
Simultaneous Genetic Ablation of PD-1, LAG-3, and TIM-3 in CD8 T Cells Delays Tumor Growth and Improves Survival Outcome

 ,
,
Abstract
:1. Introduction
2. Results
2.1. CRISPR/Cas9 Gene Editing Induces Reduction in PD-1, LAG-3, and TIM-3 Expression
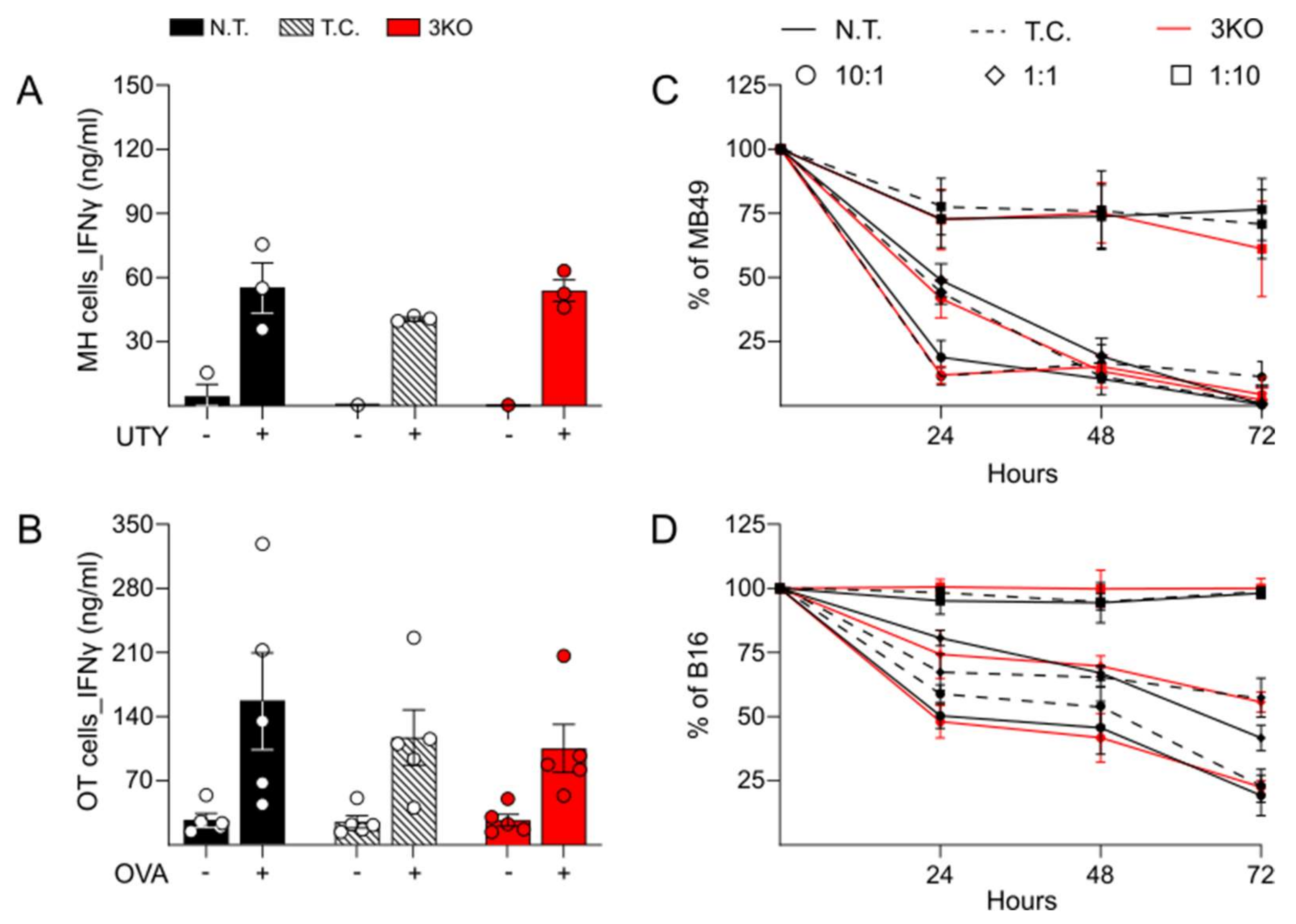
2.2. Comparable Immune Function and Anti-Tumor Activity between Triple CRISPR/Cas9 Genetically Edited CD8+ T Cells and Control
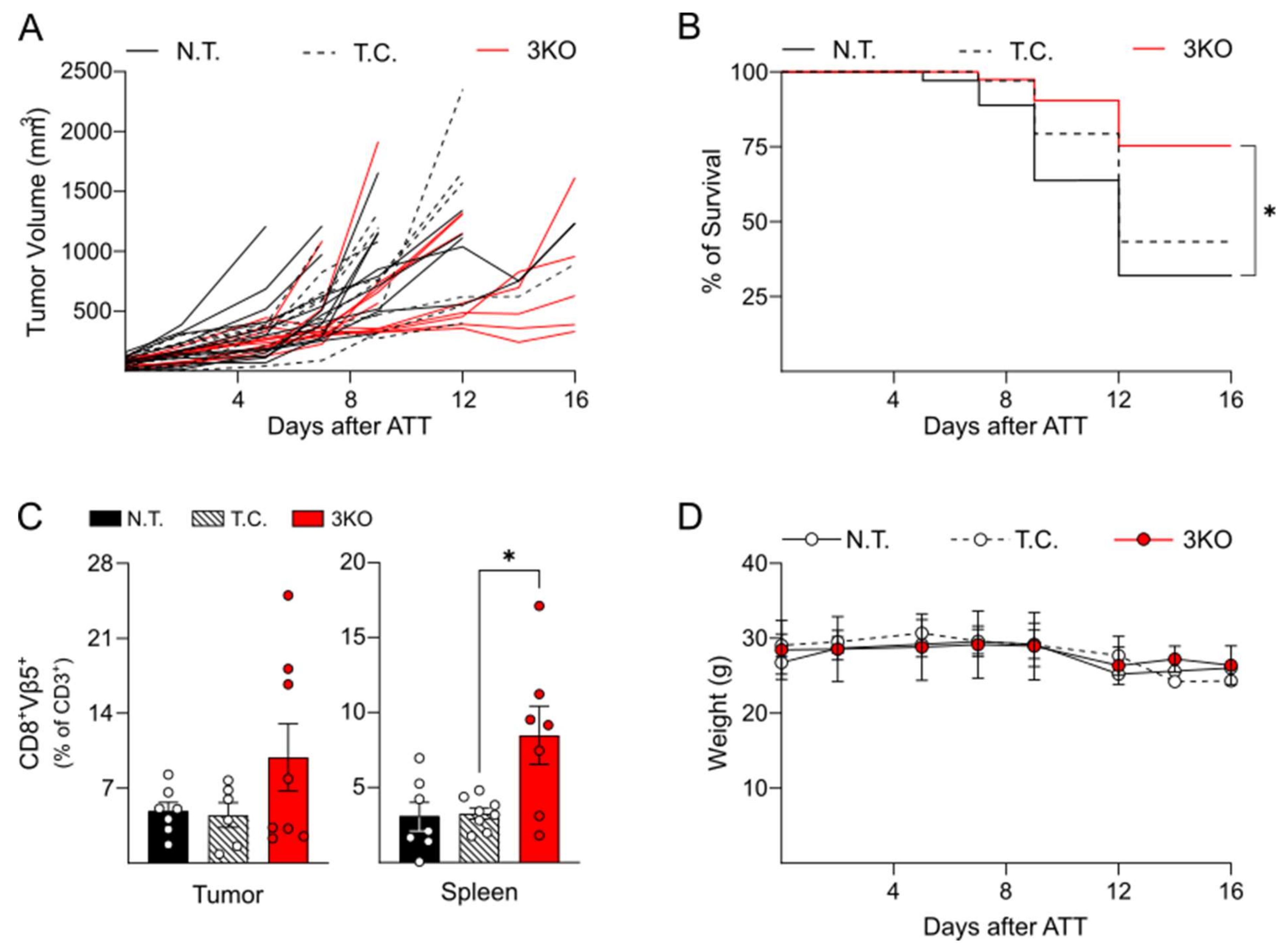
2.3. Triple Editing of PD-1, LAG-3, and TIM-3 Improves the Anti-Tumor Activity of OT-1 CD8+ T Cells In Vivo
3. Discussion
4. Material and Methods
4.1. CD8+ T Cell Isolation
4.2. CRISPR/Cas9 and Nucleofection of EL4, MH and OT-1 CD8+ T Cells
4.3. Long CD3/CD28 and Peptide-Mediated CD8+ T Cell Activation
4.4. Measurement of IFNγ Production
4.5. Killing Assay
4.6. Mice and OVA Tumor Model
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chai, L.F.; Prince, E.; Pillarisetty, V.G.; Katz, S.C. Challenges in assessing solid tumor responses to immunotherapy. Cancer Gene Ther. 2019, 27, 528–538. [Google Scholar] [CrossRef] [PubMed]
- Ai, L.; Chen, J.; Yan, H.; He, Q.; Luo, P.; Xu, Z.; Yang, X. Research status and outlook of PD-1/PD-L1 inhibitors for cancer therapy. Drug Des. Devel. Ther. 2020, 14, 3625–3649. [Google Scholar] [CrossRef] [PubMed]
- Deleuze, A.; Saout, J.; Dugay, F.; Peyronnet, B.; Mathieu, R.; Verhoest, G.; Bensalah, K.; Crouzet, L.; Laguerre, B.; Belaud-Rotureau, M.-A.; et al. Immunotherapy in renal cell carcinoma: The future is now. Int. J. Mol. Sci. 2020, 21, 2532. [Google Scholar] [CrossRef] [Green Version]
- Yap, T.A.; Parkes, E.E.; Peng, W.; Moyers, J.T.; Curran, M.A.; Tawbi, H.A. Development of immunotherapy combination strategies in cancer. Cancer Discov. 2021, 11, 1368–1397. [Google Scholar] [CrossRef] [PubMed]
- Dobosz, P.; Dzieciątkowski, T. The intriguing history of cancer immunotherapy. Front. Immunol. 2019, 10, 2965. [Google Scholar] [CrossRef] [Green Version]
- Twomey, J.D.; Zhang, B. Cancer immunotherapy update: FDA-approved checkpoint inhibitors and companion diagnostics. AAPS J. 2021, 23, 39. [Google Scholar] [CrossRef]
