
Full-Length Transcriptome and RNA-Seq Analyses Reveal the Mechanisms Underlying Waterlogging Tolerance in Kiwifruit (Actinidia valvata)

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Overview of Sequencing Data
2.2. Gene Annotation and Functional Classification
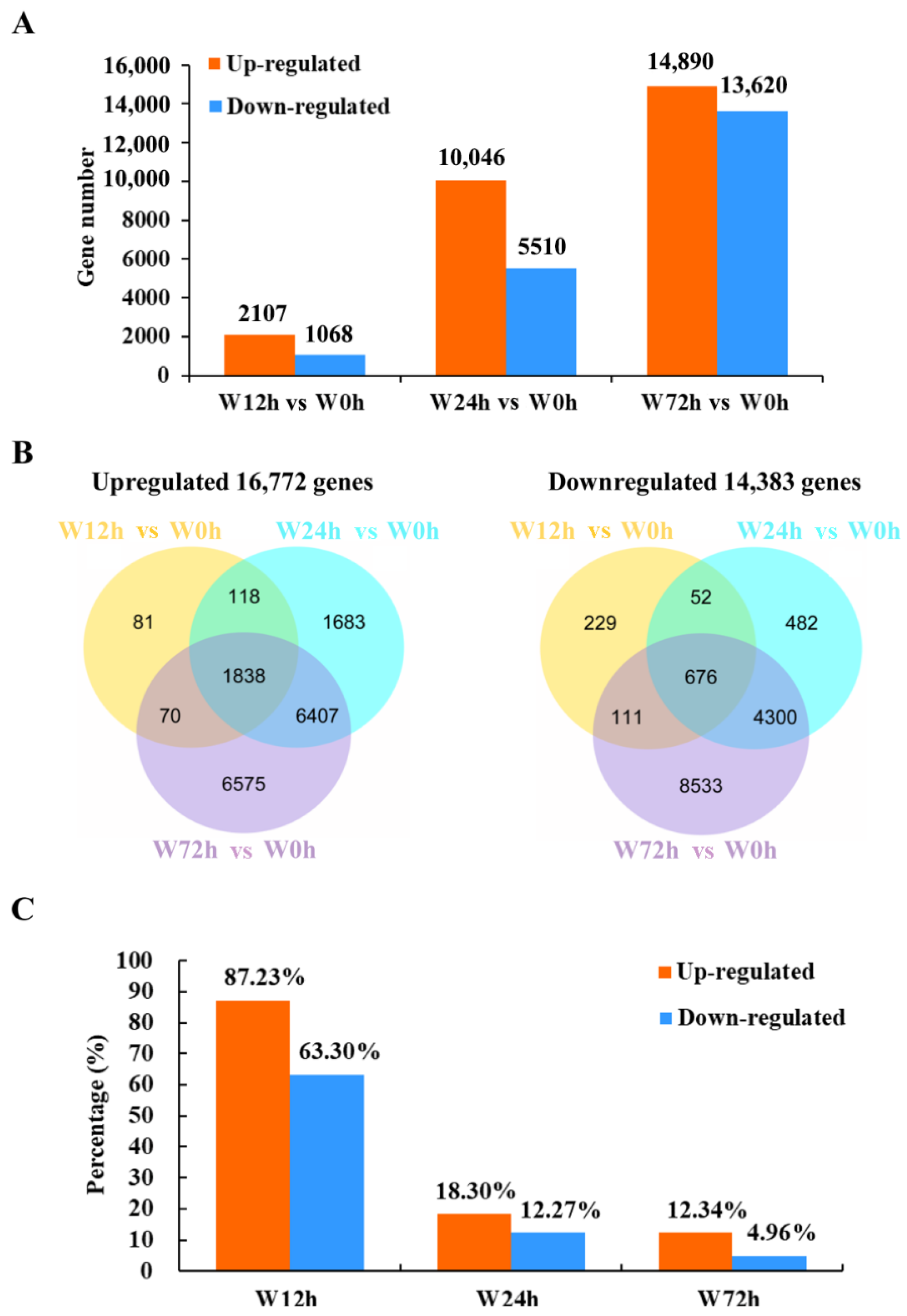
2.3. Statistical Summary of DEGs
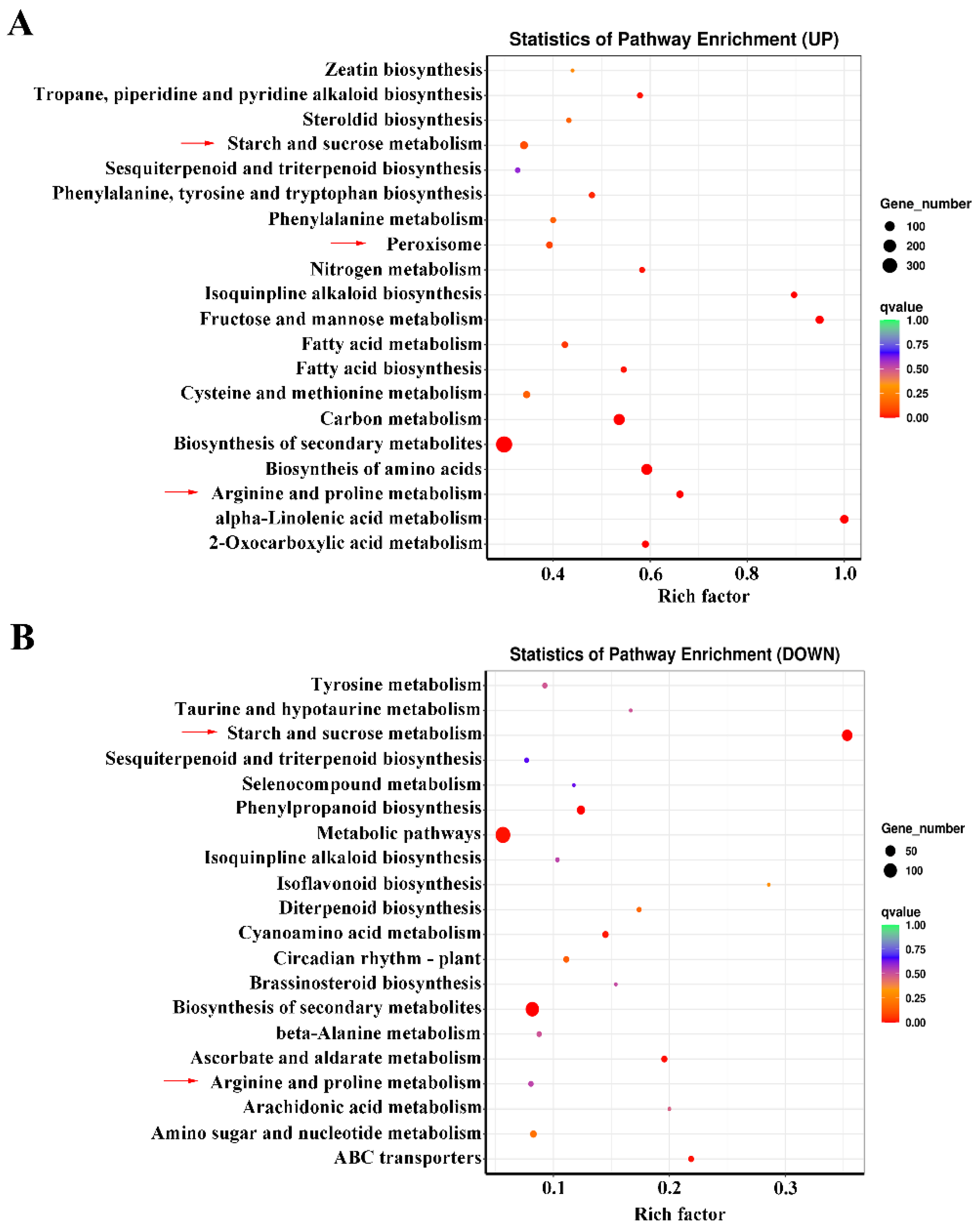
2.4. KEGG Analysis of Co-Expressed DEGs
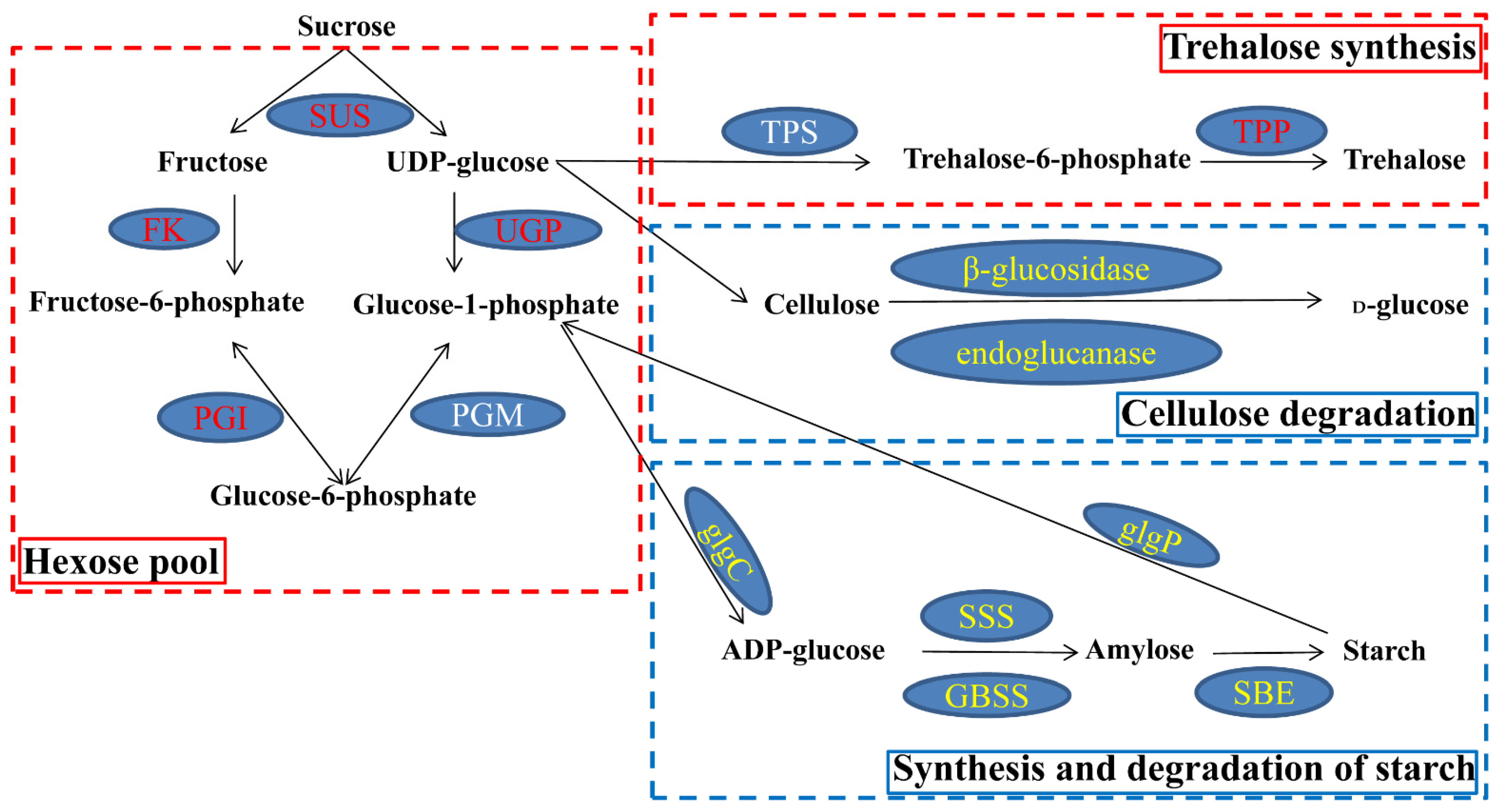
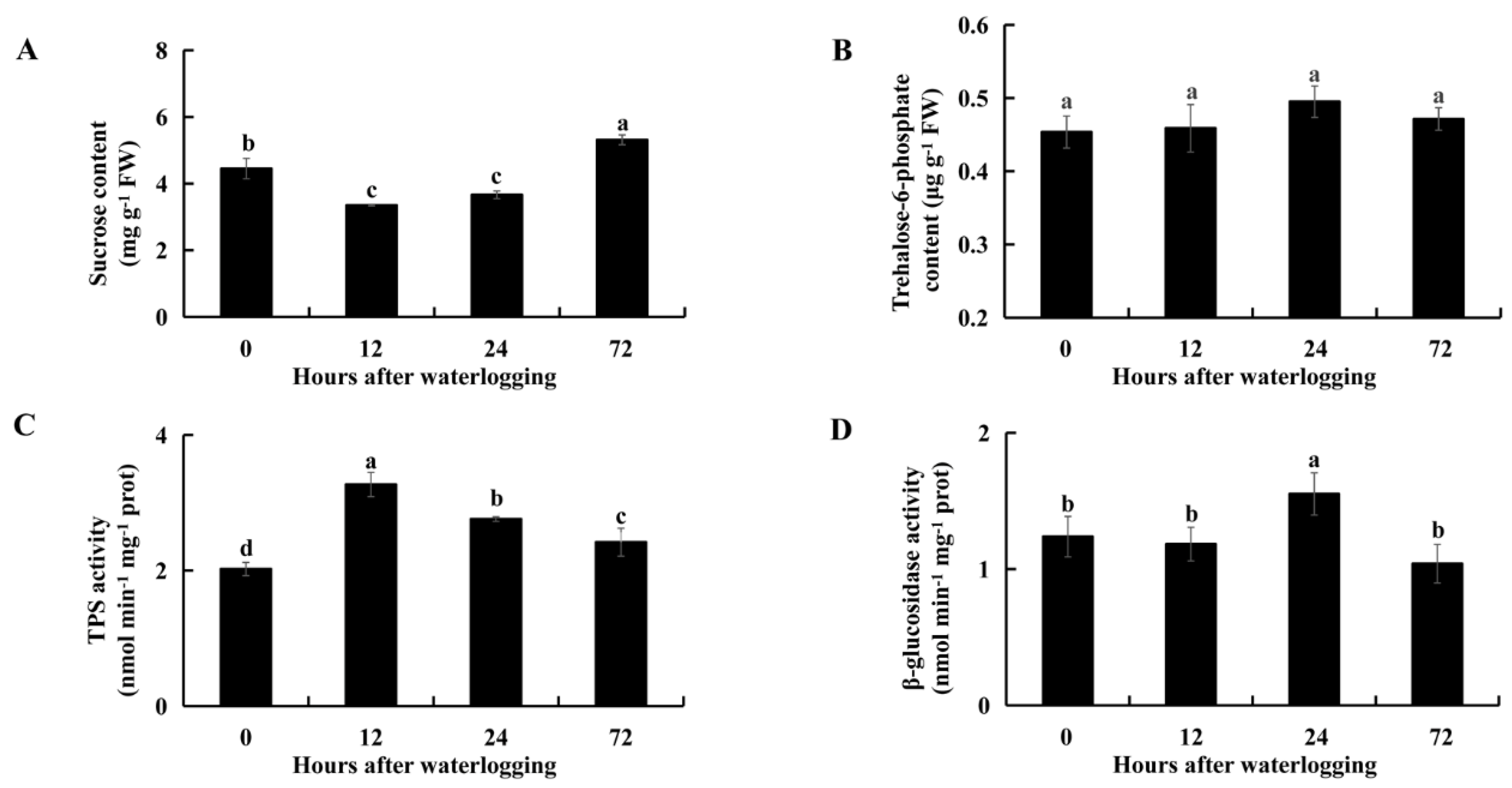
2.5. Carbohydrate Metabolism under Waterlogging Stress
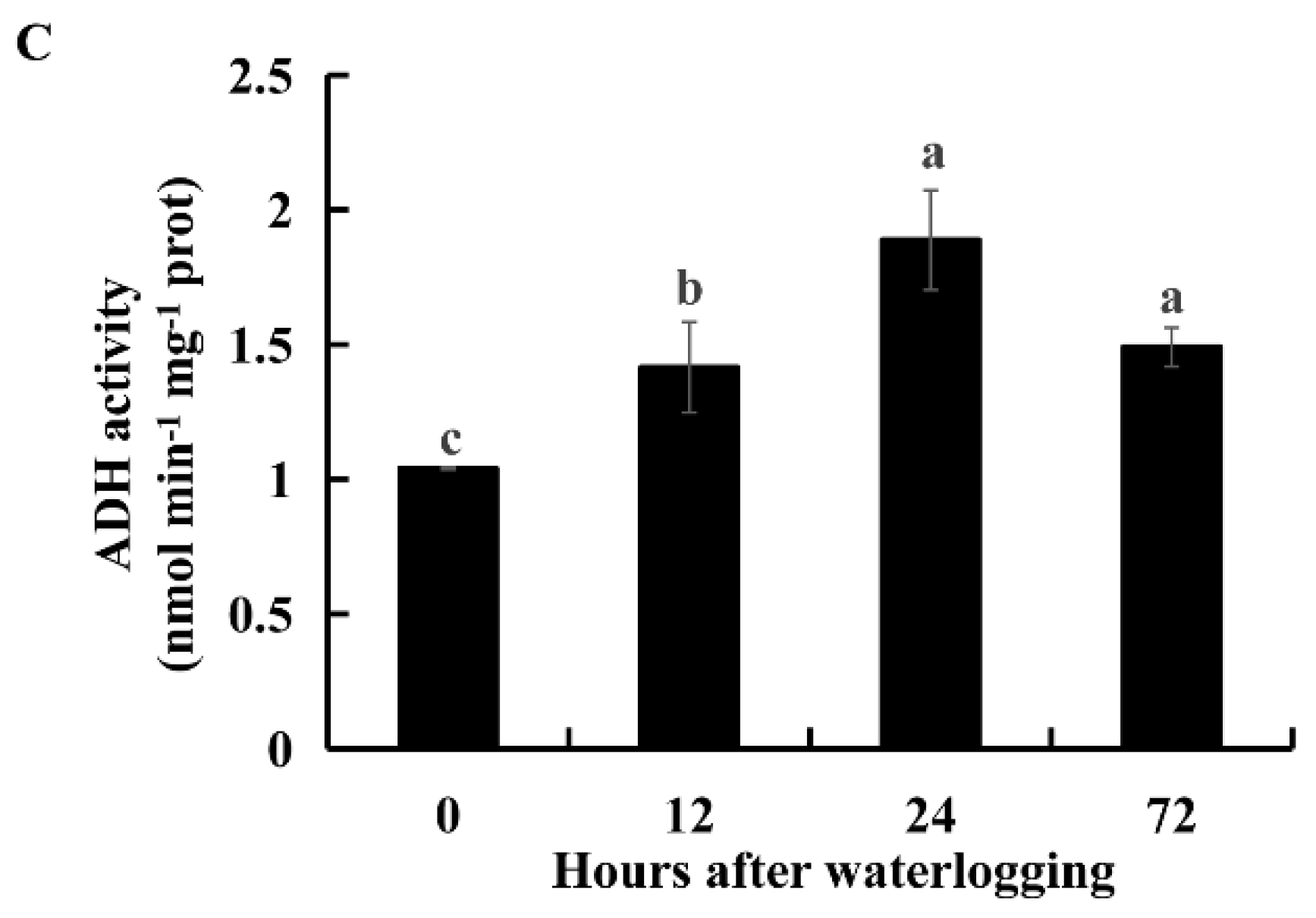
2.6. Ethanolic Fermentation under Waterlogging Stress
2.7. Free Amino Acid Metabolism under Waterlogging Stress
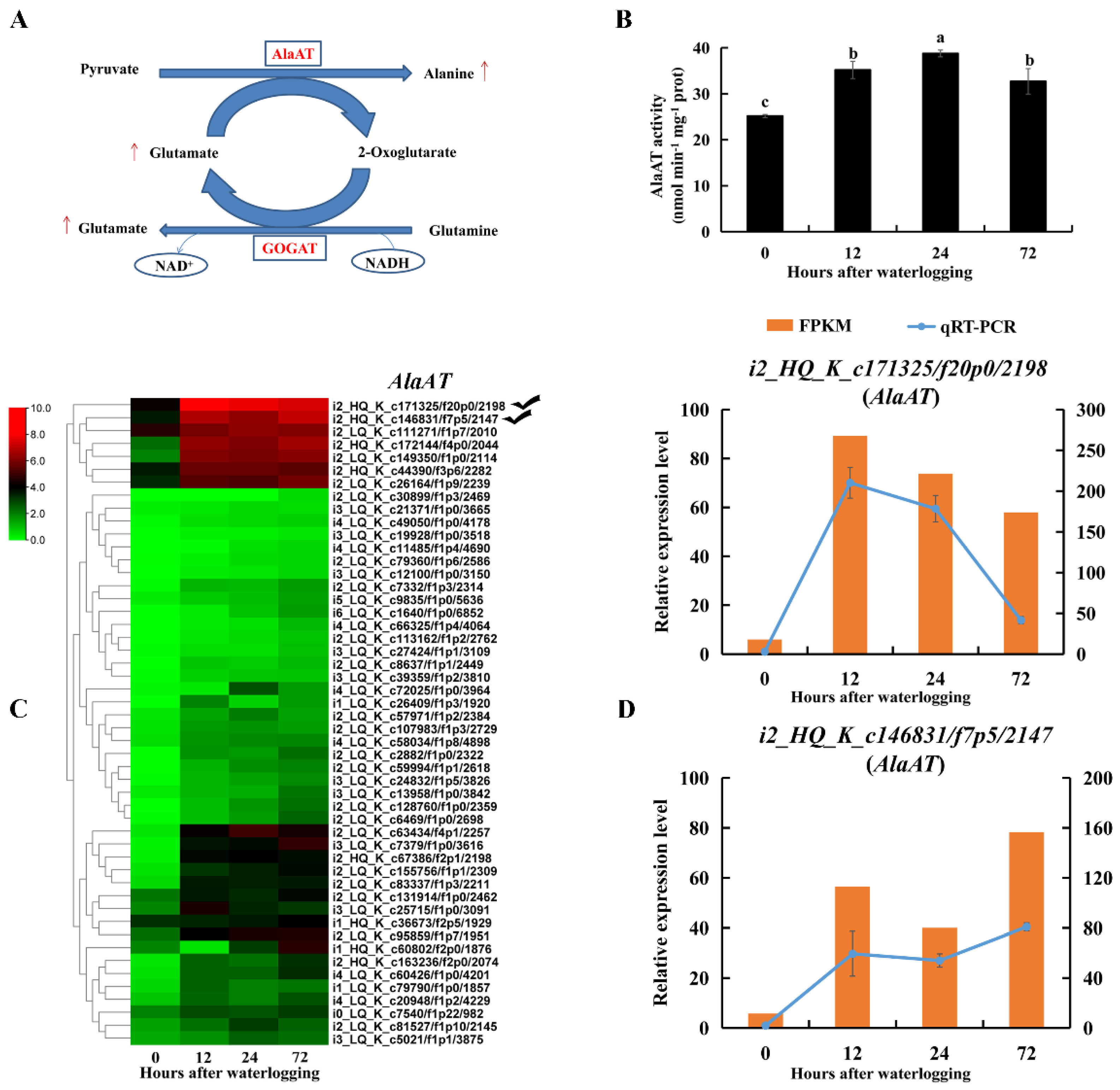
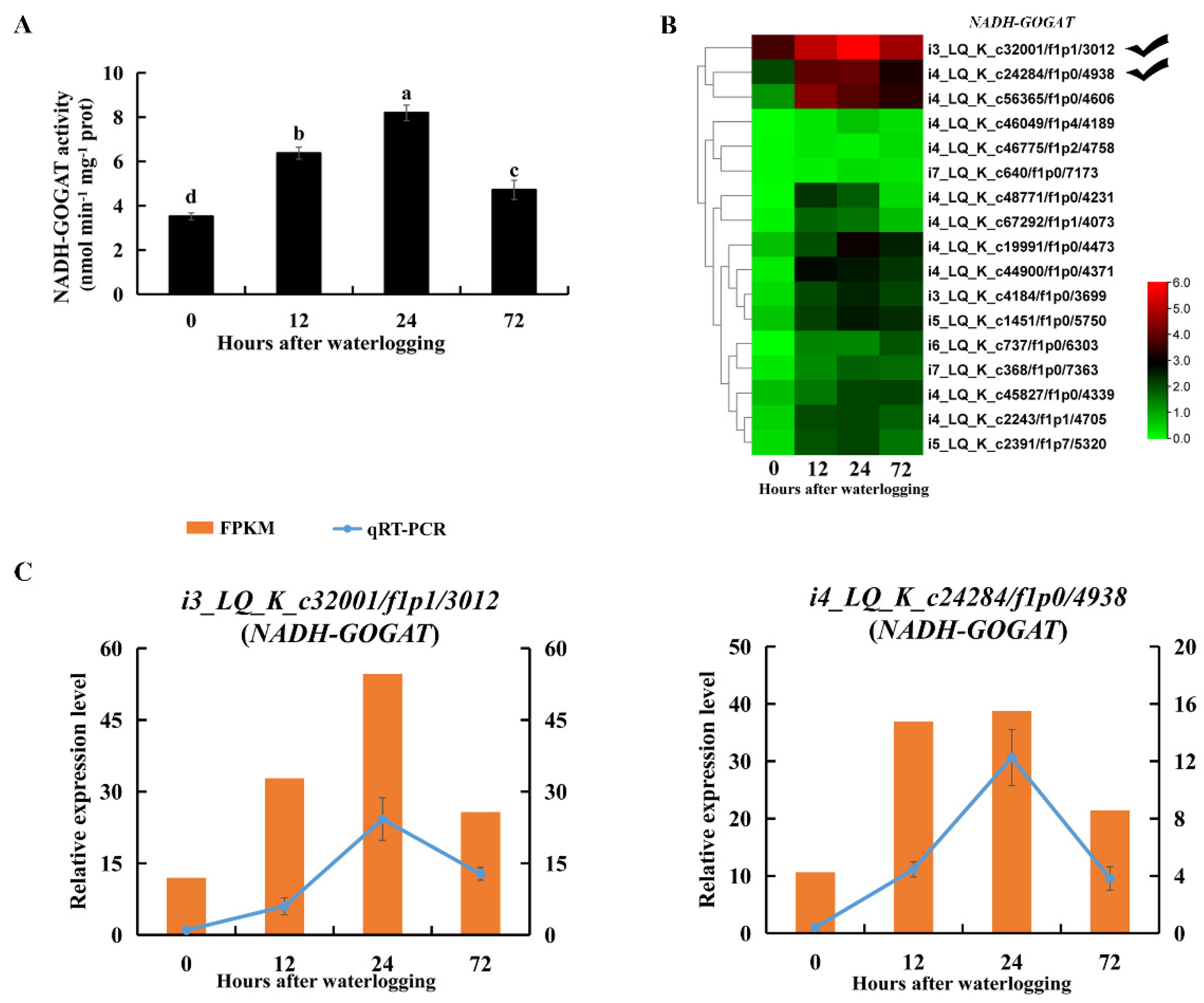
2.8. Enzymes and DEGs Related to Alanine and Glutamate Metabolism
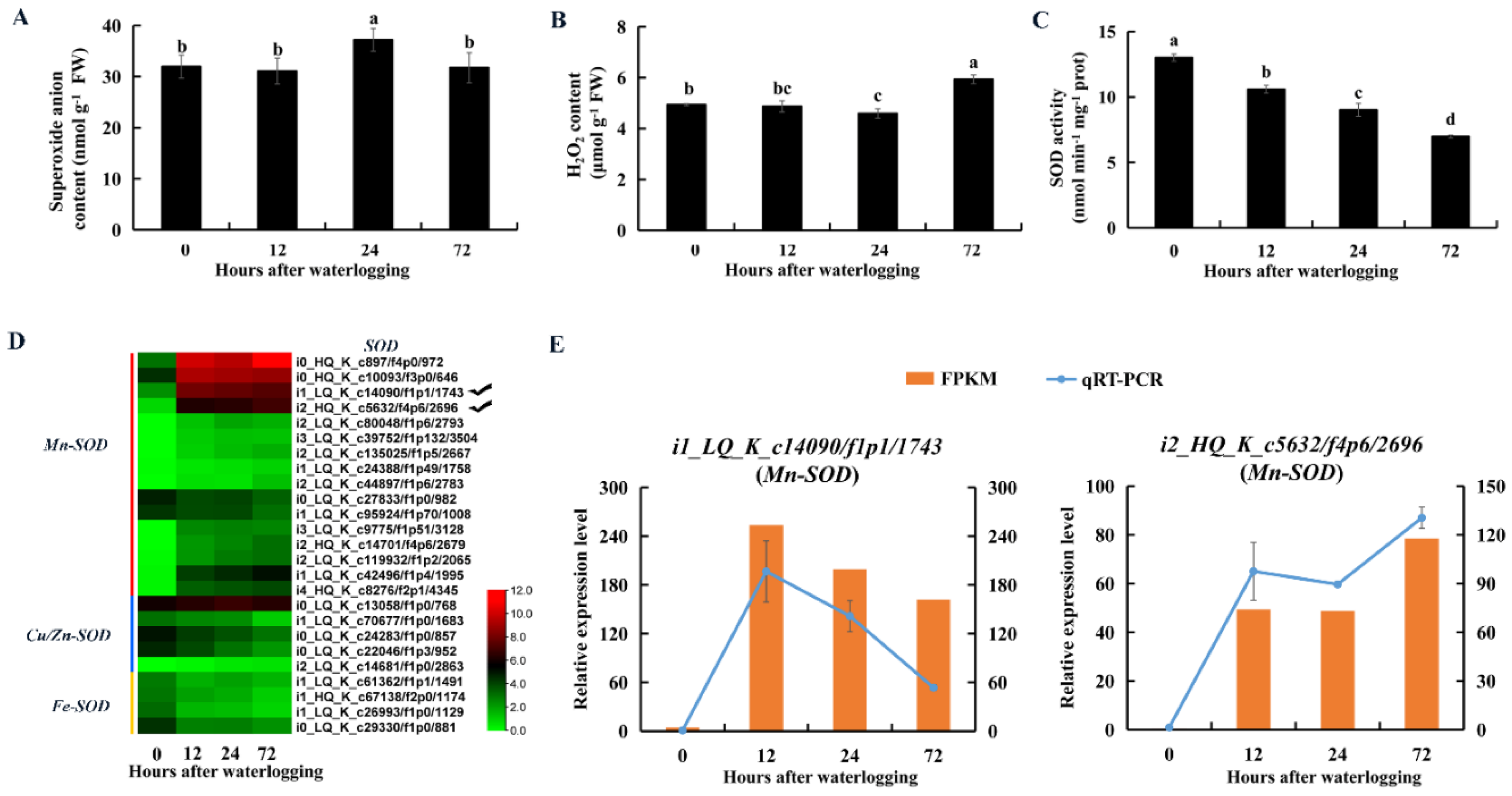
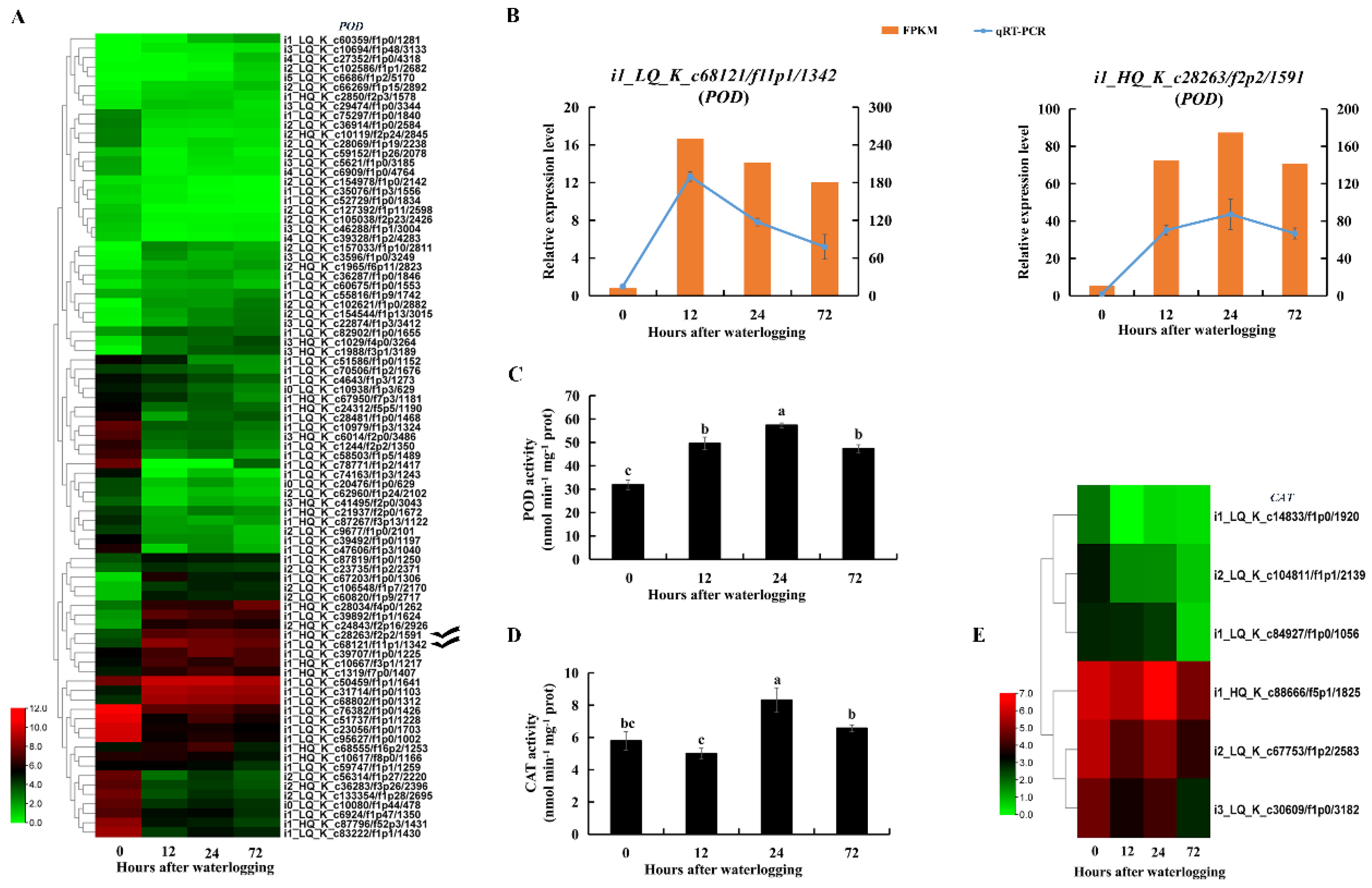
2.9. Enzymes and DEGs Related to ROS Scavenging
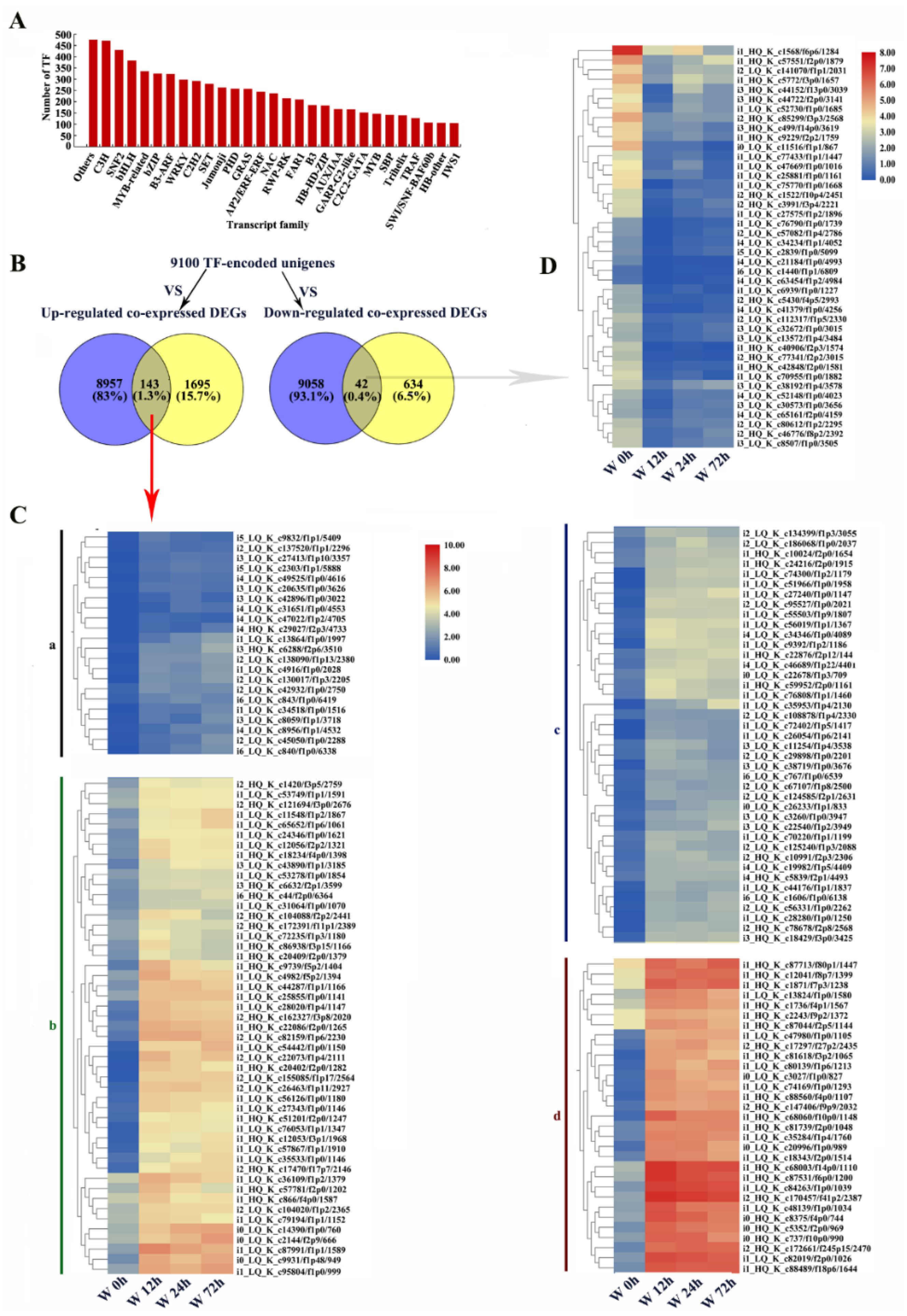
2.10. Identification of Unigenes or DEGs Encoding TFs
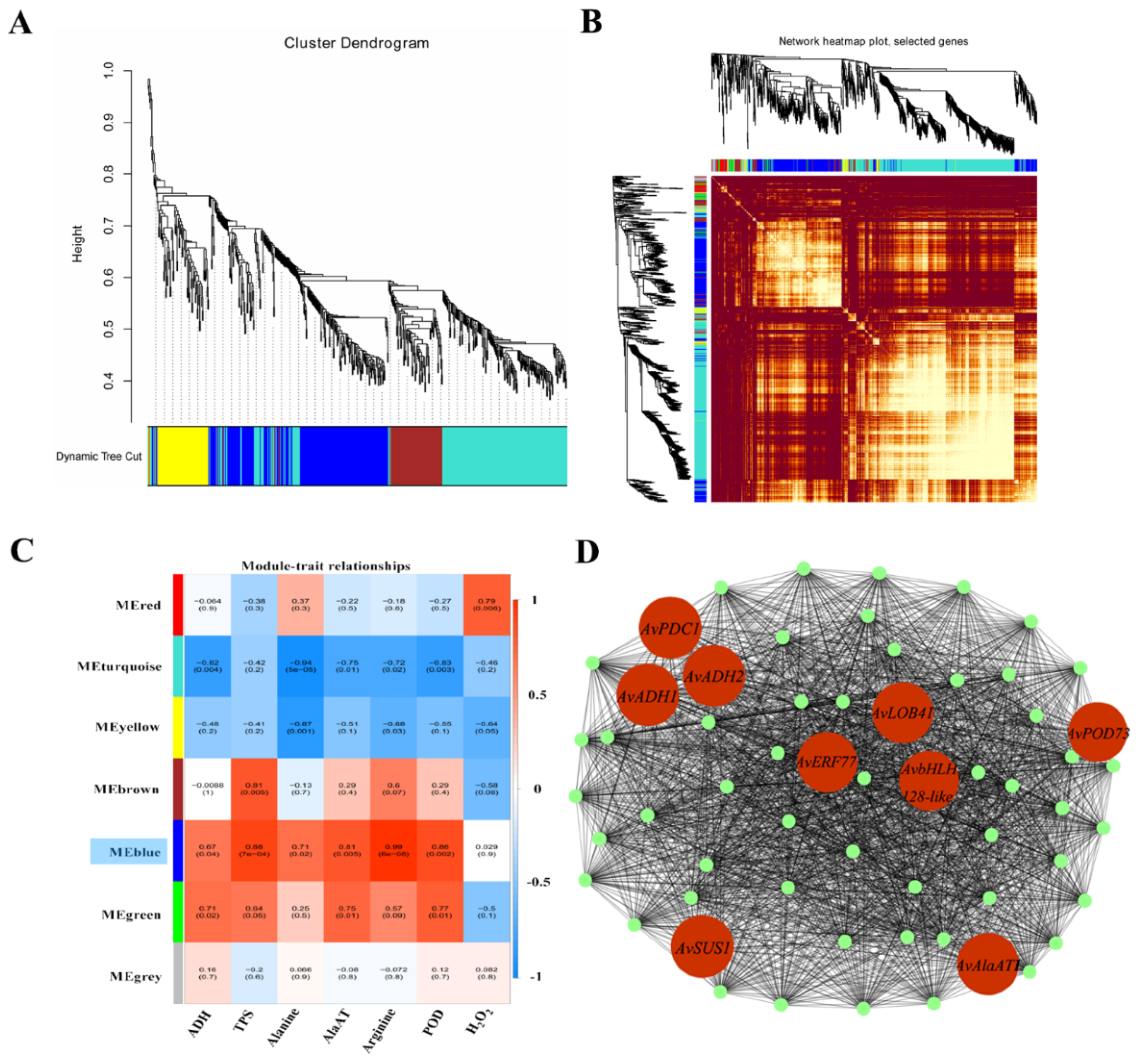
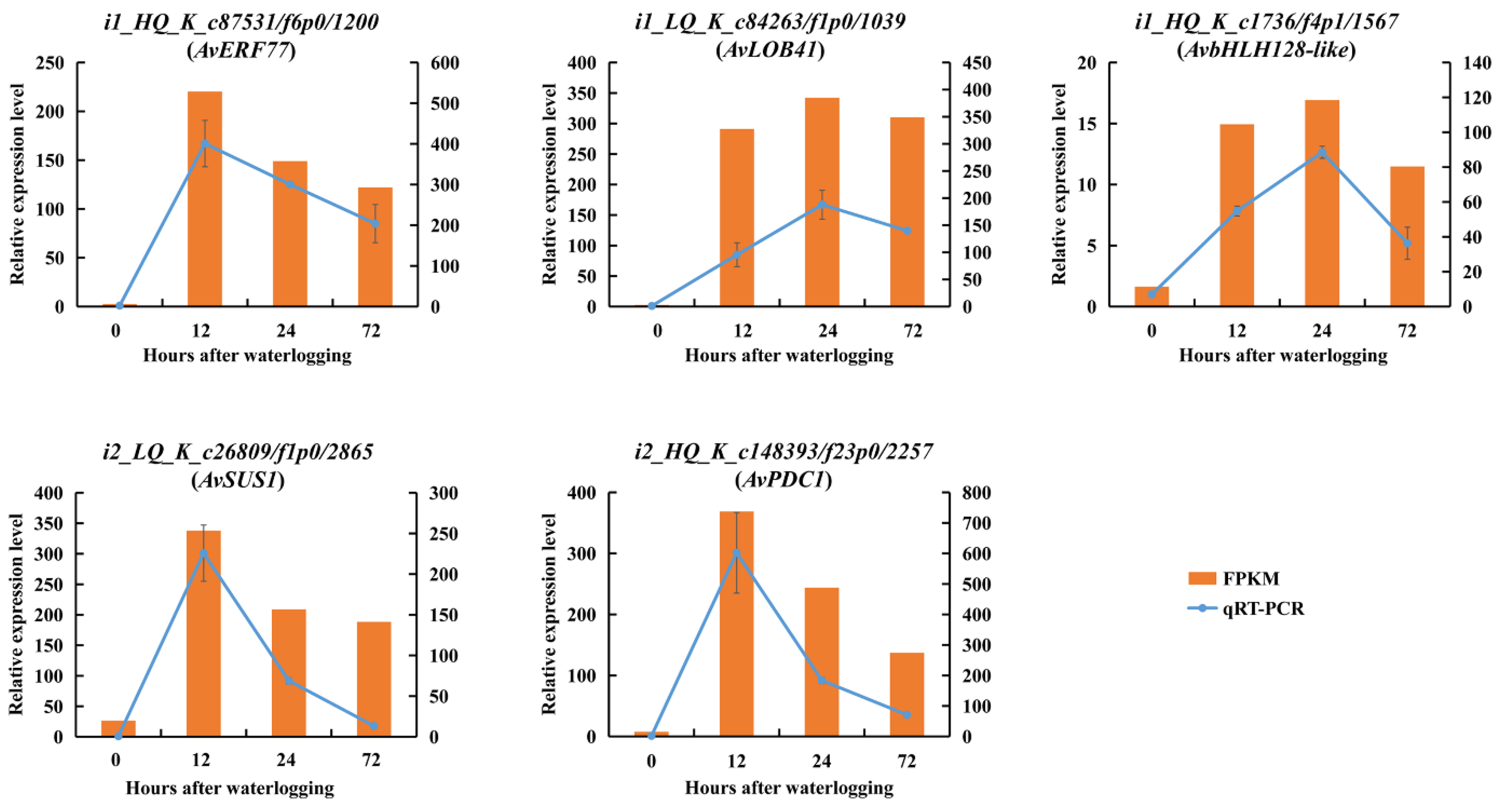
2.11. WGCNA and Validation of Hub Genes by qRT-PCR
3. Discussion
3.1. Adjustments of Carbohydrate Metabolisms under Waterlogging Stress
3.2. Adjustments of Free Amino Acid Metabolism under Waterlogging Stress
3.3. Adjustments of ROS Metabolism under Waterlogging Stress
3.4. Effects of TFs on the Regulation of Waterlogging-Responsive Genes
3.5. WGCNA of the Gene Regulatory Network for the Response to Waterlogging Stress
4. Materials and Methods
4.1. Plant Materials and Waterlogging Treatments
4.2. RNA Extraction, Illumina Library Construction, Sequencing and De Novo Assembly
4.3. PacBio Library Construction, Sequencing and Preprocessing
4.4. Functional Annotation of Transcripts and Prediction of CDSs and TFs
4.5. Identification of DEGs and qRT-PCR Analysis
4.6. Sucrose, T6P, H2O2 and Superoxide Anion Content Measurements
4.7. ADH, TPS, Beta-Glucoside, AlaAT, NADH-GOGAT, SOD, POD and CAT Activity
4.8. Free Amino Acid Content Measurements
4.9. Statistics Analyses and WGCNA
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Phukan, U.J.; Mishra, S.; Shukla, R.K. Waterlogging and submergence stress: Affects and acclimation. Crit. Rev. Biotechnol. 2016, 36, 956–966. [Google Scholar] [CrossRef] [PubMed]
- Grassini, P.; Indaco, G.V.; Pereira, M.L.; Hall, A.J.; Trápani, N. Responses to short-term waterlogging during grain filling in sunflower. Field Crops Res. 2007, 101, 352–363. [Google Scholar] [CrossRef]
- Roberts, J.; Callis, J.; Jardetzky, O.; Walbot, V.; Freeling, M. Cytoplasmic acidosis as a determinant of flooding intolerance in plants. Proc. Natl. Acad. Sci. USA 1984, 81, 6029–6033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steffens, D.; Hutsch, B.; Eschholz, T.; Losak, T.; Schubert, S. Water logging may inhibit plant growth primarily by nutrient deficiency rather than nutrient toxicity. Plant Soil Environ. 2005, 51, 545. [Google Scholar] [CrossRef] [Green Version]
- Davies, C.; Turner, D.; Dracup, M. Yellow lupin (Lupinus luteus) tolerates waterlogging better than narrow-leafed lupin (L. angustifolius) I. shoot and root growth in a controlled environment. Aust. J. Agric. Res. 2000, 51, 701–709. [Google Scholar] [CrossRef]
