
Regulation of ABCA1 by AMD-Associated Genetic Variants and Hypoxia in iPSC-RPE

 , ,
, ,  , , , and
, , , and
Abstract
:1. Introduction
2. Results
2.1. ABCA1-Deficient iRPE Cells Have Reduced Cholesterol Efflux and Increased Intracellular Lipid Accumulation
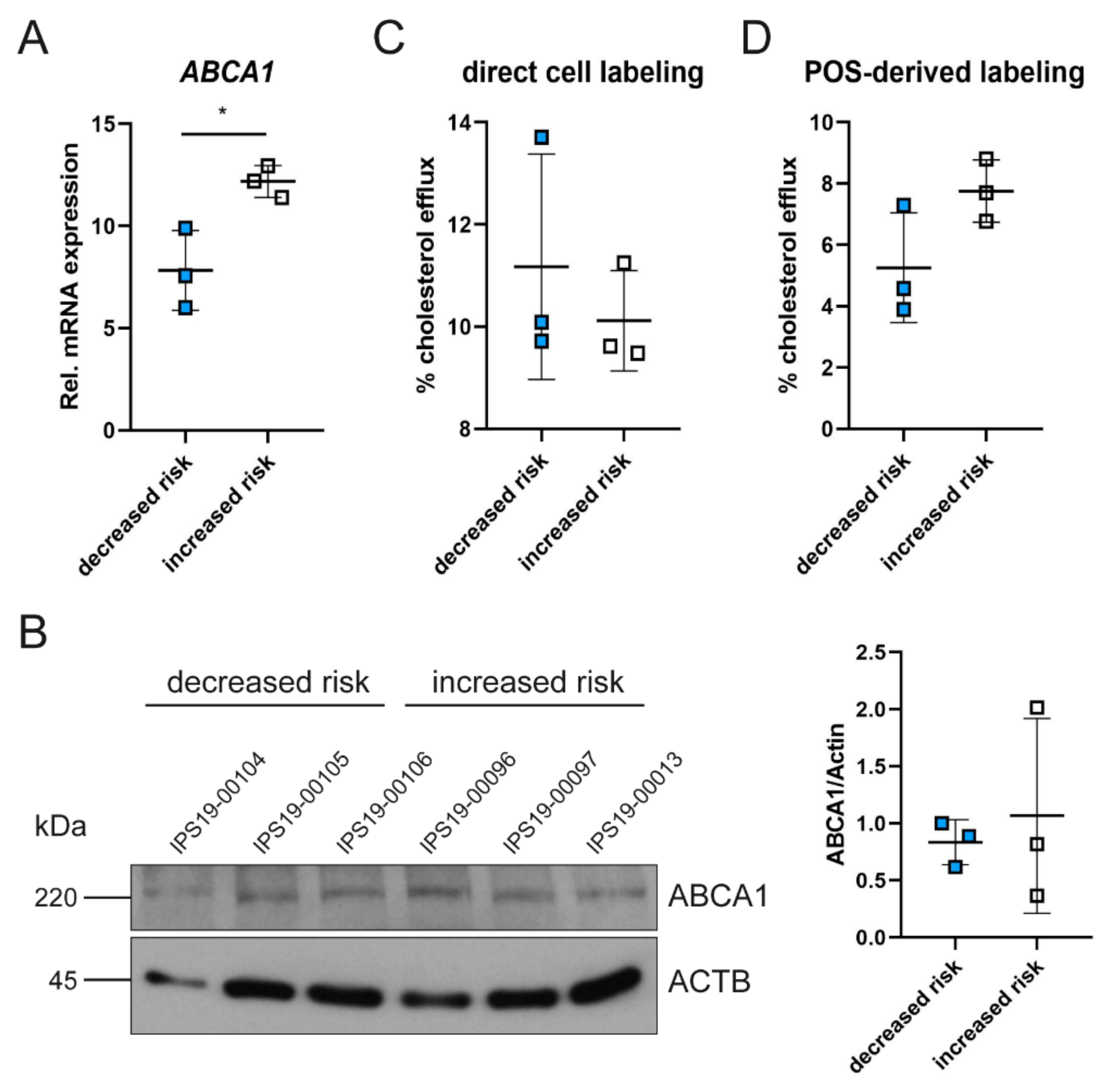
2.2. Polymorphisms in ABCA1 Are Associated with ABCA1 Expression and Cholesterol Efflux
2.3. ABCA1 Expression and Function Is Regulated by Hypoxia in RPE
3. Discussion
4. Materials and Methods
4.1. Human Subject Recruitment and iPSC Generation
4.2. Genotyping of iPSC Cell Lines for AMD-Associated Polymorphisms by Next-Generation Sequencing
4.3. Differentiation of iPSCs to iRPE Cells
4.4. Generation of ABCA1 Knockout iPSCs by CRISPR-Cas9
4.5. PCR Amplification of ABCA1 SNPs and the Region Spanning Exon 2 and Exon 3
4.6. Immunofluorescence Staining
4.7. RNA Isolation, cDNA Synthesis and qPCR
4.8. Western Blot Analysis
4.9. Cholesterol Efflux Assay
4.10. POS Phagocytosis Assay
4.11. Nile Red Staining
4.12. Animal Experiments
4.13. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wong, W.L.; Su, X.; Li, X.; Cheung, C.M.; Klein, R.; Cheng, C.Y.; Wong, T.Y. Global prevalence of age-related macular degeneration and disease burden projection for 2020 and 2040: A systematic review and meta-analysis. Lancet Glob. Health 2014, 2, e106–e116. [Google Scholar] [CrossRef] [Green Version]
- Kopitz, J.; Holz, F.G.; Kaemmerer, E.; Schutt, F. Lipids and lipid peroxidation products in the pathogenesis of age-related macular degeneration. Biochimie 2004, 86, 825–831. [Google Scholar] [CrossRef] [PubMed]
- van Leeuwen, E.M.; Emri, E.; Merle, B.M.J.; Colijn, J.M.; Kersten, E.; Cougnard-Gregoire, A.; Dammeier, S.; Meester-Smoor, M.; Pool, F.M.; de Jong, E.K.; et al. A new perspective on lipid research in age-related macular degeneration. Prog. Retin. Eye Res. 2018, 67, 56–86. [Google Scholar] [CrossRef] [PubMed]
- Gnanaguru, G.; Wagschal, A.; Oh, J.; Saez-Torres, K.L.; Li, T.; Temel, R.E.; Kleinman, M.E.; Naar, A.M.; D’Amore, P.A. Targeting of miR-33 ameliorates phenotypes linked to age-related macular degeneration. Mol. Ther. 2021, 29, 2281–2293. [Google Scholar] [CrossRef]
- Pennington, K.L.; DeAngelis, M.M. Epidemiology of age-related macular degeneration (AMD): Associations with cardiovascular disease phenotypes and lipid factors. Eye Vis. 2016, 3, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curcio, C.A.; Millican, C.L.; Bailey, T.; Kruth, H.S. Accumulation of cholesterol with age in human Bruch’s membrane. Investig. Ophthalmol. Vis. Sci. 2001, 42, 265–274. [Google Scholar]
- Anderson, D.H.; Ozaki, S.; Nealon, M.; Neitz, J.; Mullins, R.F.; Hageman, G.S.; Johnson, L.V. Local cellular sources of apolipoprotein E in the human retina and retinal pigmented epithelium: Implications for the process of drusen formation. Am. J. Ophthalmol. 2001, 131, 767–781. [Google Scholar] [CrossRef]
