
Regulatory miRNAs in Cardiovascular and Alzheimer’s Disease: A Focus on Copper


 ,
,  , , , ,
, , , , 
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Non-Coding RNAs and Cu Metabolism in Physiology
2.1. Role of microRNAs in Cell Regulation in Physiology
2.2. Cu Intake in Human Diet and Drinking Water
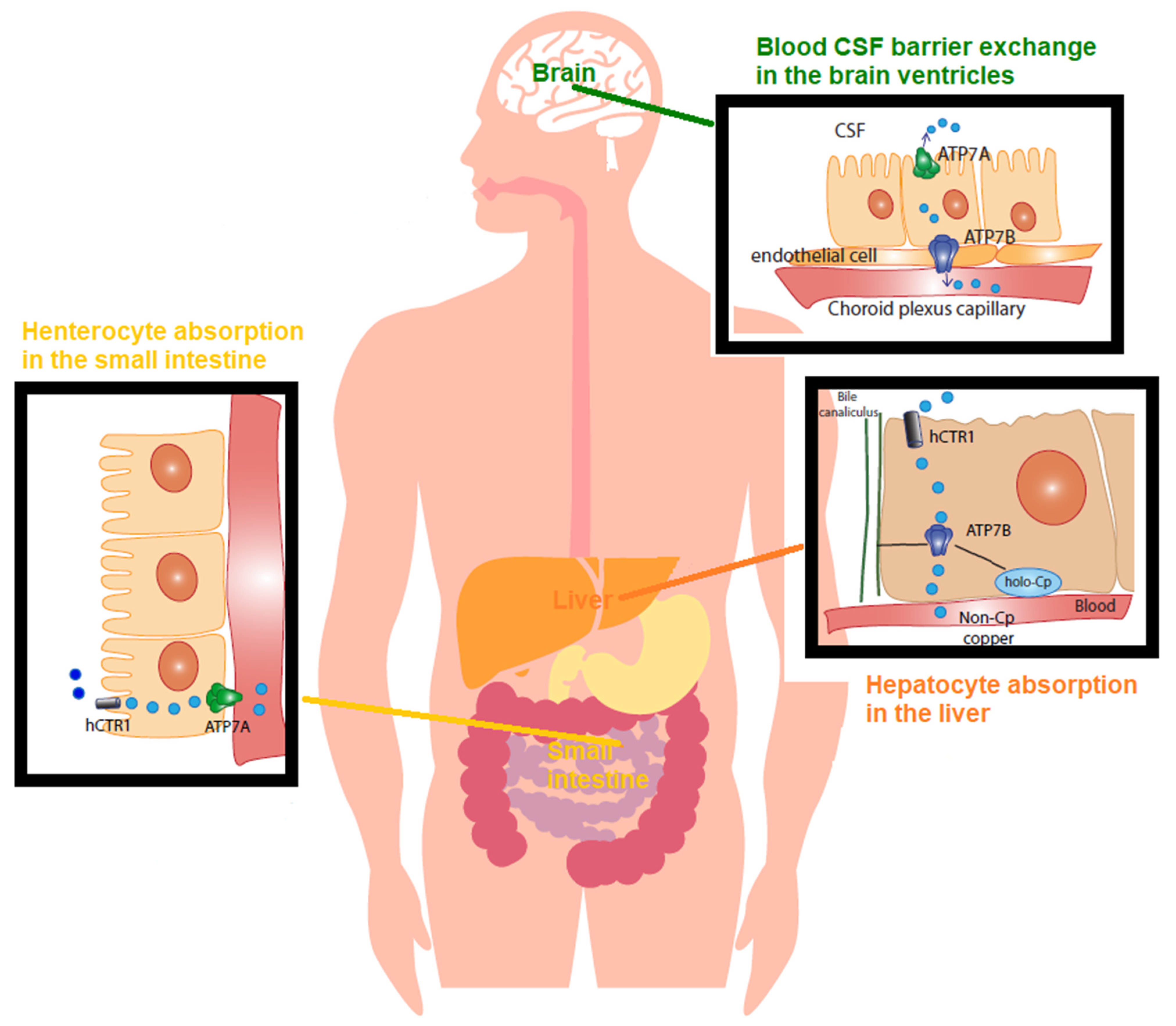
2.3. Cu Regulation in Physiology
3. MicroRNA and Cu Metabolism in Disease States
3.1. Cardiovascular Diseases
3.2. Involvement of miRNA in Cardiovascular Disease
3.3. Brief Description of Alzheimer’s Disease’s Main Features
3.4. Involvement of Cu in the Induction of MicroRNA in Alzheimer’s Disease
3.5. Involvement of Cu in Myocardial Infarction
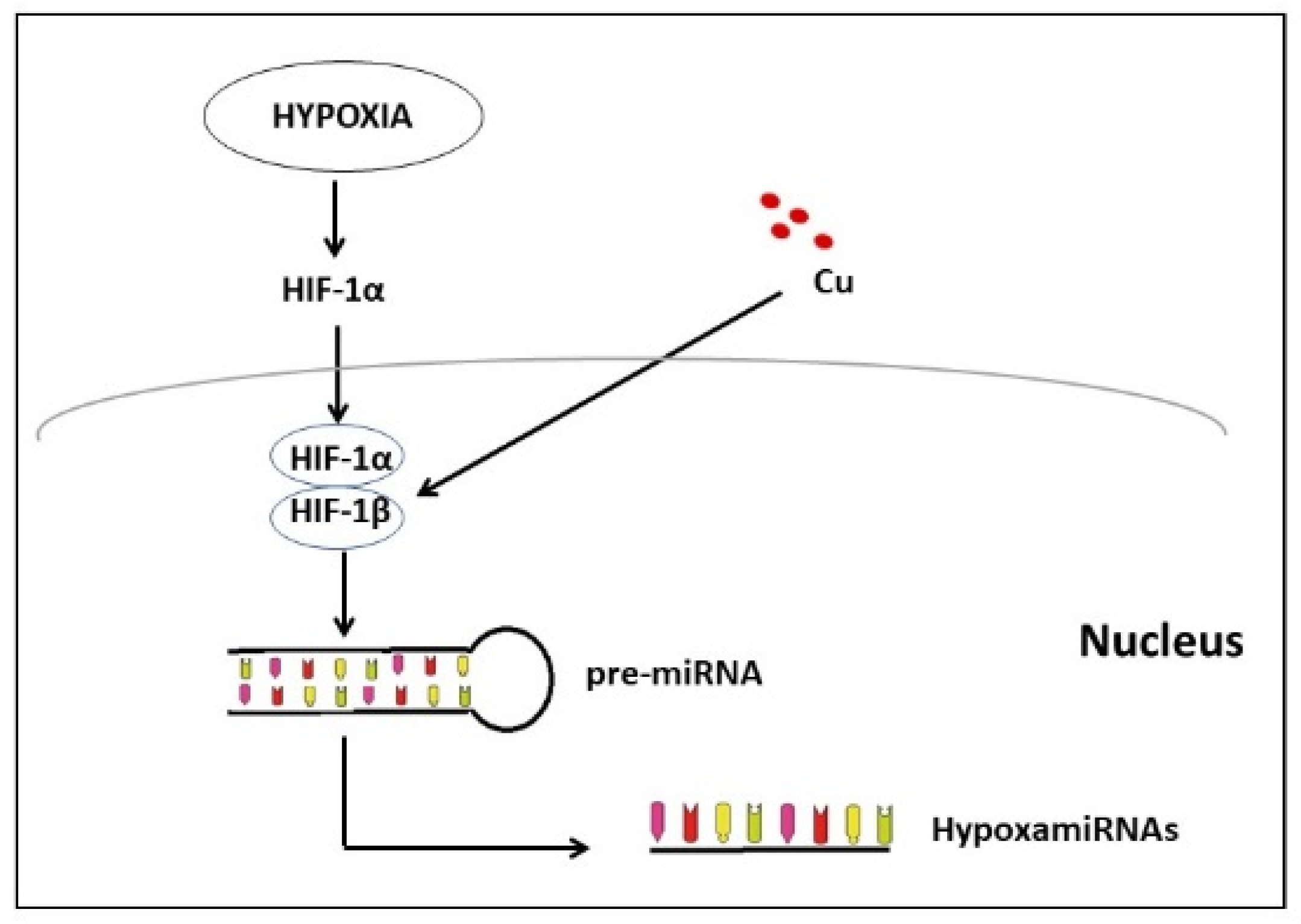
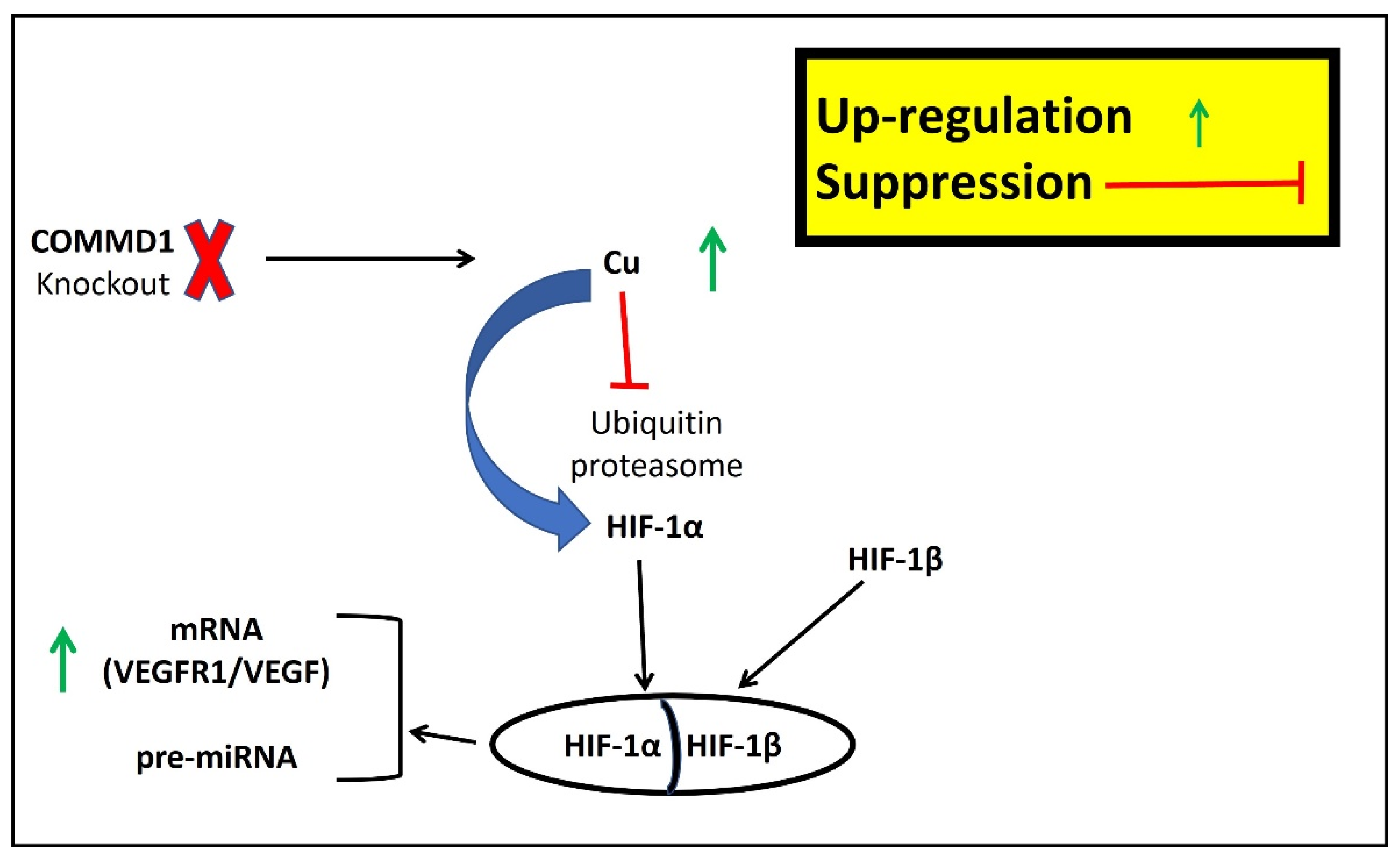
3.6. Are the miRNAs Induced by Hypoxia, the Missing Piece between Cu and Cardiovascular Disease?
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Squitti, R.; Faller, P.; Hureau, C.; Granzotto, A.; White, A.R.; Kepp, K.P. Copper Imbalance in Alzheimer’s Disease and Its Link with the Amyloid Hypothesis: Towards a Combined Clinical, Chemical, and Genetic Etiology. J. Alzheimer’s Dis. 2021, 83, 23–41. [Google Scholar] [CrossRef] [PubMed]
- Squitti, R.; Siotto, M.; Polimanti, R. Low-copper diet as a preventive strategy for Alzheimer’s disease. Neurobiol. Aging 2014, 35, S40–S50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ziello, J.E.; Jovin, I.S.; Huang, Y. Hypoxia-Inducible Factor (HIF)-1 Regulatory Pathway and its Potential for Therapeutic Intervention in Malignancy and Ischemia. Yale J. Biol. Med. 2007, 80, 51–60. [Google Scholar] [PubMed]
- Feng, W.; Ye, F.; Xue, W.; Zhou, Z.; Kang, Y.J. Copper Regulation of Hypoxia-Inducible Factor-1 Activity. Mol. Pharmacol. 2008, 75, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Hypoxia-Inducible Factor 1 and Cardiovascular Disease. Annu. Rev. Physiol. 2014, 76, 39–56. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, H.; Drouin, R.; Holmquist, G.P.; Akman, S.A. A Hot Spot for Hydrogen Peroxide-Induced Damage in the Human Hypoxia-Inducible Factor 1 Binding Site of thePGK 1Gene. Arch. Biochem. Biophys. 1997, 338, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Squitti, R.; Siotto, M.; Salustri, C.; Polimanti, R. Metal dysfunction in Alzheimer’s disease. In Studies on Alzheimer’s Disease; Springer: Berlin/Heidelberg, Germany, 2013; pp. 73–97. [Google Scholar]
- Alexander, G.C.; Knopman, D.S.; Emerson, S.S.; Ovbiagele, B.; Kryscio, R.J.; Perlmutter, J.S.; Kesselheim, A.S. Revisiting FDA Approval of Aducanumab. N. Engl. J. Med. 2021, 385, 769–771. [Google Scholar] [CrossRef]
- Ayton, S.; Portbury, S.; Kalinowski, P.; Agarwal, P.; Diouf, I.; Schneider, J.A.; Morris, M.C.; Bush, A.I. Regional brain iron associated with deterioration in Alzheimer’s disease: A large cohort study and theoretical significance. Alzheimer’s Dement. 2021, 17, 1244–1256. [Google Scholar] [CrossRef]
- Cheignon, C.; Tomas, M.; Bonnefont-Rousselot, D.; Faller, P.; Hureau, C.; Collin, F. Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox Biol. 2018, 14, 450–464. [Google Scholar] [CrossRef]
- Squitti, R.; Ventriglia, M.; Simonelli, I.; Bonvicini, C.; Costa, A.; Perini, G.; Binetti, G.; Benussi, L.; Ghidoni, R.; Koch, G.; et al. Copper Imbalance in Alzheimer’s Disease: Meta-Analysis of Serum, Plasma, and Brain Specimens, and Replication Study Evaluating ATP7B Gene Variants. Biomolecules 2021, 11, 960. [Google Scholar] [CrossRef]