- Xu, X.W.; Wang, H.H.; Qi, X.H.; Xu, Q.; Chen, X.H. Waterlogging-induced increase in fermentation and related gene expression in the root of cucumber (Cucumis sativus L.). Sci. Hortic. 2014, 179, 388–395. [Google Scholar] [CrossRef]
- Qi, X.H.; Li, Q.Q.; Ma, X.T.; Qian, C.L.; Wang, H.H.; Ren, N.N.; Shen, C.X.; Huang, S.M.; Xu, X.W.; Xu, Q.; et al. Waterlogging-induced adventitious root formation in cucumber is regulated by ethylene and auxin through reactive oxygen species signalling. Plant Cell Environ. 2019, 42, 1458–1470. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Ma, L.; He, S.; Hao, F. AtrbohD functions downstream of ROP2 and positively regulates waterlogging response in Arabidopsis. Plant Signal. Behav. 2018, 13, e1513300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christianson, J.A.; Llewellyn, D.J.; Dennis, E.S.; Wilson, I.W. Global gene expression responses to waterlogging in roots and leaves of cotton (Gossypium hirsutum L.). Plant Cell Physiol. 2010, 51, 21–37. [Google Scholar] [CrossRef] [Green Version]
- Dossa, K.; You, J.; Wang, L.H.; Zhang, Y.X.; Li, D.H.; Zhou, R.; Yu, J.Y.; Wei, X.; Zhu, X.D.; Jiang, S.Y.; et al. Transcriptomic profiling of sesame during waterlogging and recovery. Sci. Data 2019, 6, 204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, F.; Tan, Z.D.; Fang, T.; Tang, K.Y.; Liang, K.; Qiu, F.Z. A comprehensive transcriptomics analysis reveals long non-coding RNA to be involved in the key metabolic pathway in response to waterlogging stress in maize. Genes 2020, 11, 267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, M.M.; Zhang, Q.X.; Xu, J.G.; Bao, M.Y.; Zhang, D.L.; Xie, A.Q.; Sun, X. Effects of waterlogging stress on the physiological characteristics and secondary metabolites of Herbaceous Peony (Paeonia lactiflora Pall.). Am. J. Plant Sci. 2021, 12, 536. [Google Scholar] [CrossRef]
- Fukao, T.; Bailey-Serres, J. Plant responses to hypoxia-is survival a balancing act? Trends Plant Sci. 2004, 9, 449–456. [Google Scholar] [CrossRef] [PubMed]
- Ren, C.G.; Kong, C.C.; Yan, K.; Zhang, H.; Luo, Y.M.; Xie, Z.H. Elucidation of the molecular responses to waterlogging in Sesbania cannabina roots by transcriptome profiling. Sci. Rep. 2017, 7, 9256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nanjo, Y.; Maruyama, K.; Yasue, H.; Yamaguchi-Shinozaki, K.; Shinozaki, K.; Komatsu, S. Transcriptional responses to flooding stress in roots including hypocotyl of soybean seedlings. Plant Mol. Biol. 2011, 77, 129–144. [Google Scholar] [CrossRef] [PubMed]
- Narsai, R.; Rocha, M.; Geigenberger, P.; Whelan, J.; van Dongen, J.T. Comparative analysis between plant species of transcriptional and metabolic responses to hypoxia. New Phytol. 2011, 190, 472–487. [Google Scholar] [CrossRef] [PubMed]
- Branco-Price, C.; Kawaguchi, R.; Ferreira, R.B.; Bailey-Serres, J. Genome-wide analysis of transcript abundance and translation in arabidopsis seedlings subjected to oxygen deprivation. Ann. Bot. 2005, 96, 647–660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibbs, J.; Greenway, H. Mechanisms of anoxia tolerance in plants. I. growth, survival and anaerobic catabolism. Funct. Plant Biol. 2003, 30, 1–47. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.M.; Chen, Y.L.; Hu, W.; Wang, S.S.; Snider, J.L.; Zhou, Z.G. Carbohydrate metabolism in the subtending leaf cross-acclimates to waterlogging and elevated temperature stress and influences boll biomass in cotton (Gossypium hirsutum). Physiol. Plant. 2017, 161, 339–354. [Google Scholar] [CrossRef] [PubMed]