- Newsome, D.A.; Huh, W.; Green, W.R. Bruch’s membrane age-related changes vary by region. Curr. Eye Res. 1987, 6, 1211–1221. [Google Scholar] [CrossRef]
- Bird, A.C. Bruch’s membrane change with age. Br. J. Ophthalmol. 1992, 76, 166–168. [Google Scholar] [CrossRef] [Green Version]
- Grunwald, J.E.; Metelitsina, T.I.; Dupont, J.C.; Ying, G.S.; Maguire, M.G. Reduced foveolar choroidal blood flow in eyes with increasing AMD severity. Investig. Ophthalmol. Vis. Sci. 2005, 46, 1033–1038. [Google Scholar] [CrossRef] [Green Version]
- Coleman, D.J.; Silverman, R.H.; Rondeau, M.J.; Lloyd, H.O.; Khanifar, A.A.; Chan, R.V. Age-related macular degeneration: Choroidal ischaemia? Br. J. Ophthalmol. 2013, 97, 1020–1023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Apte, R.S. Targeting Tissue Lipids in Age-related Macular Degeneration. EBioMedicine 2016, 5, 26–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curcio, C.A.; Messinger, J.D.; Sloan, K.R.; McGwin, G.; Medeiros, N.E.; Spaide, R.F. Subretinal drusenoid deposits in non-neovascular age-related macular degeneration: Morphology, prevalence, topography, and biogenesis model. Retina 2013, 33, 265–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stefansson, E.; Geirsdottir, A.; Sigurdsson, H. Metabolic physiology in age related macular degeneration. Prog. Retin. Eye Res. 2011, 30, 72–80. [Google Scholar] [CrossRef]
- Ershov, A.V.; Bazan, N.G. Photoreceptor phagocytosis selectively activates PPARgamma expression in retinal pigment epithelial cells. J. NeuroSci. Res. 2000, 60, 328–337. [Google Scholar] [CrossRef]
- Anderson, D.H.; Fisher, S.K.; Steinberg, R.H. Mammalian cones: Disc shedding, phagocytosis, and renewal. Investig. Ophthalmol. Vis. Sci. 1978, 17, 117–133. [Google Scholar]
- Lakkaraju, A.; Finnemann, S.C.; Rodriguez-Boulan, E. The lipofuSci.n fluorophore A2E perturbs cholesterol metabolism in retinal pigment epithelial cells. Proc. Natl. Acad. Sci. USA 2007, 104, 11026–11031. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, I.R.; Alam, S.; Lee, J.W. Cytotoxicity of oxidized low-density lipoprotein in cultured RPE cells is dependent on the formation of 7-ketocholesterol. Investig. Ophthalmol. Vis. Sci. 2004, 45, 2830–2837. [Google Scholar] [CrossRef]
- Fliesler, S.J.; Bretillon, L. The ins and outs of cholesterol in the vertebrate retina. J. Lipid Res. 2010, 51, 3399–3413. [Google Scholar] [CrossRef] [Green Version]
- Zekavat, S.M.; Lu, J.; Maugeais, C.; Mazer, N.A. An in silico model of retinal cholesterol dynamics (RCD model): Insights into the pathophysiology of dry AMD. J. Lipid Res. 2017, 58, 1325–1337. [Google Scholar] [CrossRef] [Green Version]