- Ferris, R.L.; Blumenschein, G., Jr.; Fayette, J.; Guigay, J.; Colevas, A.D.; Licitra, L.; Harrington, K.; Kasper, S.; Vokes, E.E.; Even, C.; et al. Nivolumab for recurrent squamous-cell carcinoma of the head and neck. N. Engl. J. Med. 2016, 375, 1856–1867. [Google Scholar] [CrossRef] [Green Version]
- Garon, E.B.; Rizvi, N.A.; Hui, R.; Leighl, N.; Balmanoukian, A.S.; Eder, J.P.; Patnaik, A.; Aggarwal, C.; Gubens, M.; Horn, L.; et al. Pembrolizumab for the treatment of non–small-cell lung cancer. N. Engl. J. Med. 2015, 372, 2018–2028. [Google Scholar] [CrossRef]
- Motzer, R.J.; Escudier, B.; McDermott, D.F.; George, S.; Hammers, H.J.; Srinivas, S.; Tykodi, S.S.; Sosman, J.A.; Procopio, G.; Plimack, E.R.; et al. Nivolumab versus everolimus in advanced renal-cell carcinoma. N. Engl. J. Med. 2015, 373, 1803–1813. [Google Scholar] [CrossRef]
- Rosenberg, J.E.; Hoffman-Censits, J.; Powles, T.; van der Heijden, M.S.; Balar, A.V.; Necchi, A.; Dawson, N.; O’Donnell, P.H.; Balmanoukian, A.; Loriot, Y.; et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: A single-arm, multicentre, phase 2 trial. Lancet 2016, 387, 1909–1920. [Google Scholar] [CrossRef] [Green Version]
- El-Khoueiry, A.B.; Sangro, B.; Yau, T.; Crocenzi, T.S.; Kudo, M.; Hsu, C.; Kim, T.-Y.; Choo, S.-P.; Trojan, J.; Welling, T.H., 3rd; et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): An open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet 2017, 389, 2492–2502. [Google Scholar] [CrossRef]
- Barrueto, L.; Caminero, F.; Cash, L.; Makris, C.; Lamichhane, P.; Deshmukh, R.R. Resistance to checkpoint inhibition in cancer immunotherapy. Transl. Oncol. 2020, 13, 100738. [Google Scholar] [CrossRef] [PubMed]
- Barbari, C.; Fontaine, T.; Parajuli, P.; Lamichhane, N.; Jakubski, S.; Lamichhane, P.; Deshmukh, R.R. Immunotherapies and combination strategies for immuno-oncology. Int. J. Mol. Sci. 2020, 21, 5009. [Google Scholar] [CrossRef] [PubMed]
- Shayan, G.; Srivastava, R.; Li, J.; Schmitt, N.; Kane, L.P.; Ferris, R.L. Adaptive resistance to anti-PD1 therapy by Tim-3 up-regulation is mediated by the PI3K-Akt pathway in head and neck cancer. Oncoimmunology 2017, 6, e1261779. [Google Scholar] [CrossRef] [PubMed]
- Anderson, A.C.; Anderson, D.E.; Bregoli, L.; Hastings, W.D.; Kassam, N.; Lei, C.; Chandwaskar, R.; Karman, J.; Su, E.W.; Hirashima, M.; et al. Promotion of tissue inflammation by the immune receptor tim-3 expressed on innate immune cells. Science 2007, 318, 1141–1143. [Google Scholar] [CrossRef]