- Tublin, J.M.; Adelstein, J.M.; Del Monte, F.; Combs, C.K.; Wold, L.E. Getting to the Heart of Alzheimer Disease. Circ. Res. 2019, 124, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Squitti, R.; Mendez, A.; Ricordi, C.; Siotto, M.; Goldberg, R. Copper in Glucose Intolerance, Cognitive Decline, and Alzheimer Disease. Alzheimer Dis. Assoc. Disord. 2019, 33, 77–85. [Google Scholar] [CrossRef]
- Mijajlović, M.D.; Pavlović, A.; Brainin, M.; Heiss, W.-D.; Quinn, T.J.; Ihle-Hansen, H.B.; Hermann, D.M.; Ben Assayag, E.; Richard, E.; Thiel, A.; et al. Post-stroke dementia–a comprehensive review. BMC Med. 2017, 15, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, I.; Sagare, A.P.; Coma, M.; Perlmutter, D.; Gelein, R.; Bell, R.D.; Deane, R.J.; Zhong, E.; Parisi, M.; Ciszewski, J.; et al. Low levels of copper disrupt brain amyloid-β homeostasis by altering its production and clearance. Proc. Natl. Acad. Sci. USA 2013, 110, 14771–14776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, H.-W.; Rodriguez-Ortiz, C.J.; Lim, S.L.; Zumkehr, J.; Kilian, J.G.; Vidal, J.; Kitazawa, M. Copper-Induced Upregulation of MicroRNAs Directs the Suppression of Endothelial LRP1 in Alzheimer’s Disease Model. Toxicol. Sci. 2019, 170, 144–156. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Li, J.; Cairns, M.J. Identifying miRNAs, targets and functions. Brief. Bioinform. 2012, 15, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Abdelfattah, A.M.; Park, C.; Choi, M.Y. Update on non-canonical microRNAs. Biomol. Concepts 2014, 5, 275–287. [Google Scholar] [CrossRef] [Green Version]
- Wahid, F.; Shehzad, A.; Khan, T.; Kim, Y.Y. MicroRNAs: Synthesis, mechanism, function, and recent clinical trials. Biochim. Et Biophys. Acta (BBA)-Mol. Cell Res. 2010, 1803, 1231–1243. [Google Scholar] [CrossRef] [Green Version]
- Bronze-Da-Rocha, E. MicroRNAs Expression Profiles in Cardiovascular Diseases. BioMed Res. Int. 2014, 2014, 985408. [Google Scholar] [CrossRef] [Green Version]
- Creemers, E.E.; Tijsen, A.J.; Pinto, Y.M. Circulating microRNAs: Novel biomarkers and extracellular communicators in cardiovascular disease? Circ. Res. 2012, 110, 483–495. [Google Scholar] [CrossRef]
- Miao, L.; Yao, H.; Li, C.; Pu, M.; Yao, X.; Yang, H.; Qi, X.; Ren, J.; Wang, Y. A dual inhibition: MicroRNA-552 suppresses both transcription and translation of cytochrome P450 2E1. Biochim. Et Biophys. Acta 2016, 1859, 650–662. [Google Scholar] [CrossRef] [PubMed]
- Nair, A.A.; Tang, X.; Thompson, K.J.; Vedell, P.T.; Kalari, K.R.; Subramanian, S. Frequency of MicroRNA Response Elements Identifies Pathologically Relevant Signaling Pathways in Triple-Negative Breast Cancer. iScience 2020, 23, 101249. [Google Scholar] [CrossRef] [PubMed]
- Hoesel, B.; Schmid, J.A. The complexity of NF-κB signaling in inflammation and cancer. Mol. Cancer 2013, 12, 86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serocki, M.; Bartoszewska, S.; Janaszak-Jasiecka, A.; Ochocka, R.J.; Collawn, J.F.; Bartoszewski, R. miRNAs regulate the HIF switch during hypoxia: A novel therapeutic target. Angiogenesis 2018, 21, 183–202. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Marin-Muller, C.; Bharadwaj, U.; Chow, K.-H.; Yao, Q.; Chen, C. MicroRNAs: Control and Loss of Control in Human Physiology and Disease. World J. Surg. 2008, 33, 667–684. [Google Scholar] [CrossRef] [Green Version]
- Kepp, K.P.; Squitti, R. Copper imbalance in Alzheimer’s disease: Convergence of the chemistry and the clinic. Co-Ord. Chem. Rev. 2019, 397, 168–187. [Google Scholar] [CrossRef]
- Mattick, J.S.; Makunin, I.V. Non-coding RNA. Hum. Mol. Genet. 2006, 15 (Suppl. 1), R17–R29. [Google Scholar] [CrossRef] [Green Version]
