamar, R.; et al. MammaPrint guides treatment decisions in breast Cancer: Results of the IMPACt trial. BMC Cancer 2020, 20, 81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davey, M.G.; Hynes, S.O.; Kerin, M.J.; Miller, N.; Lowery, A.J. Ki-67 as a Prognostic Biomarker in Invasive Breast Cancer. Cancers 2021, 13, 4455. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S. Comparison and evaluation of pathway-level aggregation methods of gene expression data. BMC Genom. 2012, 13 (Suppl. 7), S26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenblum, S.I.; Efroni, S.; Schaefer, C.F.; Buetow, K.H. The PathOlogist: An automated tool for pathway-centric analysis. BMC Bioinform. 2011, 12, 133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buzdin, A.; Sorokin, M.; Garazha, A.; Sekacheva, M.; Kim, E.; Zhukov, N.; Wang, Y.; Li, X.; Kar, S.; Hartmann, C.; et al. Molecular pathway activation—New type of biomarkers for tumor morphology and personalized selection of target drugs. Semin. Cancer Biol. 2018, 53, 110–124. [Google Scholar] [CrossRef] [PubMed]
- Nordström, A.; Lewensohn, R. Metabolomics: Moving to the Clinic. J. Neuroimmune Pharmacol. 2010, 5, 4–17. [Google Scholar] [CrossRef]
- Johnson, C.H.; Ivanisevic, J.; Siuzdak, G. Metabolomics: Beyond biomarkers and towards mechanisms. Nat. Rev. Mol. Cell Biol. 2016, 17, 451–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ward, T.; Larson, J.; Meulemans, J.; Hillmann, B.; Lynch, J.; Sidiropoulos, D.; Spear, J.R.; Caporaso, G.; Blekhman, R.; Knight, R.; et al. BugBase predicts organism-level microbiome phenotypes. bioRxiv 2017, 133462. [Google Scholar] [CrossRef]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G.I. PIC-RUSt2: An improved and customizable approach for metagenome inference. bioRxiv 2019, 672295. [Google Scholar] [CrossRef] [Green Version]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Yu, Z.-K.; Xie, R.-L.; You, R.; Liu, Y.-P.; Chen, X.-Y.; Chen, M.-Y.; Huang, P.-Y. The role of the bacterial microbiome in the treatment of cancer. BMC Cancer 2021, 21, 934. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Sun, T.; Xu, J. Tumor-related Microbiome in the Breast Microenvironment and Breast Cancer. J. Cancer 2021, 12, 4841–4848. [Google Scholar] [CrossRef]
- Urbaniak, C.; Cummins, J.; Brackstone, M.; Macklaim, J.M.; Gloor, G.B.; Baban, C.K.; Scott, L.; O’Hanlon, D.M.; Burton, J.P.; Francis, K.P.; et al. Microbiota of Human Breast Tissue. Appl. Environ. Microbiol. 2014, 80, 3007–3014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tzeng, A.; Sangwan, N.; Jia, M.; Liu, C.-C.; Keslar, K.S.; Downs-Kelly, E.; Fairchild, R.L.; Al-Hilli, Z.; Grobmyer, S.R.; Eng, C. Human breast microbiome correlates with prognostic features and immunological signatures in breast cancer. Genome Med. 2021, 13, 60. [Google Scholar] [CrossRef] [PubMed]
- Chan, A.A.; Bashir, M.; Rivas, M.N.; Duvall, K.; Sieling, P.A.; Pieber, T.R.; Vaishampayan, P.A.; Love, S.M.; Lee, D.J. Characterization of the microbiome of nipple aspirate fluid of breast cancer survivors. Sci. Rep. 2016, 6, 28061. [Google Scholar] [CrossRef] [Green Version]