- Du, W.; Yang, M.; Turner, A.; Xu, C.; Ferris, R.L.; Huang, J.; Kane, L.P.; Lu, B. TIM-3 as a target for cancer immunotherapy and mechanisms of action. Int. J. Mol. Sci. 2017, 18, 645. [Google Scholar] [CrossRef]
- Hafler, D.A.; Kuchroo, V. TIMs: Central regulators of immune responses. J. Exp. Med. 2008, 205, 2699–2701. [Google Scholar] [CrossRef] [Green Version]
- Fourcade, J.; Sun, Z.; Benallaoua, M.; Guillaume, P.; Luescher, I.F.; Sander, C.; Kirkwood, J.M.; Kuchroo, V.; Zarour, H.M. Upregulation of Tim-3 and PD-1 expression is associated with tumor antigen–specific CD8+ T cell dysfunction in melanoma patients. J. Exp. Med. 2010, 207, 2175–2186. [Google Scholar] [CrossRef]
- Sakuishi, K.; Apetoh, L.; Sullivan, J.M.; Blazar, B.R.; Kuchroo, V.K.; Anderson, A.C. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J. Exp. Med. 2010, 207, 2187–2194, Correction in J. Exp. Med. 2011, 208, 1331. [Google Scholar] [CrossRef]
- Kim, J.E.; Patel, M.; Mangraviti, A.; Kim, E.S.; Theodros, D.; Velarde, E.; Liu, A.; Sankey, E.W.; Tam, A.; Xu, H.; et al. Combination therapy with anti-PD-1, anti-TIM-3, and focal radiation results in regression of murine gliomas. Clin. Cancer Res. 2016, 23, 124–136. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Du, Q.; Jin, J.; Wei, Y.; Lu, Y.; Li, Q. LAG3 and its emerging role in cancer immunotherapy. Clin. Transl. Med. 2021, 11, e365. [Google Scholar] [CrossRef] [PubMed]
- Woo, S.-R.; Turnis, M.E.; Goldberg, M.V.; Bankoti, J.; Selby, M.; Nirschl, C.J.; Bettini, M.L.; Gravano, D.M.; Vogel, P.; Liu, C.L.; et al. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate t-cell function to promote tumoral immune escape. Cancer Res. 2012, 72, 917–927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuzaki, J.; Gnjatic, S.; Mhawech-Fauceglia, P.; Beck, A.; Miller, A.; Tsuji, T.; Eppolito, C.; Qian, F.; Lele, S.; Shrikant, P.; et al. Tumor-infiltrating NY-ESO-1–specific CD8 + T cells are negatively regulated by LAG-3 and PD-1 in human ovarian cancer. Proc. Natl. Acad. Sci. USA 2010, 107, 7875–7880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oki, E.; Makiyama, A.; Miyamoto, Y.; Kotaka, M.; Kawanaka, H.; Miwa, K.; Kabashima, A.; Noguchi, T.; Yuge, K.; Kashiwada, T.; et al. Trifluridine/tipiracil plus bevacizumab as a first-line treatment for elderly patients with metastatic colorectal cancer (KSCC1602): A multicenter phase II trial. Cancer Med. 2020, 10, 454–461. [Google Scholar] [CrossRef] [PubMed]