- Palazzo, A.F.; Lee, E.S. Non-coding RNA: What is functional and what is junk? Front. Genet. 2015, 6, 2. [Google Scholar] [CrossRef] [Green Version]
- Dhanoa, J.K.; Sethi, R.S.; Verma, R.; Arora, J.S.; Mukhopadhyay, C.S. Long non-coding RNA: Its evolutionary relics and biological implications in mammals: A review. J. Anim. Sci. Technol. 2018, 60, 25. [Google Scholar] [CrossRef] [Green Version]
- Romano, G.; Veneziano, D.; Acunzo, M.; Croce, C.M. Small non-coding RNA and cancer. Carcinogenesis 2017, 38, 485–491. [Google Scholar] [CrossRef] [Green Version]
- O’Connell, R.M.; Rao, D.S.; Chaudhuri, A.A.; Baltimore, D. Physiological and pathological roles for microRNAs in the immune system. Nat. Rev. Immunol. 2010, 10, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Grosshans, H. Active turnover modulates mature microRNA activity in Caenorhabditis elegans. Nature 2009, 461, 546–549. [Google Scholar] [CrossRef] [PubMed]
- Davis-Dusenbery, B.; Hilyard, A.C.; Lagna, G.; Hata, A. SMAD proteins control DROSHA-mediated microRNA maturation. Nature 2008, 454, 56–61. [Google Scholar] [CrossRef] [Green Version]
- Gruber, J.J.; Zatechka, D.S.; Sabin, L.R.; Yong, J.; Lum, J.J.; Kong, M.; Zong, W.-X.; Zhang, Z.; Lau, C.-K.; Rawlings, J.; et al. Ars2 Links the Nuclear Cap-Binding Complex to RNA Interference and Cell Proliferation. Cell 2009, 138, 328–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdi, J.; Rastgoo, N.; Li, L.; Chen, W.; Chang, H.; Abdi, J.; Rastgoo, N.; Li, L.; Chen, W.; Chang, H. Role of tumor suppressor p53 and micro-RNA interplay in multiple myeloma pathogenesis. J. Hematol. Oncol. 2017, 10, 169. [Google Scholar] [CrossRef] [Green Version]
- Feng, Z.; Zhang, C.; Wu, R.; Hu, W. Tumor suppressor p53 meets microRNAs. J. Mol. Cell Biol. 2011, 3, 44–50. [Google Scholar] [CrossRef]
- Treiber, T.; Treiber, N.; Plessmann, U.; Harlander, S.; Daiß, J.-L.; Eichner, N.; Lehmann, G.; Schall, K.; Urlaub, H.; Meister, G. A Compendium of RNA-Binding Proteins that Regulate MicroRNA Biogenesis. Mol. Cell 2017, 66, 270–284.e13. [Google Scholar] [CrossRef] [Green Version]
- Michlewski, G.; Guil, S.; Cáceres, J.F. Stimulation of pri-miR-18a processing by hnRNP A1. Regul. Micrornas 2010, 700, 28–35. [Google Scholar]
- Nussbacher, J.K.; Yeo, G.W. Systematic Discovery of RNA Binding Proteins that Regulate MicroRNA Levels. Mol. Cell 2018, 69, 1005–1016.e7. [Google Scholar] [CrossRef] [Green Version]
- Lee, I.; Ajay, S.S.; Yook, J.I. New class of microRNA targets containing simultaneous 5′-UTR and 3′-UTR interaction sites. Genome Res. 2009, 19, 1175–1183. [Google Scholar] [CrossRef] [Green Version]
- Catalanotto, C.; Cogoni, C.; Zardo, G. MicroRNA in Control of Gene Expression: An Overview of Nuclear Functions. Int. J. Mol. Sci. 2016, 17, 1712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meijer, H.A.; Kong, Y.W.; Lu, W.T.; Wilczynska, A.; Spriggs, R.V.; Robinson, S.W.; Godfrey, J.D.; Willis, A.E.; Bushell, M. Translational Repression and eIF4A2 Activity Are Critical for MicroRNA-Mediated Gene Regulation. Science 2013, 340, 82–85. [Google Scholar] [CrossRef] [PubMed]
- Gagnon, K.T.; Li, L.; Chu, Y.; Janowski, B.A.; Corey, D.R. RNAi Factors Are Present and Active in Human Cell Nuclei. Cell Rep. 2014, 6, 211–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, H.; Sun, S.; Tu, K. A splicing-independent function of SF2/ASF in microRNA processing. Mol. Cell 2010, 38, 67–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gebert, L.F.R.; Macrae, I.J. Regulation of microRNA function in animals. Nat. Rev. Mol. Cell Biol. 2019, 20, 21–37. [Google Scholar] [CrossRef] [PubMed]