- Kumutha, D.; Sairam, R.; Ezhilmathi, K.; Chinnusamy, V.; Meena, R. Effect of waterlogging on carbohydrate metabolism in pigeon pea (Cajanus cajan L.): Upregulation of sucrose synthase and alcohol dehydrogenase. Plant Sci. 2008, 175, 706–716. [Google Scholar] [CrossRef]
- Kovacs, Z.; Simon-Sarkadi, L.; Vashegyi, I.; Kocsy, G. Different accumulation of free amino acids during short- and long-term osmotic stress in wheat. Sci. World J. 2012, 2012, 216521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.Y.; Yuan, B.; Xu, Y.; Huang, B.R. Differential responses of amino acids and soluble proteins to heat stress associated with genetic variations in heat tolerance for hard fescue. J. Am. Soc. Hortic. Sci. 2018, 143, 45–55. [Google Scholar] [CrossRef] [Green Version]
- Dellero, Y. Manipulating amino acid metabolism to improve crop nitrogen use efficiency for a sustainable agriculture. Front. Plant Sci. 2020, 11, 1857. [Google Scholar] [CrossRef] [PubMed]
- Anee, T.I.; Nahar, K.; Rahman, A.; Al Mahmud, J.; Bhuiyan, T.F.; Ul Alam, M.; Fujita, M.; Hasanuzzaman, M. Oxidative damage and antioxidant defense in Sesamum indicum after different waterlogging durations. Plants 2019, 8, 196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, B.; Xu, S.Z.; Zou, X.L.; Zheng, Y.L.; Qiu, F.Z. Changes of antioxidative enzymes and lipid peroxidation in leaves and roots of waterlogging-tolerant and waterlogging-sensitive maize genotypes at seedling stage. Agric. Sci. China 2010, 9, 651–661. [Google Scholar] [CrossRef]
- Gomez, J.M.; Hernandez, J.A.; Jimenez, A.; del Rio, L.A.; Sevilla, F. Differential response of antioxidative enzymes of chloroplasts and mitochondria to long-term NaCl stress of pea plants. Free Radic. Res. 1999, 31, S11–S18. [Google Scholar] [CrossRef] [PubMed]
- van Veen, H.; Vashisht, D.; Akman, M.; Girke, T.; Mustroph, A.; Reinen, E.; Hartman, S.; Kooiker, M.; van Tienderen, P.; Schranz, M.E.; et al. Transcriptomes of eight Arabidopsis thaliana accessions reveal core conserved, genotype- and organ-specific responses to flooding stress. Plant Physiol. 2016, 172, 668–689. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.X.; Yarra, R.; Jin, L.F.; Cao, H.X. Genome-wide identification and expression analysis of MYB gene family in oil palm (Elaeis guineensis Jacq.) under abiotic stress conditions. Environ. Exp. Bot. 2020, 180, 104245. [Google Scholar] [CrossRef]
- Cui, X.; Wang, Y.X.; Liu, Z.W.; Wang, W.L.; Li, H.; Zhuang, J. Transcriptome-wide identification and expression profile analysis of the bHLH family genes in Camellia sinensis. Funct. Integr. Genom. 2018, 18, 489–503. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.Y.; Huang, S.N.; Mo, Z.H.; Xuan, J.P.; Jia, X.D.; Wang, G.; Guo, Z.R. De novo transcriptome sequencing and comparative analysis of differentially expressed genes in kiwifruit under waterlogging stress. Mol. Breed. 2015, 35, 208. [Google Scholar] [CrossRef]
- Bai, D.F.; Li, Z.; Qi, X.J.; Chen, J.Y.; Gu, H.; Huang, W.Q.; Ren, J.J.; Zhong, Y.P.; Fang, J.B. Physiological responses and tolerance evaluation of four species of Actinidia to waterlogging stress. J. Fruit Sci. 2019, 36, 163–173. [Google Scholar] [CrossRef]