- Repa, J.J.; Turley, S.D.; Lobaccaro, J.A.; Medina, J.; Li, L.; Lustig, K.; Shan, B.; Heyman, R.A.; Dietschy, J.M.; Mangelsdorf, D.J. Regulation of absorption and ABC1-mediated efflux of cholesterol by RXR heterodimers. Science 2000, 289, 1524–1529. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, E.J.; Anthanont, P.; Diffenderfer, M.R.; Polisecki, E.; Asztalos, B.F. Diagnosis and treatment of high density lipoprotein deficiency. Prog. Cardiovasc. Dis. 2016, 59, 97–106. [Google Scholar] [CrossRef] [Green Version]
- Storti, F.; Klee, K.; Todorova, V.; Steiner, R.; Othman, A.; van der Velde-Visser, S.; Samardzija, M.; Meneau, I.; Barben, M.; Karademir, D.; et al. Impaired ABCA1/ABCG1-mediated lipid efflux in the mouse retinal pigment epithelium (RPE) leads to retinal degeneration. eLife 2019, 8, e45100. [Google Scholar] [CrossRef] [PubMed]
- Vedhachalam, C.; Duong, P.T.; Nickel, M.; Nguyen, D.; Dhanasekaran, P.; Saito, H.; Rothblat, G.H.; Lund-Katz, S.; Phillips, M.C. Mechanism of ATP-binding cassette transporter A1-mediated cellular lipid efflux to apolipoprotein A-I and formation of high density lipoprotein particles. J. Biol. Chem. 2007, 282, 25123–25130. [Google Scholar] [CrossRef] [Green Version]
- Fritsche, L.G.; Igl, W.; Bailey, J.N.; Grassmann, F.; Sengupta, S.; Bragg-Gresham, J.L.; Burdon, K.P.; Hebbring, S.J.; Wen, C.; Gorski, M.; et al. A large genome-wide association study of age-related macular degeneration highlights contributions of rare and common variants. Nat. Genet. 2016, 48, 134–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peter, I.; Huggins, G.S.; Ordovas, J.M.; Haan, M.; Seddon, J.M. Evaluation of new and established age-related macular degeneration susceptibility genes in the Women’s Health Initiative Sight Exam (WHI-SE) Study. Am. J. Ophthalmol. 2011, 152, 1005–1013.e1001. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Stambolian, D.; Edwards, A.O.; Branham, K.E.; Othman, M.; Jakobsdottir, J.; Tosakulwong, N.; Pericak-Vance, M.A.; Campochiaro, P.A.; Klein, M.L.; et al. Genetic variants near TIMP3 and high-density lipoprotein-associated loci influence susceptibility to age-related macular degeneration. Proc. Natl. Acad. Sci. USA 2010, 107, 7401–7406. [Google Scholar] [CrossRef] [Green Version]
- Fauser, S.; Smailhodzic, D.; Caramoy, A.; van de Ven, J.P.; Kirchhof, B.; Hoyng, C.B.; Klevering, B.J.; Liakopoulos, S.; den Hollander, A.I. Evaluation of serum lipid concentrations and genetic variants at high-density lipoprotein metabolism loci and TIMP3 in age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2011, 52, 5525–5528. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Reynolds, R.; Fagerness, J.; Rosner, B.; Daly, M.J.; Seddon, J.M. Association of variants in the LIPC and ABCA1 genes with intermediate and large drusen and advanced age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2011, 52, 4663–4670. [Google Scholar] [CrossRef] [Green Version]