- Xuan, C.; Shamonki, J.M.; Chung, A.; DiNome, M.; Chung, M.; Sieling, P.A.; Lee, D.J. Microbial Dysbiosis Is Associated with Human Breast Cancer. PLoS ONE 2014, 9, e83744. [Google Scholar] [CrossRef] [Green Version]
- Dieleman, S.; Aarnoutse, R.; Ziemons, J.; Kooreman, L.; Boleij, A.; Smidt, M. Exploring the Potential of Breast Microbiota as Biomarker for Breast Cancer and Therapeutic Response. Am. J. Pathol. 2021, 191, 968–982. [Google Scholar] [CrossRef]
- Eslami-S, Z.; Majidzadeh-A, K.; Halvaei, S.; Babapirali, F.; Esmaeili, R. Microbiome and Breast Cancer: New Role for an Ancient Population. Front. Oncol. 2020, 10, 120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bodai, B. Breast Cancer: Lifestyle, the Human Gut Microbiota/Microbiome, and Survivorship. Perm. J. 2020, 24. [Google Scholar] [CrossRef]
- Fessler, J.; Matson, V.; Gajewski, T.F. Exploring the emerging role of the microbiome in cancer immunotherapy. J. Immunother. Cancer 2019, 7, 108. [Google Scholar] [CrossRef]
- Strouse, C.; Mangalam, A.; Zhang, J. Bugs in the system: Bringing the human microbiome to bear in cancer immunotherapy. Gut Microbes 2019, 10, 109–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Havel, J.J.; Chowell, D.; Chan, T.A. The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nat. Rev. Cancer 2019, 19, 133–150. [Google Scholar] [CrossRef] [PubMed]
- Rea, D.; Coppola, G.; Palma, G.; Barbieri, A.; Luciano, A.; Del Prete, P.; Rossetti, S.; Berretta, M.; Facchini, G.; Perdonà, S.; et al. Microbiota effects on cancer: From risks to therapies. Oncotarget 2018, 9, 17915–17927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKee, A.M.; Kirkup, B.M.; Madgwick, M.; Fowler, W.J.; Price, C.A.; Dreger, S.A.; Ansorge, R.; Makin, K.A.; Caim, S.; Le Gall, G.; et al. Antibiotic-induced disturbances of the gut microbiota result in accelerated breast tumor growth. iScience 2021, 24, 103012. [Google Scholar] [CrossRef]
- Bedada, T.L.; Feto, T.K.; Awoke, K.S.; Garedew, A.D.; Yifat, F.T.; Birri, D.J. Probiotics for cancer alternative prevention and treatment. Biomed. Pharmacother. 2020, 129, 110409. [Google Scholar] [CrossRef]
- Marteau, P. Safety aspects of probiotic products. Näringsforskning 2001, 45, 22–24. [Google Scholar] [CrossRef] [Green Version]
- Fan, Y.; Pedersen, O. Gut microbiota in human metabolic health and disease. Nat. Rev. Microbiol. 2021, 19, 55–71. [Google Scholar] [CrossRef]
- Mogensen, T.H. Pathogen Recognition and Inflammatory Signaling in Innate Immune Defenses. Clin. Microbiol. Rev. 2009, 22, 240–273. [Google Scholar] [CrossRef] [Green Version]