- Tan, C.; Liu, H.X.; Ren, J.; Ye, X.L.; Feng, H.; Liu, Z.Y. Single-molecule real-time sequencing facilitates the analysis of transcripts and splice isoforms of anthers in Chinese cabbage (Brassica rapa L. ssp. pekinensis). BMC Plant Biol. 2019, 19, 517. [Google Scholar] [CrossRef] [PubMed]
- Cui, G.W.; Chai, H.; Yin, H.; Yang, M.; Hu, G.F.; Guo, M.Y.; Yi, R.; Zhang, P. Full-length transcriptome sequencing reveals the low-temperature-tolerance mechanism of Medicago falcata roots. BMC Plant Biol. 2019, 19, 575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Min, X.Y.; Lin, X.S.; Ndayambaza, B.; Wang, Y.R.; Liu, W.X. Coordinated mechanisms of leaves and roots in response to drought stress underlying full-length transcriptome profiling in Vicia sativa L. BMC Plant Biol. 2020, 20, 165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, L.; Zhou, Y.; Chen, Q.; Xiao, S. New insights into the role of lipids in plant hypoxia responses. Prog. Lipid Res. 2021, 81, 101072. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Pan, R.; Zhou, M.; Xu, Y.; Zhang, W. Lipid remodelling plays an important role in wheat (Triticum aestivum) hypoxia stress. Funct. Plant Biol. 2020, 47, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Tan, W.; Xie, L.; Qi, H.; Yang, Y.; Huang, L.; Lai, Y.; Tan, Y.; Zhou, D.; Yu, L.; et al. Polyunsaturated linolenoyl-CoA modulates ERF-VII-mediated hypoxia signaling in Arabidopsis. J. Integr. Plant Biol. 2020, 62, 330–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, L.; Chen, Q.; Chen, M.; Yu, L.; Huang, L.; Chen, L.; Wang, F.; Xia, F.; Zhu, T.; Wu, J.; et al. Unsaturation of very-long-chain ceramides protects plant from hypoxia-induced damages by modulating ethylene signaling in Arabidopsis. PLoS Genet. 2015, 11, e1005143. [Google Scholar] [CrossRef]
- Xie, L.; Yu, L.; Chen, Q.; Wang, F.; Huang, L.; Xia, F.; Zhu, T.; Wu, J.; Yin, J.; Liao, B.; et al. Arabidopsis acyl-CoA-binding protein ACBP3 participates in plant response to hypoxia by modulating very-long-chain fatty acid metabolism. Plant J. 2015, 81, 53–67. [Google Scholar] [CrossRef] [PubMed]
- Shockey, J.M.; Fulda, M.S.; Browse, J.A. Arabidopsis contains nine long-chain acyl-coenzyme a synthetase genes that participate in fatty acid and glycerolipid metabolism. Plant Physiol. 2002, 129, 1710–1722. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, R.R.; Fulda, M.; Paul, M.V.; Anders, M.; Plum, F.; Weits, D.A.; Kosmacz, M.; Larson, T.R.; Graham, I.A.; Beemster, G.T. Low-oxygen response is triggered by an ATP-dependent shift in oleoyl-CoA in Arabidopsis. Proc. Natl. Acad. Sci. USA 2018, 115, E12101–E12110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, L.; Tan, W.; Yang, Y.; Tan, Y.; Zhou, Y.; Zhou, D.; Xiao, S.; Chen, Q. Long-chain acyl-CoA synthetase LACS2 contributes to submergence tolerance by modulating cuticle permeability in Arabidopsis. Plants 2020, 9, 262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Upchurch, R.G. Fatty acid unsaturation, mobilization, and regulation in the response of plants to stress. Biotechnol. Lett. 2008, 30, 967–977. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.; Xue, L.; Lu, L.; Song, G.; Xie, M.; Zhang, H. Characteristics of the response of Actinidia polygama to long-term waterlogging stress. J. Fruit Sci. 2019, 36, 327–337. [Google Scholar] [CrossRef]