- Berenberg, T.L.; Metelitsina, T.I.; Madow, B.; Dai, Y.; Ying, G.S.; Dupont, J.C.; Grunwald, L.; Brucker, A.J.; Grunwald, J.E. The association between drusen extent and foveolar choroidal blood flow in age-related macular degeneration. Retina 2012, 32, 25–31. [Google Scholar] [CrossRef] [Green Version]
- Parathath, S.; Mick, S.L.; Feig, J.E.; Joaquin, V.; Grauer, L.; Habiel, D.M.; Gassmann, M.; Gardner, L.B.; Fisher, E.A. Hypoxia is present in murine atherosclerotic plaques and has multiple adverse effects on macrophage lipid metabolism. Circ. Res. 2011, 109, 1141–1152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmitz, G.; Langmann, T. Transcriptional regulatory networks in lipid metabolism control ABCA1 expression. Biochim. Biophys. Acta 2005, 1735, 1–19. [Google Scholar] [CrossRef] [PubMed]
- See, R.H.; Caday-Malcolm, R.A.; Singaraja, R.R.; Zhou, S.; Silverston, A.; Huber, M.T.; Moran, J.; James, E.R.; Janoo, R.; Savill, J.M.; et al. Protein kinase A site-specific phosphorylation regulates ATP-binding cassette A1 (ABCA1)-mediated phospholipid efflux. J. Biol. Chem. 2002, 277, 41835–41842. [Google Scholar] [CrossRef] [Green Version]
- Sene, A.; Khan, A.A.; Cox, D.; Nakamura, R.E.; Santeford, A.; Kim, B.M.; Sidhu, R.; Onken, M.D.; Harbour, J.W.; Hagbi-Levi, S.; et al. Impaired cholesterol efflux in senescent macrophages promotes age-related macular degeneration. Cell Metab. 2013, 17, 549–561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, Y.T.; Li, Y.; Ryu, J.; Su, P.Y.; Cheng, C.H.; Wu, W.H.; Li, Y.S.; Quinn, P.M.J.; Leong, K.W.; Tsang, S.H. Impaired cholesterol efflux in retinal pigment epithelium of individuals with juvenile macular degeneration. Am. J. Hum. Genet. 2021, 108, 903–918. [Google Scholar] [CrossRef] [PubMed]
- Zimmer, A.D.; Walbrecq, G.; Kozar, I.; Behrmann, I.; Haan, C. Phosphorylation of the pyruvate dehydrogenase complex precedes HIF-1-mediated effects and pyruvate dehydrogenase kinase 1 upregulation during the first hours of hypoxic treatment in hepatocellular carcinoma cells. Hypoxia 2016, 4, 135–145. [Google Scholar] [CrossRef] [Green Version]
- Ebner, L.J.A.; Samardzija, M.; Storti, F.; Todorova, V.; Karademir, D.; Behr, J.; Simpson, F.; Thiersch, M.; Grimm, C. Transcriptomic analysis of the mouse retina after acute and chronic normobaric and hypobaric hypoxia. Sci. Rep. 2021, 11, 16666. [Google Scholar] [CrossRef]
- Kurihara, T.; Westenskow, P.D.; Gantner, M.L.; Usui, Y.; Schultz, A.; Bravo, S.; Aguilar, E.; Wittgrove, C.; Friedlander, M.; Paris, L.P.; et al. Hypoxia-induced metabolic stress in retinal pigment epithelial cells is sufficient to induce photoreceptor degeneration. eLife 2016, 5, e14319. [Google Scholar] [CrossRef]