- DeVita, M.A.; Aulisio, M.P. The Ethics of Medical Mistakes: Historical, Legal, and Institutional Perspectives. Kennedy Inst. Ethic-J. 2001, 11, 115–116. [Google Scholar] [CrossRef]
- World Health Organization. Trace Elements in Human Nutrition and Health; World Health Organization: Geneva, Switzerland, 1996. [Google Scholar]
- de Romaña, D.L.; Olivares, M.; Uauy, R.; Araya, M. Risks and benefits of copper in light of new insights of copper homeostasis. J. Trace Elem. Med. Biol. 2011, 25, 3–13. [Google Scholar] [CrossRef]
- Bertrand, E.; Lewandowska, E.; Szpak, G.M.; Hoogenraad, T.; Blaauwgers, H.G.; Członkowska, A.; Dymecki, J. Neuropathological analysis of pathological forms of astroglia in Wilson′s disease. Folia Neuropathol. 2001, 39, 73–79. [Google Scholar]
- Turnlund, J.R.; Keyes, W.R.; Kim, S.K.; Domek, J.M. Long-term high copper intake: Effects on copper absorption, retention, and homeostasis in men. Am. J. Clin. Nutr. 2005, 81, 822–828. [Google Scholar] [CrossRef]
- Kim, H.-J.; Lee, J.Y.; Paik, K.-W.; Koh, K.-W.; Won, J.; Choe, S.; Lee, J.; Moon, J.-T.; Park, Y.-J. Effects of Cu/Al intermetallic compound (IMC) on copper wire and aluminum pad bondability. IEEE Trans. Compon. Packag. Technol. 2003, 26, 367–374. [Google Scholar] [CrossRef] [Green Version]
- Siotto, M.; Squitti, R. Copper imbalance in Alzheimer’s disease: Overview of the exchangeable copper component in plasma and the intriguing role albumin plays. Co-Ord. Chem. Rev. 2018, 371, 86–95. [Google Scholar] [CrossRef]
- Walshe, J.M. Wilson′s disease: The importance of measuring serum caeruloplasmin non-immunologically. Ann. Clin. Biochem. Int. J. Lab. Med. 2003, 40, 115–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sensi, S.L.; Granzotto, A.; Siotto, M.; Squitti, R. Copper and Zinc Dysregulation in Alzheimer’s Disease. Trends Pharmacol. Sci. 2018, 39, 1049–1063. [Google Scholar] [CrossRef] [PubMed]
- Bucossi, S.; Ventriglia, M.; Panetta, V. Copper in Alzheimer’s disease: A meta-analysis of serum, plasma, and cerebrospinal fluid studies. J. Alzheimer’s Dis. 2011, 24, 175–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, B.-S.; Zheng, W. Copper transport to the brain by the blood-brain barrier and blood-CSF barrier. Brain Res. 2009, 1248, 14–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Squitti, R.; Barbati, G.; Rossi, L.; Ventriglia, M.; Forno, G.D.; Cesaretti, S.; Moffa, F.; Caridi, I.; Cassetta, E.; Pasqualetti, P.; et al. Excess of nonceruloplasmin serum copper in AD correlates with MMSE, CSF-amyloid, and h-tau. Neurology 2006, 67, 76–82. [Google Scholar] [CrossRef]
- Zischka, H.; Einer, C. Mitochondrial copper homeostasis and its derailment in Wilson disease. Int. J. Biochem. Cell Biol. 2018, 102, 71–75. [Google Scholar] [CrossRef]
- Bandmann, O.; Weiss, K.H.; Kaler, S.G. Wilson′s disease and other neurological copper disorders. Lancet Neurol. 2015, 14, 103–113. [Google Scholar] [CrossRef] [Green Version]
- Roberts, E.A.; Schilsky, M.L.; American Association for Study of Liver Diseases (AASLD). Diagnosis and treatment of Wilson disease: An update. Hepatology 2008, 47, 2089–2111. [Google Scholar] [CrossRef]
- Ameh, T.; Sayes, C.M. The potential exposure and hazards of copper nanoparticles: A review. Environ. Toxicol. Pharmacol. 2019, 71, 103220. [Google Scholar] [CrossRef]
- Virani, S.S.; Alonso, A.; Aparicio, H.J. Heart disease and stroke statistics—2021 update: A report from the American Heart Association. Circulation 2021, 143, e254–e743. [Google Scholar] [CrossRef] [PubMed]