- Klinkenberg, J.; Faist, H.; Saupe, S.; Lambertz, S.; Krischke, M.; Stingl, N.; Fekete, A.; Mueller, M.J.; Feussner, I.; Hedrich, R. Two fatty acid desaturases, STEAROYL-ACYL CARRIER PROTEIN Ɗ9-DESATURASE6 and FATTY ACID DESATURASE3, are involved in drought and hypoxia stress signaling in Arabidopsis crown galls. Plant Physiol. 2014, 164, 570–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soto, G.; Stritzler, M.; Lisi, C.; Alleva, K.; Pagano, M.E.; Ardila, F.; Mozzicafreddo, M.; Cuccioloni, M.; Angeletti, M.; Ayub, N.D. Acetoacetyl-CoA thiolase regulates the mevalonate pathway during abiotic stress adaptation. J. Exp. Bot. 2011, 62, 5699–5711. [Google Scholar] [CrossRef] [PubMed]
- Niu, M.; Yan, H.; Xiong, Y.; Zhang, Y.; Zhang, X.; Li, Y.; da Silva, J.A.T.; Ma, G. Cloning, characterization, and functional analysis of acetyl-CoA C-acetyltransferase and 3-hydroxy-3-methylglutaryl-CoA synthase genes in Santalum album. Sci. Rep. 2021, 11, 1082. [Google Scholar] [CrossRef] [PubMed]
- Yan, M.; Xue, C.; Xiong, Y.; Meng, X.; Li, B.; Shen, R.; Lan, P. Proteomic dissection of the similar and different responses of wheat to drought, salinity and submergence during seed germination. J. Proteom. 2020, 220, 103756. [Google Scholar] [CrossRef] [PubMed]
- Feussner, I.; Wasternack, C. The lipoxygenase pathway. Annu. Rev. Plant Biol. 2002, 53, 275–297. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Yao, Q.; Patil, G.B.; Agarwal, G.; Deshmukh, R.K.; Lin, L.; Wang, B.; Wang, Y.; Prince, S.J.; Song, L. Identification and comparative analysis of differential gene expression in soybean leaf tissue under drought and flooding stress revealed by RNA-Seq. Front. Plant Sci. 2016, 7, 1044. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.; Ren, B.; Dong, S.; Liu, P.; Zhao, B.; Zhang, J. Comparative proteomic analysis reveals that exogenous 6-benzyladenine (6-BA) improves the defense system activity of waterlogged summer maize. BMC Plant Biol. 2020, 20, 44. [Google Scholar] [CrossRef] [PubMed]
- Yuan, L.B.; Dai, Y.S.; Xie, L.J.; Yu, L.J.; Zhou, Y.; Lai, Y.X.; Yang, Y.C.; Xu, L.; Chen, Q.F.; Xiao, S. Jasmonate regulates plant responses to postsubmergence reoxygenation through transcriptional activation of antioxidant synthesis. Plant Physiol. 2017, 173, 1864–1880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bailey-Serres, J.; Voesenek, L.A.C.J. Flooding stress: Acclimations and genetic diversity. Annu. Rev. Plant Biol. 2008, 59, 313–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arora, K.; Panda, K.K.; Mittal, S.; Mallikarjuna, M.G.; Rao, A.R.; Dash, P.K.; Thirunavukkarasu, N. RNAseq revealed the important gene pathways controlling adaptive mechanisms under waterlogged stress in maize. Sci. Rep. 2017, 7, 10950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, H.T.; Zhang, J.Y.; Wang, G.; Jia, Z.H.; Huang, S.N.; Wang, T.; Guo, Z.R. Functional characterization of waterlogging and heat stresses tolerance gene pyruvate decarboxylase 2 from Actinidia deliciosa. Int. J. Mol. Sci. 2017, 18, 2377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.Y.; Huang, S.N.; Wang, G.; Xuan, J.P.; Guo, Z.R. Overexpression of Actinidia deliciosa pyruvate decarboxylase 1 gene enhances waterlogging stress in transgenic Arabidopsis thaliana. Plant Physiol. Biochem. 2016, 106, 244–252. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.Y.; Huang, S.N.; Chen, Y.H.; Wang, G.; Guo, Z.R. Identification and characterization of two waterlogging responsive alcohol dehydrogenase genes (AdADH1 and AdADH2) in Actinidia deliciosa. Mol. Breed. 2017, 37, 52. [Google Scholar] [CrossRef]