- Ascierto, P.A.; Bono, P.; Bhatia, S.; Melero, I.; Nyakas, M.S.; Svane, I.-M.; Larkin, J.; Gomez-Roca, C.; Schadendorf, D.; Dummer, R.; et al. Efficacy of BMS-986016, a monoclonal antibody that targets lymphocyte activation gene-3 (LAG-3), in combination with nivolumab in pts with melanoma who progressed during prior anti–PD-1/PD-L1 therapy (mel prior IO) in all-comer and bi-omarker-enriched populations. Ann. Oncol. 2017, 28, V611–V612. [Google Scholar]
- Hong, D.S.; Schoffski, P.; Calvo, A.; Sarantopoulos, J.; De Olza, M.O.; Carvajal, R.D.; Prawira, A.; Kyi, C.; Esaki, T.; Akerley, W.L.; et al. Phase I/II study of LAG525 ± spartalizumab (PDR001) in patients (pts) with advanced malignancies. J. Clin. Oncol. 2018, 36, 3012. [Google Scholar] [CrossRef]
- Lecocq, Q.; Keyaerts, M.; Devoogdt, N.; Breckpot, K. The next-generation immune checkpoint LAG-3 and its therapeutic potential in oncology: Third time’s a charm. Int. J. Mol. Sci. 2020, 22, 75. [Google Scholar] [CrossRef]
- Ruffo, E.; Wu, R.C.; Bruno, T.C.; Workman, C.J.; Vignali, D.A. Lymphocyte-activation gene 3 (LAG3): The next immune checkpoint receptor. Semin. Immunol. 2019, 42, 101305. [Google Scholar] [CrossRef]
- Wolf, Y.; Anderson, A.C.; Kuchroo, V.K. TIM3 comes of age as an inhibitory receptor. Nat. Rev. Immunol. 2019, 20, 173–185. [Google Scholar] [CrossRef]
- Kennedy, L.B.; Salama, A. A review of cancer immunotherapy toxicity. CA A Cancer J. Clin. 2020, 70, 86–104. [Google Scholar] [CrossRef] [Green Version]
- Schnell, A.; Bod, L.; Madi, A.; Kuchroo, V.K. The yin and yang of co-inhibitory receptors: Toward anti-tumor immunity without autoimmunity. Cell Res. 2020, 30, 285–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okazaki, T.; Okazaki, I.-M.; Wang, J.; Sugiura, D.; Nakaki, F.; Yoshida, T.; Kato, Y.; Fagarasan, S.; Muramatsu, M.; Eto, T.; et al. PD-1 and LAG-3 inhibitory co-receptors act synergistically to prevent autoimmunity in mice. J. Exp. Med. 2011, 208, 395–407. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Zhang, X.; Cheng, C.; Mu, W.; Liu, X.; Li, N.; Wei, X.; Liu, X.; Xia, C.; Wang, H. CRISPR-Cas9 mediated LAG-3 disruption in CAR-T cells. Front. Med. 2017, 11, 554–562. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Li, Y.; Shao, Y.; Zhang, Y. Gene modification strategies for next-generation CAR T cells against solid cancers. J. Hematol. Oncol. 2020, 13, 54. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Liu, X.; Fang, C.; Jiang, S.; June, C.H.; Zhao, Y. Multiplex genome editing to generate universal CAR T cells resistant to PD1 inhibition. Clin. Cancer Res. 2016, 23, 2255–2266. [Google Scholar] [CrossRef] [Green Version]
- McGowan, E.; Lin, Q.; Ma, G.; Yin, H.; Chen, S.; Lin, Y. PD-1 disrupted CAR-T cells in the treatment of solid tumors: Promises and challenges. Biomed. Pharmacother. 2019, 121, 109625. [Google Scholar] [CrossRef]
- Zou, F.; Lu, L.; Liu, J.; Xia, B.; Zhang, W.; Hu, Q.; Liu, W.; Zhang, Y.; Lin, Y.; Jing, S.; et al. Engineered triple inhibitory receptor resistance improves anti-tumor CAR-T cell performance via CD56. Nat. Commun. 2019, 10, 4109. [Google Scholar] [CrossRef]
- Valujskikh, A.; Lantz, O.; Celli, S.; Matzinger, P.; Heeger, P.S. Cross-primed CD8+ T cells mediate graft rejection via a distinct effector pathway. Nat. Immunol. 2002, 3, 844–851. [Google Scholar] [CrossRef]
- Hogquist, K.A.; Jameson, S.C.; Heath, W.R.; Howard, J.L.; Bevan, M.J.; Carbone, F.R. T cell receptor antagonist peptides induce positive selection. Cell 1994, 76, 17–27. [Google Scholar] [CrossRef]
- Herda, S.; Heimann, A.; Obermayer, B.; Ciraolo, E.; Althoff, S.; Russ, J.; Grunert, C.; Busse, A.; Bullinger, L.; Pezzutto, A.; et al. Long-term in vitro expansion ensures increased yield of central memory T cells as per-spective for manufacturing challenges. Int. J. Cancer 2021, 148, 3097–3110. [Google Scholar] [CrossRef]
- Wherry, E.J.; Kurachi, M. Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 2015, 15, 486–499. [Google Scholar] [CrossRef] [PubMed]
- Roth, T.L.; Puig-Saus, C.; Yu, R.; Shifrut, E.; Carnevale, J.; Li, P.J.; Hiatt, J.; Saco, J.; Krystofinski, P.; Li, H.; et al. Reprogramming human T cell function and specificity with non-viral genome targeting. Nature 2018, 559, 405–409. [Google Scholar] [CrossRef] [PubMed]