- Westerterp, M.; Murphy, A.J.; Wang, M.; Pagler, T.A.; Vengrenyuk, Y.; Kappus, M.S.; Gorman, D.J.; Nagareddy, P.R.; Zhu, X.; Abramowicz, S.; et al. Deficiency of ATP-binding cassette transporters A1 and G1 in macrophages increases inflammation and accelerates atherosclerosis in mice. Circ. Res. 2013, 112, 1456–1465. [Google Scholar] [CrossRef]
- Oram, J. Tangier disease and ABCA1. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2000, 1529, 321–330. [Google Scholar] [CrossRef]
- Winder, A.F.; Alexander, R.; Garner, A.; Johnston, D.; Vallance, D.; McCreanor, G.; Frohlich, J. The pathology of cornea in Tangier disease (familial high density lipoprotein deficiency). J. Clin. Pathol. 1996, 49, 407–410. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Collins, H.L.; Ranalletta, M.; Fuki, I.V.; Billheimer, J.T.; Rothblat, G.H.; Tall, A.R.; Rader, D.J. Macrophage ABCA1 and ABCG1, but not SR-BI, promote macrophage reverse cholesterol transport in vivo. J. Clin. Investig. 2007, 117, 2216–2224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ranalletta, M.; Wang, N.; Han, S.; Yvan-Charvet, L.; Welch, C.; Tall, A.R. Decreased atherosclerosis in low-density lipoprotein receptor knockout mice transplanted with Abcg1−/− bone marrow. Arter. Thromb. Vasc. Biol. 2006, 26, 2308–2315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hallam, D.; Collin, J.; Bojic, S.; Chichagova, V.; Buskin, A.; Xu, Y.; Lafage, L.; Otten, E.G.; Anyfantis, G.; Mellough, C.; et al. An Induced Pluripotent Stem Cell Patient Specific Model of Complement Factor H (Y402H) Polymorphism Displays Characteristic Features of Age-Related Macular Degeneration and Indicates a Beneficial Role for UV Light Exposure. Stem Cells 2017, 35, 2305–2320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebeling, M.C.; Geng, Z.; Kapphahn, R.J.; Roehrich, H.; Montezuma, S.R.; Dutton, J.R.; Ferrington, D.A. Impaired Mitochondrial Function in iPSC-Retinal Pigment Epithelium with the Complement Factor H Polymorphism for Age-Related Macular Degeneration. Cells 2021, 10, 789. [Google Scholar] [CrossRef]
- Yang, J.; Li, Y.; Chan, L.; Tsai, Y.T.; Wu, W.H.; Nguyen, H.V.; Hsu, C.W.; Li, X.; Brown, L.M.; Egli, D.; et al. Validation of genome-wide association study (GWAS)-identified disease risk alleles with patient-specific stem cell lines. Hum. Mol. Genet. 2014, 23, 3445–3455. [Google Scholar] [CrossRef] [Green Version]
- Saini, J.S.; Corneo, B.; Miller, J.D.; Kiehl, T.R.; Wang, Q.; Boles, N.C.; Blenkinsop, T.A.; Stern, J.H.; Temple, S. Nicotinamide Ameliorates Disease Phenotypes in a Human iPSC Model of Age-Related Macular Degeneration. Cell Stem Cell 2017, 20, 635–647.e637. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Wang, M.; Han, Y.; Zhang, R.; Ma, L. ABCA1 rs1883025 polymorphism and risk of age-related macular degeneration. Graefe’s Arch. Clin. Exp. Ophthalmol. 2016, 254, 323–332. [Google Scholar] [CrossRef]