- Iordachescu, M.; Imai, R. Trehalose biosynthesis in response to abiotic stresses. J. Integr. Plant Biol. 2008, 50, 1223–1229. [Google Scholar] [CrossRef] [PubMed]
- Geigenberger, P.; Fernie, A.R.; Gibon, Y.; Christ, M.; Stitt, M. Metabolic activity decreases as an adaptive response to low internal oxygen in growing potato tubers. Biol. Chem. 2000, 381, 723–740. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.H.; Zhang, Y.X.; Qi, X.Q.; Li, D.H.; Wei, W.L.; Zhang, X.R. Global gene expression responses to waterlogging in roots of sesame (Sesamum indicum L.). Acta Physiol. Plant. 2012, 34, 2241–2249. [Google Scholar] [CrossRef]
- Chen, Y.L.; Wang, H.M.; Hu, W.; Wang, S.S.; Snider, J.L.; Zhou, Z.G. Co-occurring elevated temperature and waterlogging stresses disrupt cellulose synthesis by altering the expression and activity of carbohydrate balance-associated enzymes during fiber development in cotton. Environ. Exp. Bot. 2017, 135, 106–117. [Google Scholar] [CrossRef]
- Singhania, R.R.; Patel, A.K.; Sukumaran, R.K.; Larroche, C.; Pandey, A. Role and significance of beta-glucosidases in the hydrolysis of cellulose for bioethanol production. Bioresour. Technol. 2013, 127, 500–507. [Google Scholar] [CrossRef]
- Christianson, J.A.; Llewellyn, D.J.; Dennis, E.S.; Wilson, I.W. Comparisons of early transcriptome responses to low-oxygen environments in three dicotyledonous plant species. Plant Signal. Behav. 2010, 5, 1006–1009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hildebrandt, T.M. Synthesis versus degradation: Directions of amino acid metabolism during Arabidopsis abiotic stress response. Plant Mol. Biol. 2018, 98, 121–135. [Google Scholar] [CrossRef] [PubMed]
- Szabados, L.; Savouré, A. Proline: A multifunctional amino acid. Trends Plant Sci. 2010, 15, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Alcázar, R.; Marco, F.; Cuevas, J.C.; Patron, M.; Ferrando, A.; Carrasco, P.; Tiburcio, A.F.; Altabella, T. Involvement of polyamines in plant response to abiotic stress. Biotechnol. Lett. 2006, 28, 1867–1876. [Google Scholar] [CrossRef] [PubMed]
- Branco-Price, C.; Kaiser, K.A.; Jang, C.J.H.; Larive, C.K.; Bailey-Serres, J. Selective mRNA translation coordinates energetic and metabolic adjustments to cellular oxygen deprivation and reoxygenation in Arabidopsis thaliana. Plant J. 2008, 56, 743–755. [Google Scholar] [CrossRef] [PubMed]
- Ricoult, C.; Echeverria, L.O.; Cliquet, J.B.; Limami, A.M. Characterization of alanine aminotransferase (AlaAT) multigene family and hypoxic response in young seedlings of the model legume Medicago truncatula. J. Exp. Bot. 2006, 57, 3079–3089. [Google Scholar] [CrossRef] [PubMed]
- Limami, A.M.; Glevarec, G.; Ricoult, C.; Cliquet, J.B.; Planchet, E. Concerted modulation of alanine and glutamate metabolism in young Medicago truncatula seedlings under hypoxic stress. J. Exp. Bot. 2008, 59, 2325–2335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bailey-Serres, J.; Fukao, T.; Gibbs, D.J.; Holdsworth, M.J.; Lee, S.C.; Licausi, F.; Perata, P.; Voesenek, L.A.C.J.; van Dongen, J.T. Making sense of low oxygen sensing. Trends Plant Sci. 2012, 17, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Reggiani, R.; Nebuloni, M.; Mattana, M.; Brambilla, I. Anaerobic accumulation of amino acids in rice roots: Role of the glutamine synthetase/glutamate synthase cycle. Amino Acids 2000, 18, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Slocum, R.D. Genes, enzymes and regulation of arginine biosynthesis in plants. Plant Physiol. Biochem. 