- Arenas de Larriva, A.P.; Limia-Pérez, L.; Alcalá-Díaz, J.F.; Alonso, A.; López-Miranda, J.; Delgado-Lista, J. Ceruloplasmin and Coronary Heart Disease—A Systematic Review. Nutrients 2020, 12, 3219. [Google Scholar] [CrossRef] [PubMed]
- Pal, A.; Squitti, R.; Picozza, M.; Pawar, A.; Rongioletti, M.; Dutta, A.K.; Sahoo, S.; Goswami, K.; Sharma, P.; Prasad, R. Zinc and COVID-19: Basis of Current Clinical Trials. Biol. Trace Element Res. 2020, 199, 2882–2892. [Google Scholar] [CrossRef] [PubMed]
- Squitti, R.; Ventriglia, M.; Granzotto, A.; Sensi, S.L.; Rongioletti, M.C.A. Non-Ceruloplasmin Copper as a Stratification Biomarker of Alzheimer’s Disease Patients: How to Measure and Use It. Curr. Alzheimer Res. 2021, 18, 533–545. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhang, W.; Wu, Z.; Yang, Y.; Kang, Y.J. Copper levels affect targeting of hypoxia-inducible factor 1α to the promoters of hypoxia-regulated genes. J. Biol. Chem. 2018, 293, 14669–14677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cummins, E.P.; Keogh, C.E.; Crean, D.; Taylor, C.T. The role of HIF in immunity and inflammation. Mol. Asp. Med. 2016, 47–48, 24–34. [Google Scholar] [CrossRef]
- Prasad, S.V.N.; Duan, Z.-H.; Gupta, M.K.; Surampudi, V.S.K.; Volinia, S.; Calin, G.; Liu, C.-G.; Kotwal, A.; Moravec, C.S.; Starling, R.C.; et al. Unique MicroRNA Profile in End-stage Heart Failure Indicates Alterations in Specific Cardiovascular Signaling Networks. J. Biol. Chem. 2009, 284, 27487–27499. [Google Scholar] [CrossRef] [Green Version]
- Enes Coşkun, M.; Kervancıoğlu, M.; Öztuzcu, S. Plasma microRNA profiling of children with idiopathic dilated cardiomyopathy. Biomarkers 2016, 21, 56–61. [Google Scholar] [CrossRef]
- Zeng, X.; Li, X.; Wen, H. Expression of circulating microRNA-182, CITED2 and HIF-1 in ischemic cardiomyopathy and their correlation. J. Clin. Cardiol. 2017, 33, 119–122. [Google Scholar]
- Olson, E.; Van Rooij, E. Dual Targeting of MIR-208 and MIR-499 in the Treatment of Cardiac Disorders. U.S. Patent 8,629,119, 14 January 2014. [Google Scholar]
- Li, X.; Liu, C.Y.; Li, Y.S. Deep RNA sequencing elucidates microRNA-regulated molecular pathways in ischemic cardiomyopathy and nonischemic cardiomyopathy. Genet. Mol. Res. 2016, 15, 9. [Google Scholar] [CrossRef]
- Shyu, K.-G.; Wang, B.-W.; Cheng, W.-P.; Lo, H.-M. MicroRNA-208a Increases Myocardial Endoglin Expression and Myocardial Fibrosis in Acute Myocardial Infarction. Can. J. Cardiol. 2015, 31, 679–690. [Google Scholar] [CrossRef] [PubMed]
- Cengiz, M.; Karatas, O.F.; Koparir, E.; Yavuzer, S.; Ali, C.; Yavuzer, H.; Kirat, E.; Karter, Y.; Ozen, M. Differential Expression of Hypertension-Associated MicroRNAs in the Plasma of Patients With White Coat Hypertension. Medicine 2015, 94, e693. [Google Scholar] [CrossRef] [PubMed]
- Li, J.-Z.; Tang, X.-N.; Li, T.-T.; Liu, L.-J.; Yu, S.-Y.; Zhou, G.-Y.; Shao, Q.-R.; Sun, H.-P.; Wu, C.; Yang, Y. Paeoniflorin inhibits doxorubicin-induced cardiomyocyte apoptosis by downregulating microRNA-1 expression. Exp. Ther. Med. 2016, 11, 2407–2412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, E.L.; Baker, A.H.; Brittan, M. Dissecting the transcriptome in cardiovascular disease. Cardiovasc. Res. 2021, 118. [Google Scholar] [CrossRef]
- Eulalio, A.; Mano, M.; Ferro, M.D.; Zentilin, L.; Sinagra, G.; Zacchigna, S.; Giacca, M. Functional screening identifies miRNAs inducing cardiac regeneration. Nature 2012, 492, 376–381. [Google Scholar] [CrossRef]
- Tian, Y.; Liu, Y.; Wang, T. A microRNA-Hippo pathway that promotes cardiomyocyte proliferation and cardiac regeneration in mice. Sci. Transl. Med. 2015, 7, 279ra38. [Google Scholar] [CrossRef] [Green Version]