- Mehravar, M.; Shirazi, A.; Nazari, M.; Banan, M. Mosaicism in CRISPR/Cas9-mediated genome editing. Dev. Biol. 2018, 445, 156–162. [Google Scholar] [CrossRef] [PubMed]
- Brinkman, E.K.; Chen, T.; Amendola, M.; Van Steensel, B. Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res. 2014, 42, e168. [Google Scholar] [CrossRef] [PubMed]
- Yen, S.-T.; Zhang, M.; Deng, J.M.; Usman, S.J.; Smith, C.N.; Parker-Thornburg, J.; Swinton, P.G.; Martin, J.F.; Behringer, R.R. Somatic mosaicism and allele complexity induced by CRISPR/Cas9 RNA injections in mouse zygotes. Dev. Biol. 2014, 393, 3–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, R.-Y.; Eppolito, C.; Lele, S.; Shrikant, P.; Matsuzaki, J.; Odunsi, K. LAG3 and PD1 co-inhibitory molecules collaborate to limit CD8+ T cell signaling and dampen antitumor immunity in a murine ovarian cancer model. Oncotarget 2015, 6, 27359–27377. [Google Scholar] [CrossRef]
- Eyles, J.E.; Vessillier, S.; Jones, A.; Stacey, G.; Schneider, C.K.; Price, J. Cell therapy products: Focus on issues with manufacturing and quality control of chimeric antigen receptor T-cell therapies. J. Chem. Technol. Biotechnol. 2018, 94, 1008–1016. [Google Scholar] [CrossRef]
- Hollyman, D.; Stefanski, J.; Przybylowski, M.; Bartido, S.; Borquez-Ojeda, O.; Taylor, C.; Yeh, R.; Capacio, V.; Olszewska, M.; Hosey, J.; et al. Manufacturing validation of biologically functional t cells targeted to CD19 antigen for autologous adoptive cell therapy. J. Immunother. 2009, 32, 169–180. [Google Scholar] [CrossRef] [Green Version]
- Ngiow, S.F.; von Scheidt, B.; Akiba, H.; Yagita, H.; Teng, M.W.; Smyth, M.J. Anti-TIM3 antibody promotes T cell IFN-gamma-mediated antitumor immunity and suppresses established tumors. Cancer Res. 2011, 71, 3540–3551. [Google Scholar] [CrossRef] [Green Version]
- Van Hooren, L.; Sandin, L.C.; Moskalev, I.; Ellmark, P.; Dimberg, A.; Black, P.; Tötterman, T.H.; Mangsbo, S.M. Local checkpoint inhibition of CTLA-4 as a monotherapy or in combination with anti-PD1 prevents the growth of murine bladder cancer. Eur. J. Immunol. 2016, 47, 385–393. [Google Scholar] [CrossRef]