2005, 43, 729–745. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.; Nawata, E.; Hosokawa, M.; Domae, Y.; Sakuratani, T. Alterations in photosynthesis and some antioxidant enzymatic activities of mungbean subjected to waterlogging. Plant Sci. 2002, 163, 117–123. [Google Scholar] [CrossRef]
- Yan, B.; Dai, Q.J.; Liu, X.Z.; Huang, S.B.; Wang, Z.X. Flooding-induced membrane damage, lipid oxidation and activated oxygen generation in corn leaves. Plant Soil. 1996, 179, 261–268. [Google Scholar] [CrossRef]
- Prasad, T.K.; Anderson, M.D.; Stewart, C.R. Localization and characterization of peroxidases in the mitochondria of chilling-acclimated maize seedlings. Plant Physiol. 1995, 108, 1597–1605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, G.P.; Tanakamaru, K.; Abe, J.; Morita, S. Influence of waterlogging on some anti-oxidative enzymatic activities of two barley genotypes differing in anoxia tolerance. Acta Physiol. Plant. 2007, 29, 171–176. [Google Scholar] [CrossRef]
- He, Z.Q.; Su, Y.J.; Wang, T. Full-length transcriptome analysis of four different tissues of Cephalotaxus oliveri. Int. J. Mol. Sci. 2021, 22, 787. [Google Scholar] [CrossRef] [PubMed]
- Dawood, T.; Yang, X.P.; Visser, E.J.W.; te Beek, T.A.H.; Kensche, P.R.; Cristescu, S.M.; Lee, S.; Flokova, K.; Duy, N.; Mariani, C.; et al. A co-opted hormonal cascade activates dormant adventitious root primordia upon flooding in Solanum dulcamara. Plant Physiol. 2016, 170, 2351–2364. [Google Scholar] [CrossRef] [Green Version]
- Voesenek, L.A.C.J.; Sasidharan, R.; Visser, E.J.W.; Bailey-Serres, J. Flooding stress signaling through perturbations in oxygen, ethylene, nitric oxide and light. New Phytol. 2016, 209, 39–43. [Google Scholar] [CrossRef] [Green Version]
- Du, H.W.; Huang, M.; Liu, L. The genome wide analysis of GT transcription factors that respond to drought and waterlogging stresses in maize. Euphytica 2016, 208, 113–122. [Google Scholar] [CrossRef]
- Thirunavukkarasu, N.; Hossain, F.; Mohan, S.; Shiriga, K.; Mittal, S.; Sharma, R.; Singh, R.K.; Gupta, H.S. Genome-wide expression of transcriptomes and their co-expression pattern in subtropical maize (Zea mays L.) under waterlogging stress. PLoS ONE 2013, 8, e70433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandey, D.M.; Kim, S.R. Identification and expression analysis of hypoxia stress inducible CCCH-type zinc finger protein genes in rice. J. Plant Biol. 2012, 55, 489–497. [Google Scholar] [CrossRef]
- Yang, A.; Dai, X.Y.; Zhang, W.H. A R2R3-type MYB gene, OsMYB2, is involved in salt, cold, and dehydration tolerance in rice. J. Exp. Bot. 2012, 63, 2541–2556. [Google Scholar] [CrossRef] [PubMed]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Y.; Yu, J.; Cang, J.; Liu, L.J.; Mu, Y.C.; Wang, J.H.; Zhang, D. Detection of sugar accumulation and expression levels of correlative key enzymes in winter wheat (Triticum aestivum) at low temperatures. Biosci. Biotechnol. Biochem. 2011, 75, 681–687. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, S. Heat and chilling induced disruption of redox homeostasis and its regulation by hydrogen peroxide in germinating rice seeds (Oryza sativa L., Cultivar Ratna). Physiol. Mol. Biol. Plants 2013, 19, 199–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, Y.S.; Wu, X.L.; Sun, M.X.; Peng, F.T. Hydrogen sulfide alleviates waterlogging-induced damage in peach seedlings via enhancing antioxidative system and inhibiting ethylene synthesis. Front. Plant Sci. 2020, 11, 696. [Google Scholar] [CrossRef] [PubMed]