- Porrello, E.R.; Mahmoud, A.I.; Simpson, E.; Johnson, B.A.; Grinsfelder, D.; Canseco, D.; Mammen, P.P.; Rothermel, B.A.; Olson, E.N.; Sadek, H.A. Regulation of neonatal and adult mammalian heart regeneration by the miR-15 family. Proc. Natl. Acad. Sci. USA 2013, 110, 187–192. [Google Scholar] [CrossRef] [Green Version]
- Turunen, T.A.; Roberts, T.C.; Laitinen, P.; Väänänen, M.-A.; Korhonen, P.; Malm, T.; Ylä-Herttuala, S.; Turunen, M.P. Changes in nuclear and cytoplasmic microRNA distribution in response to hypoxic stress. Sci. Rep. 2019, 9, 10332. [Google Scholar] [CrossRef] [Green Version]
- Porrello, E.R.; Mahmoud, A.I.; Simpson, E.; Hill, J.A.; Richardson, J.A.; Olson, E.N.; Sadek, H.A. Transient Regenerative Potential of the Neonatal Mouse Heart. Science 2011, 331, 1078–1080. [Google Scholar] [CrossRef] [Green Version]
- Bergmann, O.; Zdunek, S.; Felker, A.; Salehpour, M.; Alkass, K.; Bernard, S.; Sjostrom, S.L.; Szewczykowska, M.; Jackowska, T.; Dos Remedios, C. Dynamics of Cell Generation and Turnover in the Human Heart. Cell 2015, 161, 1566–1575. [Google Scholar] [CrossRef] [Green Version]
- Fasanaro, P.; D’Alessandra, Y.; Di Stefano, V.; Melchionna, R.; Romani, S.; Pompilio, G.; Capogrossi, M.C.; Martelli, F. MicroRNA-210 Modulates Endothelial Cell Response to Hypoxia and Inhibits the Receptor Tyrosine Kinase Ligand Ephrin-A3. J. Biol. Chem. 2008, 283, 15878–15883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaccagnini, G.; Greco, S.; Voellenkle, C.; Gaetano, C.; Martelli, F. miR-210 hypoxamiR in Angiogenesis and Diabetes. Antioxid. Redox Signal. 2021. [Google Scholar] [CrossRef] [PubMed]
- Noman, M.Z.; Buart, S.; Romero, P.; Ketari, S.; Janji, B.; Mari, B.; Mami-Chouaib, F.; Chouaib, S. Hypoxia-Inducible miR-210 Regulates the Susceptibility of Tumor Cells to Lysis by Cytotoxic T Cells. Cancer Res. 2012, 72, 4629–4641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haque, I.; Banerjee, S.; Mehta, S. Cysteine-rich 61-connective tissue growth factor-nephroblastoma-overexpressed 5 (CCN5)/Wnt-1-induced signaling protein-2 (WISP-2) regulates microRNA-10b via hypoxia-inducible factor-1α-TWIST signaling networks in human breast cancer cells. J. Biol. Chem. 2011, 286, 43475–43485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, Y.C.; Khanna, S.; Roy, S.; Sen, C.K. miR-200b Targets Ets-1 and Is Down-regulated by Hypoxia to Induce Angiogenic Response of Endothelial Cells. J. Biol. Chem. 2011, 286, 2047–2056. [Google Scholar] [CrossRef] [Green Version]
- Lei, Z.; Li, B.; Yang, Z.; Fang, H.; Zhang, G.-M.; Feng, Z.-H.; Huang, B. Regulation of HIF-1α and VEGF by miR-20b Tunes Tumor Cells to Adapt to the Alteration of Oxygen Concentration. PLoS ONE 2009, 4, e7629. [Google Scholar] [CrossRef] [Green Version]
- Vegter, E.L.; Van Der Meer, P.; De Windt, L.J.; Pinto, Y.M.; Voors, A.A. MicroRNAs in heart failure: From biomarker to target for therapy. Eur. J. Heart Fail. 2016, 18, 457–468. [Google Scholar] [CrossRef] [Green Version]
- Aguirre, A.; Montserrat, N.; Zacchigna, S.; Nivet, E.; Hishida, T.; Krause, M.N.; Kurian, L.; Ocampo, A.; Vazquez-Ferrer, E.; Rodriguez-Esteban, C.; et al. In Vivo Activation of a Conserved MicroRNA Program Induces Mammalian Heart Regeneration. Cell Stem Cell 2014, 15, 589–604. [Google Scholar] [CrossRef] [Green Version]
- Gomes, C.P.D.C.; Schroen, B.; Kuster, G.M. Regulatory RNAs in heart failure. Circulation 2020, 141, 313–328. [Google Scholar] [CrossRef]
- Sassi, Y.; Avramopoulos, P.; Ramanujam, D.P.; Grüter, L.; Werfel, S.; Giosele, S.; Brunner, A.-D.; Esfandyari, D.; Papadopoulou, A.-S.; De Strooper, B. Cardiac myocyte miR-29 promotes pathological remodeling of the heart by activating Wnt signaling. Nat. Commun. 2017, 8, 1614. [Google Scholar] [CrossRef]