- Liang, B.; Wang, X.; Song, X.; Bai, R.; Yang, H.; Yang, Z.; Xiao, C.; Bian, Y. MicroRNA-20a/b regulates cholesterol efflux through post-transcriptional repression of ATP-binding cassette transporter A1. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2017, 1862, 929–938. [Google Scholar] [CrossRef]
- Lin, S.C.; Wang, C.C.; Wu, M.H.; Yang, S.H.; Li, Y.H.; Tsai, S.J. Hypoxia-induced microRNA-20a expression increases ERK phosphorylation and angiogenic gene expression in endometriotic stromal cells. J. Clin. Endocrinol. Metab. 2012, 97, E1515–E1523. [Google Scholar] [CrossRef] [Green Version]
- Tabas, I. Consequences of cellular cholesterol accumulation: Basic concepts and physiological implications. J. Clin. Investig. 2002, 110, 905–911. [Google Scholar] [CrossRef] [PubMed]
- Chertov, A.O.; Holzhausen, L.; Kuok, I.T.; Couron, D.; Parker, E.; Linton, J.D.; Sadilek, M.; Sweet, I.R.; Hurley, J.B. Roles of glucose in photoreceptor survival. J. Biol. Chem. 2011, 286, 34700–34711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ristau, T.; Ersoy, L.; Lechanteur, Y.; den Hollander, A.I.; Daha, M.R.; Hahn, M.; Hoyng, C.B.; Fauser, S. Allergy is a protective factor against age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2014, 55, 210–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maggi, J.; Koller, S.; Bahr, L.; Feil, S.; Kivrak Pfiffner, F.; Hanson, J.V.M.; Maspoli, A.; Gerth-Kahlert, C.; Berger, W. Long-Range PCR-Based NGS Applications to Diagnose Mendelian Retinal Diseases. Int. J. Mol. Sci. 2021, 22, 1508. [Google Scholar] [CrossRef] [PubMed]
- Regent, F.; Morizur, L.; Lesueur, L.; Habeler, W.; Plancheron, A.; Ben M’Barek, K.; Monville, C. Automation of human pluripotent stem cell differentiation toward retinal pigment epithelial cells for large-scale productions. Sci. Rep. 2019, 9, 10646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reichman, S.; Slembrouck, A.; Gagliardi, G.; Chaffiol, A.; Terray, A.; Nanteau, C.; Potey, A.; Belle, M.; Rabesandratana, O.; Duebel, J.; et al. Generation of Storable Retinal Organoids and Retinal Pigmented Epithelium from Adherent Human iPS Cells in Xeno-Free and Feeder-Free Conditions. Stem Cells 2017, 35, 1176–1188. [Google Scholar] [CrossRef] [Green Version]
- Parinot, C.; Rieu, Q.; Chatagnon, J.; Finnemann, S.C.; Nandrot, E.F. Large-scale purification of porcine or bovine photoreceptor outer segments for phagocytosis assays on retinal pigment epithelial cells. J. Vis. Exp. 2014, 94, e52100. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | ABCA1 rs1883025 (Risk: C) | ABCA1 rs2740488 (Risk: A) | CFH Y402H rs1061170 (Risk: C) | ARMS2 A69S rs10490924 (Risk: T) | C3 R102G rs2230199 (Risk: G) | CFH I62V rs800292 (Protective: A) | CFHR3/1 del rs12144939 (Protective: T) |