- Zong, J.Q.; Gao, Y.Z.; Chen, J.B.; Guo, H.L.; Wang, Y.; Meng, F.; Jiang, Y.W.; Liu, J. Growth and enzymatic activity of four warm-season turfgrass species exposed to waterlogging. J. Am. Soc. Hortic. Sci. 2015, 140, 151–162. [Google Scholar] [CrossRef] [Green Version]
- Ventura, I.; Brunello, L.; Iacopino, S.; Valeri, M.C.; Novi, G.; Dornbusch, T.; Perata, P.; Loreti, E. Arabidopsis phenotyping reveals the importance of alcohol dehydrogenase and pyruvate decarboxylase for aerobic plant growth. Sci. Rep. 2020, 10, 16669. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.L.; Hou, Z.H.; Huang, C.Y.; Chen, Q.; Gao, W.; Zhang, J.X. Cloning, purification and characterization of trehalose-6-phosphate synthase from Pleurotus tuoliensis. PeerJ 2018, 6, e5230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veena, J.; Singla, N.K.; Sunita, J.; Kaushalya, G. Activities of enzymes of fermentation pathways in the leaves and roots of contrasting cultivars of sorghum (Sorghum bicolor L.) during flooding. Physiol. Mol. Biol. Plants 2010, 16, 241–247. [Google Scholar] [CrossRef] [Green Version]
- Liu, N.; Lin, Z.F.; Guan, L.L.; Gaughan, G.; Lin, G.Z. Antioxidant enzymes regulate reactive oxygen species during pod elongation in Pisum sativum and Brassica chinensis. PLoS ONE 2014, 9, e87588. [Google Scholar] [CrossRef] [Green Version]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Zheng, G.H.; Jin, W.W.; Fan, P.; Feng, X.N.; Bai, Y.; Tao, T.; Yu, L.J. A novel method for detecting amino acids derivatized with phenyl isothiocyanate by high-performance liquid chromatography-electrospray ionization mass spectrometry. Int. J. Mass Spectrom. 2015, 392, 1–6. [Google Scholar] [CrossRef]















Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Z.; Bai, D.; Zhong, Y.; Lin, M.; Sun, L.; Qi, X.; Hu, C.; Fang, J. Full-Length Transcriptome and RNA-Seq Analyses Reveal the Mechanisms Underlying Waterlogging Tolerance in Kiwifruit (Actinidia valvata). Int. J. Mol. Sci. 2022, 23, 3237. https://doi.org/10.3390/ijms23063237
Li Z, Bai D, Zhong Y, Lin M, Sun L, Qi X, Hu C, Fang J. Full-Length Transcriptome and RNA-Seq Analyses Reveal the Mechanisms Underlying Waterlogging Tolerance in Kiwifruit (Actinidia valvata). International Journal of Molecular Sciences. 2022; 23(6):3237. https://doi.org/10.3390/ijms23063237
Chicago/Turabian StyleLi, Zhi, Danfeng Bai, Yunpeng Zhong, Miaomiao Lin, Leiming Sun, Xiujuan Qi, Chungen Hu, and Jinbao Fang. 2022. "Full-Length Transcriptome and RNA-Seq Analyses Reveal the Mechanisms Underlying Waterlogging Tolerance in Kiwifruit (Actinidia valvata)" International Journal of Molecular Sciences 23, no. 6: 3237. https://doi.org/10.3390/ijms23063237
APA StyleLi, Z., Bai, D., Zhong, Y., Lin, M., Sun, L., Qi, X., Hu, C., & Fang, J. (2022). Full-Length Transcriptome and RNA-Seq Analyses Reveal the Mechanisms Underlying Waterlogging Tolerance in Kiwifruit (Actinidia valvata). International Journal of Molecular Sciences, 23(6), 3237. https://doi.org/10.3390/ijms23063237