- Huang, Y.; Li, W.; Wu, J.; Han, M.; Li, B. The diagnostic value of circulating microRNAs in heart failure (Review). Exp. Ther. Med. 2019, 17, 1985–2003. [Google Scholar] [CrossRef] [Green Version]
- Zeisel, J.; Bennett, K.; Fleming, R. World Alzheimer Report 2020: Design, dignity, dementia: Dementia-related design and the built environment. Published online. 2020.
- Alzheimer’s Association. Alzheimer’s Disease Facts and Figures. Alzheimer Dement. 2020, 16, 391–460. [Google Scholar] [CrossRef]
- Alzheimer’s Association. Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2019, 15, 321–387. [Google Scholar] [CrossRef]
- Barnard, N.D.; Bush, A.; Ceccarelli, A.; Cooper, J.; de Jager, C.A.; Erickson, K.I.; Fraser, G.; Kesler, S.; Levin, S.M.; Lucey, B.; et al. Dietary and lifestyle guidelines for the prevention of Alzheimer’s disease. Neurobiol. Aging 2014, 35, S74–S78. [Google Scholar] [CrossRef] [Green Version]
- Baumgart, M.; Snyder, H.M.; Carrillo, M.C.; Fazio, S.; Kim, H.; Johns, H. Summary of the evidence on modifiable risk factors for cognitive decline and dementia: A population-based perspective. Alzheimer’s Dement. 2015, 11, 718–726. [Google Scholar] [CrossRef] [Green Version]
- Knopman, D.S.; Haeberlein, S.B.; Carrillo, M.C.; Hendrix, J.A.; Kerchner, G.; Margolin, R.; Maruff, P.; Miller, D.S.; Tong, G.; Tome, M.B.; et al. The National Institute on Aging and the Alzheimer’s Association Research Framework for Alzheimer’s disease: Perspectives from the Research Roundtable. Alzheimer’s Dement. 2018, 14, 563–575. [Google Scholar] [CrossRef]
- Yoon, S.; Kim, S.E.; Ko, Y.; Jeong, G.H.; Lee, K.H.; Lee, J.; Solmi, M.; Jacob, L.; Smith, L.; Stickley, A.; et al. Differential expression of MicroRNAs in Alzheimer’s disease: A systematic review and meta-analysis. Mol. Psychiatry 2022. [Google Scholar] [CrossRef]
- Bush, A.I.; Tanzi, R.E. Therapeutics for Alzheimer’s disease based on the metal hypothesis. Neurotherapeutics 2008, 5, 421–432. [Google Scholar] [CrossRef] [Green Version]
- Zafar, A.; Singh, S.; Naseem, I. Cu(II)-coumestrol interaction leads to ROS-mediated DNA damage and cell death: A putative mechanism for anticancer activity. J. Nutr. Biochem. 2016, 33, 15–27. [Google Scholar] [CrossRef]
- Lutsenko, S.; Bhattacharjee, A.; Hubbard, A.L. Copper handling machinery of the brain. Metallomics 2010, 2, 596–608. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Jiang, X.; Lin, X.; Zhang, Z.; Wu, D.; Zhou, L.; Liu, J.; Yang, X. Hippocampal Subcellular Organelle Proteomic Alteration of Copper-Treated Mice. Toxicol. Sci. 2018, 164, 250–263. [Google Scholar] [CrossRef] [PubMed]
- Magenta, A.; Cencioni, C.; Fasanaro, P.; Zaccagnini, G.; Greco, S.; Sarra-Ferraris, G.; Antonini, A.; Martelli, F.; Capogrossi, M.C. miR-200c is upregulated by oxidative stress and induces endothelial cell apoptosis and senescence via ZEB1 inhibition. Cell Death Differ. 2011, 18, 1628–1639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.G.; Wang, J.L.; Li, L.; Xue, L.X.; Zhang, Y.Q.; Wang, P.C. MicroRNA-135a and-200b, potential Biomarkers for Alzheimer’ s disease, regulate β secretase and amyloid precursor protein. Brain Res. 2014, 1583, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Guedes, J.R.; Santana, I.; Cunha, C. MicroRNA deregulation and chemotaxis and phagocytosis impairment in Alzheimer’s disease. Alzheimer’s & Dementia: Diagnosis. Assess. Dis. Monit. 2016, 3, 7–17. [Google Scholar]