- Pandey, S.; Kawai, T.; Akira, S. Microbial Sensing by Toll-Like Receptors and Intracellular Nucleic Acid Sensors. Cold Spring Harb. Perspect. Biol. 2014, 7, a016246. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Pan, Z.; Kang, X.; Yang, Y.; Kang, H.; Zhang, N.; Rosati, J.M.; Jiao, X. Amino acids 89–96 of Salmonella typhimurium flagellin represent the major domain responsible for TLR5-independent adjuvanticity in the humoral immune response. Cell. Mol. Immunol. 2014, 12, 625–632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedman, R. Drug resistance in cancer: Molecular evolution and compensatory proliferation. Oncotarget 2016, 7, 11746–11755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stringer, A.M.; Gibson, R.J.; Logan, R.M.; Bowen, J.M.; Yeoh, A.S.J.; Laurence, J.; Keefe, D.M.K. Irinotecan-induced mucositis is associated with changes in intestinal mucins. Cancer Chemother. Pharmacol. 2009, 64, 123–132. [Google Scholar] [CrossRef]
- Gonçalves, P.; Araújo, J.R.; Di Santo, J.P. A Cross-Talk Between Microbiota-Derived Short-Chain Fatty Acids and the Host Mucosal Immune System Regulates Intestinal Homeostasis and Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2018, 24, 558–572. [Google Scholar] [CrossRef] [Green Version]
- Yoo, J.Y.; Groer, M.; Dutra, S.V.O.; Sarkar, A.; McSkimming, D.I. Gut Microbiota and Immune System Interactions. Microorganisms 2020, 8, 1587. [Google Scholar] [CrossRef] [PubMed]
- Arpaia, N.; Campbell, C.; Fan, X.; Dikiy, S.; Van Der Veeken, J.; DeRoos, P.; Liu, H.; Cross, J.R.; Pfeffer, K.; Coffer, P.J.; et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature 2013, 504, 451–455. [Google Scholar] [CrossRef] [PubMed]
- Maslowski, K.M.; Vieira, A.T.; Ng, A.; Kranich, J.; Sierro, F.; Yu, D.; Schilter, H.C.; Rolph, M.S.; Mackay, F.; Artis, D.; et al. Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature 2009, 461, 1282–1286. [Google Scholar] [CrossRef]
- Zhang, Z.; Tang, H.; Chen, P.; Xie, H.; Tao, Y. Demystifying the manipulation of host immunity, metabolism, and extraintestinal tumors by the gut microbiome. Signal Transduct. Target. Ther. 2019, 4, 41. [Google Scholar] [CrossRef] [PubMed]
- Baker, J.M.; Al-Nakkash, L.; Herbst-Kralovetz, M.M. Estrogen–gut microbiome axis: Physiological and clinical implications. Maturitas 2017, 103, 45–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwa, M.; Plottel, C.S.; Blaser, M.J.; Adams, S. The Intestinal Microbiome and Estrogen Receptor–Positive Female Breast Cancer. JNCI J. Natl. Cancer Inst. 2016, 108. [Google Scholar] [CrossRef]
- Flores, R.; Shi, J.; Fuhrman, B.; Xu, X.; Veenstra, T.D.; Gail, M.H.; Gajer, P.; Ravel, J.; Goedert, J.J. Fecal microbial determinants of fecal and systemic estrogens and estrogen metabolites: A cross-sectional study. J. Transl. Med. 2012, 10, 253. [Google Scholar] [CrossRef] [Green Version]
- Thirunavukkarasan, M.; Wang, C.; Rao, A.; Hind, T.; Teo, Y.R.; Siddiquee, A.A.-M.; Goghari, M.A.I.; Kumar, A.P.; Herr, D.R. Short-chain fatty acid receptors inhibit invasive phenotypes in breast cancer cells. PLoS ONE 2017, 12, e0186334. [Google Scholar] [CrossRef]
- Hopkins, M.M.; Meier, K.E. Free Fatty Acid Receptors and Cancer: From Nutrition to Pharmacology. Handb. Exp. Pharmacol. 2016, 236, 233–251. [Google Scholar] [CrossRef]
- Huang, C.-K.; Chang, P.-H.; Kuo, W.-H.; Chen, C.-L.; Jeng, Y.-M.; Chang, K.-J.; Shew, J.-Y.; Hu, C.-M.; Lee, W.-H. Adipocytes promote malignant growth of breast tumours with monocarboxylate transporter 2 expression via β-hydroxybutyrate. Nat. Commun. 2017, 8, 14706. [Google Scholar] [CrossRef]