|---|---|---|---|---|---|---|---|
| A18945 (Thermo Fisher Scientific) | TC | CA | TT | GT | GG | GA | GG |
| IPS19-00104 (Decreased ABCA1 risk) | TT | CC | TT | GT | GC | GG | GT |
| IPS19-00105 (Decreased ABCA1 risk) | TT | CC | CT | GT | GC | GA | GG |
| IPS19-00106 (Decreased ABCA1 risk) | TT | CC | CT | GT | GC | GG | GG |
| IPS19-00096 (Increased ABCA1 risk) | CC | AA | CT | TT | GC | GG | GT |
| IPS19-00097 (Increased ABCA1 risk) | CC | AA | TT | GG | GC | GA | GG |
| IPS19-00013 (Increased ABCA1 risk) | CC | AA | CC | GG | GG | GG | GG |
| Gene Name | Forward Primer (5′ to 3′) | Reverse Primer (5′ to 3′) |
|---|---|---|
| hABCA1 | GGTCATGGCTGAGGTGAACA | TGGTCATTGTCCCTGCTGTC |
| mAbca1 | GCGTGAAGCCTGTCATCTAC | CATGAGAGGAGTGATCGACC |
| hABCG1 | CTCCTGTTCTCGGGGTTCTT | CCCTTCGAACCCATACCTGAC |
| hACTB | CCTGGCACCCAGCACAAT | GGGCCGGACTCGTCATAC |
| mActb | CAACGGCTCCGGCATGTGC | CTCTTGCTCTGGGCCTCG |
| hADM | ATCACTCTCTTAGCAGGGTCT | CCACTTATTCCACTTCTTTCG |
| hBEST1 | CTTTATGGGCTCCACCTTCA | CAGTAGTTTGGTCCTTGAGTTTG |
| hNR1H3 | TGCCCCATGGACACCTACA | TCTTGCCGCTTCAGTTTCTTC |
| hOTX2 | AGTCGAGGGTGCAGGTATGG | TTTGACCTCCATTCTGCTGTTG |
| hPDK1 | CACGCTGGGTAATGAGGATT | GGAGGTCTCAACACGAGGT |
| hRLBP1 | GCTGCTGGAGAATGAGGAAAC | TGGCTGGTGGATGAAGTGG |
| hRPE65 | TGACAAGGCTGACACAGGCA | CAAAGATGGGTTCTGATGGGTATG |
| hRPL28 | GCAATTCCTTCCGCTACAAC | TGTTCTTGCGGATCATGTGT |
| hSREBP-1C | GGAGGGGTAGGGCCAACG | CATGTCTTCGAAAGTGCAATCC |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peters, F.; Ebner, L.J.A.; Atac, D.; Maggi, J.; Berger, W.; den Hollander, A.I.; Grimm, C. Regulation of ABCA1 by AMD-Associated Genetic Variants and Hypoxia in iPSC-RPE. Int. J. Mol. Sci. 2022, 23, 3194. https://doi.org/10.3390/ijms23063194
Peters F, Ebner LJA, Atac D, Maggi J, Berger W, den Hollander AI, Grimm C. Regulation of ABCA1 by AMD-Associated Genetic Variants and Hypoxia in iPSC-RPE. International Journal of Molecular Sciences. 2022; 23(6):3194. https://doi.org/10.3390/ijms23063194
Chicago/Turabian StylePeters, Florian, Lynn J. A. Ebner, David Atac, Jordi Maggi, Wolfgang Berger, Anneke I. den Hollander, and Christian Grimm. 2022. "Regulation of ABCA1 by AMD-Associated Genetic Variants and Hypoxia in iPSC-RPE" International Journal of Molecular Sciences 23, no. 6: 3194. https://doi.org/10.3390/ijms23063194
APA StylePeters, F., Ebner, L. J. A., Atac, D., Maggi, J., Berger, W., den Hollander, A. I., & Grimm, C. (2022). Regulation of ABCA1 by AMD-Associated Genetic Variants and Hypoxia in iPSC-RPE. International Journal of Molecular Sciences, 23(6), 3194. https://doi.org/10.3390/ijms23063194