- Wei, S.C.; Duffy, C.R.; Allison, J.P. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov. 2018, 8, 1069–1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faustino-Rocha, A.; Oliveira, P.A.; Pinho-Oliveira, J.; Teixeira-Guedes, C.; Soares-Maia, R.; da Costa, R.G.; Colaco, B.; Pires, M.J.; Colaco, J.; Ferreira, R.; et al. Estimation of rat mammary tumor volume using caliper and ultrasonography measurements. Lab. Anim. 2013, 42, 217–224. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Gene | cRNA Name | RNA Sequence | PAM | Binding | CDF | Off-Targets |
|---|---|---|---|---|---|---|
| Pdcd1 | PD1_crRNA1 | ACAGCCCAAGTGAATGACCA | GGG | Exon 1 | 87 | 0-0-3-18-140 |
| Havcr2 | TIM3_crRNA2 | ATGTGACTCTGGATGACCAT | GGG | Exon 2 | 80 | 0-0-1-14-133 |
| Lag-3 | LAG3_crRNA3 | ACCCGCACCCGGTCGCTACA | CGG | Exon 3 | 98 | 0-0-0-1-16 |
| - | trRNA | AGCAUAGCAAGUUAAAAUAAGGCUAGUCCGUUAUCAACUUGAAAAAGUGGCACCGAGUCGGUGCUUU | - | - | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ciraolo, E.; Althoff, S.; Ruß, J.; Rosnev, S.; Butze, M.; Pühl, M.; Frentsch, M.; Bullinger, L.; Na, I.-K. Simultaneous Genetic Ablation of PD-1, LAG-3, and TIM-3 in CD8 T Cells Delays Tumor Growth and Improves Survival Outcome. Int. J. Mol. Sci. 2022, 23, 3207. https://doi.org/10.3390/ijms23063207
Ciraolo E, Althoff S, Ruß J, Rosnev S, Butze M, Pühl M, Frentsch M, Bullinger L, Na I-K. Simultaneous Genetic Ablation of PD-1, LAG-3, and TIM-3 in CD8 T Cells Delays Tumor Growth and Improves Survival Outcome. International Journal of Molecular Sciences. 2022; 23(6):3207. https://doi.org/10.3390/ijms23063207
Chicago/Turabian StyleCiraolo, Elisa, Stefanie Althoff, Josefine Ruß, Stanislav Rosnev, Monique Butze, Miriam Pühl, Marco Frentsch, Lars Bullinger, and Il-Kang Na. 2022. "Simultaneous Genetic Ablation of PD-1, LAG-3, and TIM-3 in CD8 T Cells Delays Tumor Growth and Improves Survival Outcome" International Journal of Molecular Sciences 23, no. 6: 3207. https://doi.org/10.3390/ijms23063207
APA StyleCiraolo, E., Althoff, S., Ruß, J., Rosnev, S., Butze, M., Pühl, M., Frentsch, M., Bullinger, L., & Na, I. -K. (2022). Simultaneous Genetic Ablation of PD-1, LAG-3, and TIM-3 in CD8 T Cells Delays Tumor Growth and Improves Survival Outcome. International Journal of Molecular Sciences, 23(6), 3207. https://doi.org/10.3390/ijms23063207