- Long, S.L.; Gahan, C.G.; Joyce, S.A. Interactions between gut bacteria and bile in health and disease. Mol. Asp. Med. 2017, 56, 54–65. [Google Scholar] [CrossRef]
- Tang, X.; Lin, C.-C.; Spasojevic, I.; Iversen, E.S.; Chi, J.-T.; Marks, J.R. A joint analysis of metabolomics and genetics of breast cancer. Breast Cancer Res. 2014, 16, 415. [Google Scholar] [CrossRef] [Green Version]
- Ridlon, J.M.; Hylemon, P.B. Identification and characterization of two bile acid coenzyme A transferases from Clostridium scindens, a bile acid 7α-dehydroxylating intestinal bacterium. J. Lipid Res. 2012, 53, 66–76. [Google Scholar] [CrossRef] [Green Version]
- Miller-Fleming, L.; Olin-Sandoval, V.; Campbell, K.; Ralser, M. Remaining Mysteries of Molecular Biology: The Role of Polyamines in the Cell. J. Mol. Biol. 2015, 427, 3389–3406. [Google Scholar] [CrossRef]
- Kovács, T.; Mikó, E.; Vida, A.; Sebő, É.; Toth, J.; Csonka, T.; Boratkó, A.; Ujlaki, G.; Lente, G.; Kovács, P.; et al. Cadaverine, a metabolite of the microbiome, reduces breast cancer aggressiveness through trace amino acid receptors. Sci. Rep. 2019, 9, 1300. [Google Scholar] [CrossRef] [Green Version]
- Petra, A.I.; Panagiotidou, S.; Hatziagelaki, E.; Stewart, J.M.; Conti, P.; Theoharides, T.C. Gut-Microbiota-Brain Axis and Its Effect on Neuropsychiatric Disorders with Suspected Immune Dysregulation. Clin. Ther. 2015, 37, 984–995. [Google Scholar] [CrossRef] [Green Version]
- Dinan, T.G.; Cryan, J.F. The Microbiome-Gut-Brain Axis in Health and Disease. Gastroenterol. Clin. N. Am. 2017, 46, 77–89. [Google Scholar] [CrossRef] [Green Version]
- Rogers, G.B.; Keating, D.; Young, R.; Wong, M.-L.; Licinio, J.; Wesselingh, S. From gut dysbiosis to altered brain function and mental illness: Mechanisms and pathways. Mol. Psychiatry 2016, 21, 738–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrosino, J.F. The microbiome in precision medicine: The way forward. Genome Med. 2018, 10, 12. [Google Scholar] [CrossRef] [PubMed]


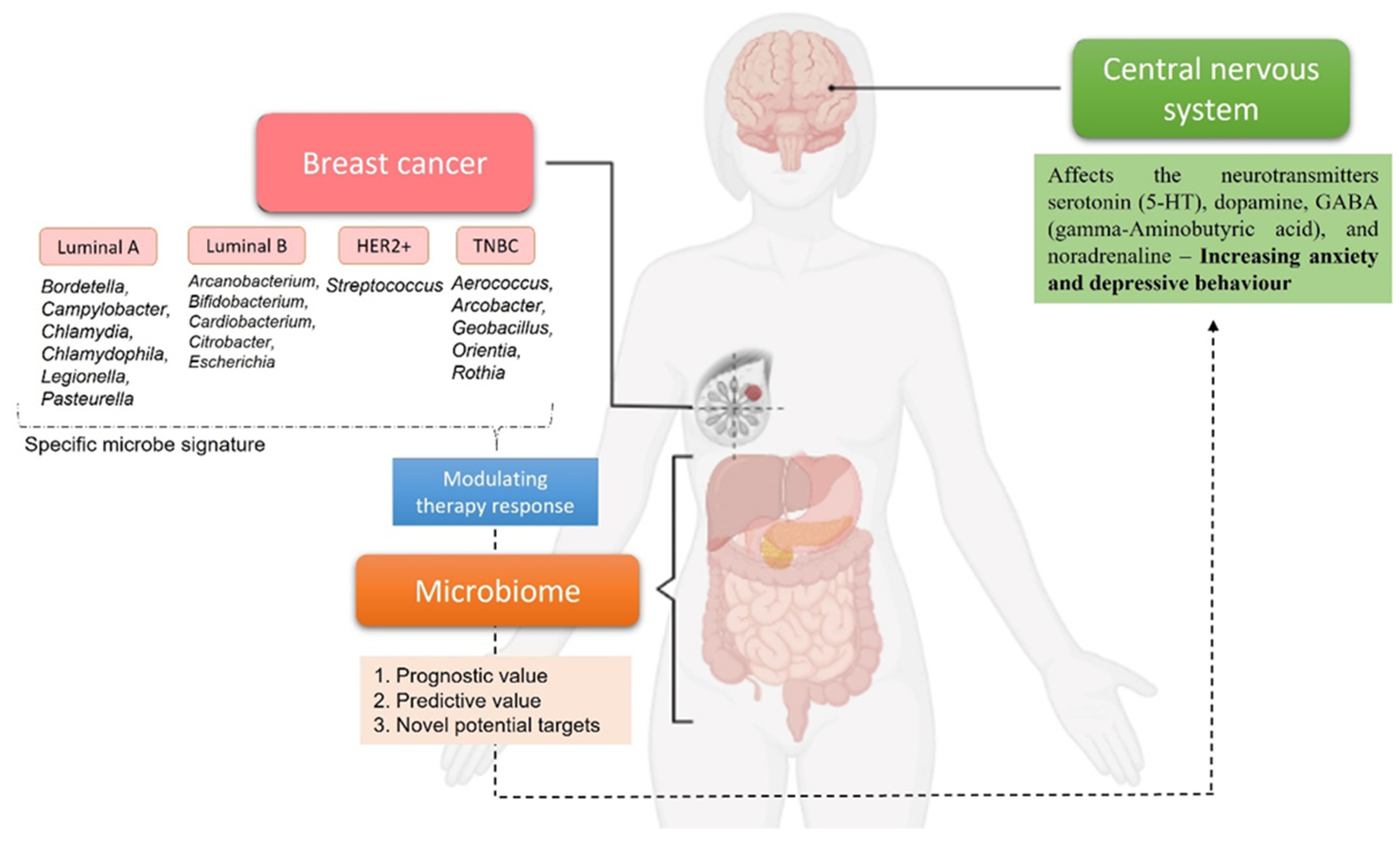{kind=link}
| Breast Cancer Subtype | Receptor Profile | Subtype Prevalence | Subcategories | Prognosis | Immune Cell Patterns |
|---|---|---|---|---|---|
| Hormone positive | ER+ or PR+ | 70% | Luminal A | When compared to other subtypes, it grows more slowly and is less aggressive. | Nk, Neutrophils Tregs, TAMs 1 and 2, Mast cells TCD8+, TCD4+, B lymphocytes |
| Luminal B | Because it has a higher grade than luminal A, it is linked to a worse prognosis. | ||||
| HER2 positive | HER2+ | 20% | - | Poor prognosis and aggressive disease progression | Tregs, Neutrophils, DCs, Mast cells, Tγδ |
| Triple-negative breast cancer | Er−, Pr−, and HER2− | 10% | Basal-like 1 and 2 (BL-1, BL-2), immunomodulatory (IM), mesenchymal (M), mesenchymal stem cell-like (MSL), and luminal androgen receptor (LAR) | It has the worst prognosis. TNBC is extremely common among black women and those who have a BRCA1 gene mutation. | Tregs, TAMs 1 and 2, Mast cells TCD8+, TCD4+, DCs |
| Pathways Associated with Metabolism | Target Proteins/Enzymes or Metabolites | Therapy |
|---|---|---|
| Glycolysis | GLUT1, Hexokinase, LDHA, Pyruvate kinase, SGLT-2 | Lapatinib, Paclitaxel, Trastuzumab, 2-deoxy-D-glucose, Dapagliflozin, Oxamate and Tamoxifen |
| Fatty acid synthesis | FASN | Adriamycin, Omeprazole, Conjugated linolic acid, Orlistat, Fasnall, Cerulenin and C75 |
| Redox metabolism | GCLC | Tamoxifen |
| Mitochondrial energy metabolism | ERRα, NQO1 | Lapatinib, Tamoxifen |
| TCA cycle | Pyruvate dehydrogenase kinase (PDK3) | siRNA, Metformin |
| Predictive Biomarkers | Predict Response to a Therapy | A Breast Cancer Patient with Extra Copies of the HER2 Gene Will Respond Favorably to the HER2 Inhibitor Trastuzumab |
|---|---|---|
| Prognostic biomarkers | Predict patient outcome | Ki-67 and proliferating cell nuclear antigen overexpression; estrogen receptor (ER) and progesterone receptor (PR) overexpression; transforming growth factor- (TGF-); apoptotic imbalance indicators, including bcl-2 overexpression and an elevated bax/bcl-2 ratio; changes in differentiation signals, such as c-myc and related protein overexpression; loss of differentiation markers, such as TGF-II receptor and retinoic acid receptor; and changes in angiogenesis proteins, such as VEGF overexpression, are all instances. |
| Diagnostic biomarkers | It helps clinicians to identify a subtype of cancer accurately | Carbohydrate antigen 15-3 (CA15-3); circulating DNA (ctDNA) and RNA (e.g., micro RNAs); circulating tumor cells and exosomes |
| Risk assessment biomarkers | Predicts the patient’s risk of developing a malignancy | Pathogenic mutations in BRCA1 and BRCA2 is a risk factor for developing breast and ovarian cancer |
| Cancer recurrence monitoring biomarkers | Surveillance marker to monitor recurrence of cancer | Chemokine receptor 9 (CCR9); miRNAs by downregulating E-cadherin and thus affecting EMT and breast cancer cell metastasis; non-cancer cell components |
| Biomarkers Involved in Cancer Drug Resistance | Identifies possible markers for drug resistance | Estrogen Receptor Alpha (ESR1) Mutation; miRNA; circRNA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Costa, B.; Vale, N. Drug Metabolism for the Identification of Clinical Biomarkers in Breast Cancer. Int. J. Mol. Sci. 2022, 23, 3181. https://doi.org/10.3390/ijms23063181
Costa B, Vale N. Drug Metabolism for the Identification of Clinical Biomarkers in Breast Cancer. International Journal of Molecular Sciences. 2022; 23(6):3181. https://doi.org/10.3390/ijms23063181
Chicago/Turabian StyleCosta, Bárbara, and Nuno Vale. 2022. "Drug Metabolism for the Identification of Clinical Biomarkers in Breast Cancer" International Journal of Molecular Sciences 23, no. 6: 3181. https://doi.org/10.3390/ijms23063181
APA StyleCosta, B., & Vale, N. (2022). Drug Metabolism for the Identification of Clinical Biomarkers in Breast Cancer. International Journal of Molecular Sciences, 23(6), 3181. https://doi.org/10.3390/ijms23063181