- Li, K.; Li, C.; Xiao, Y.; Wang, T.; Kang, Y.J. Featured Article: The loss of copper is associated with the increase in copper metabolism MURR domain 1 in ischemic hearts of mice. Exp. Biol. Med. 2018, 243, 780–785. [Google Scholar] [CrossRef] [Green Version]
- Xiao, Y.; Wang, T.; Song, X.; Yang, D.; Chu, Q.; Kang, Y.J. Copper promotion of myocardial regeneration. Exp. Biol. Med. 2020, 245, 911–921. [Google Scholar] [CrossRef]
- Li, C.; Wang, T.; Xiao, Y.; Li, K.; Meng, X.; Kang, Y.J. COMMD1 upregulation is involved in copper efflux from ischemic hearts. Exp. Biol. Med. 2020, 246, 607–616. [Google Scholar] [CrossRef]
- Opie, L.H. Heart Physiology: From Cell to Circulation; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2004. [Google Scholar]
- Cole, M.A.; Jamil, A.H.A.; Heather, L.C.; Murray, A.J.; Sutton, E.R.; Slingo, M.; Sebag-Montefiore, L.; Tan, S.C.; Aksentijević, D.; Gildea, O.S.; et al. On the pivotal role of PPARa in adaptation of the heart to hypoxia and why fat in the diet increases hypoxic injury. FASEB J. 2016, 30, 2684–2697. [Google Scholar] [CrossRef] [Green Version]
- Kerkelä, R.; Karsikas, S.; Szabo, Z.; Serpi, R.; Magga, J.; Gao, E.; Alitalo, K.; Anisimov, A.; Sormunen, R.; Pietilä, I.; et al. Activation of Hypoxia Response in Endothelial Cells Contributes to Ischemic Cardioprotection. Mol. Cell. Biol. 2013, 33, 3321–3329. [Google Scholar] [CrossRef] [Green Version]
- Fialho, M.D.L.S.; Jamil, A.H.A.; Stannard, G.A.; Heather, L.C. Hypoxia-inducible factor 1 signalling, metabolism and its therapeutic potential in cardiovascular disease. Biochim. Et. Biophys. Acta (BBA)-Mol. Basis Dis. 2018, 1865, 831–843. [Google Scholar] [CrossRef] [PubMed]
- Heriansyah, T.; Chomsy, I.N.; Kumboyono, K.; Pratiwi, P.A.; Wihastuti, T.A. Expression of Hypoxia-Inducible Factor-1α (HIF1A) and Lp-PLA2 in Low, Intermediate, and High Cardiovascular Disease Risk Population. Vasc. Health Risk Manag. 2020, 16, 507. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Hassan, H. Hypoxia in Alzheimer′s disease: Effects of hypoxia inducible factors. Neural Regen. Res. 2021, 16, 310–311. [Google Scholar] [CrossRef]
- Dingwall, C. A copper-binding site in the cytoplasmic domain of BACE1 identifies a possible link to metal homoeostasis and oxidative stress in Alzheimer′s disease. Biochem. Soc. Trans. 2007, 35, 571–573. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Targeting HIF-1 for cancer therapy. Nat. Cancer 2003, 3, 721–732. [Google Scholar] [CrossRef]
- Salceda, S.; Caro, J. Hypoxia-inducible Factor 1α (HIF-1α) Protein Is Rapidly Degraded by the Ubiquitin-Proteasome System under Normoxic Conditions. J. Biol. Chem. 1997, 272, 22642–22647. [Google Scholar] [CrossRef] [Green Version]
- Greco, S.; Gaetano, C.; Martelli, F. HypoxamiR Regulation and Function in Ischemic Cardiovascular Diseases. Antioxid. Redox Signal. 2014, 21, 1202–1219. [Google Scholar] [CrossRef] [Green Version]
- Qiao, A.; Khechaduri, A.; Mutharasan, R.K.; Wu, R.; Nagpal, V.; Ardehali, H. MicroRNA-210 Decreases heme Levels by Targeting Ferrochelatase in Cardiomyocytes. J. Am. Heart Assoc. 2013, 2, e000121. [Google Scholar] [CrossRef] [Green Version]
- Cicchillitti, L.; Di Stefano, V.; Isaia, E.; Crimaldi, L.; Fasanaro, P.; Ambrosino, V.; Antonini, A.; Capogrossi, M.C.; Gaetano, C.; Piaggio, G.; et al. Hypoxia-inducible Factor 1-α Induces miR-210 in Normoxic Differentiating Myoblasts. J. Biol. Chem. 2012, 287, 44761–44771. [Google Scholar] [CrossRef] [Green Version]
- Zeng, L.L.; He, X.S.; Liu, J.R.; Zheng, C.B.; Wang, Y.T.; Yang, G.Y. Lentivirus-mediated overexpression of microRNA-210 improves long-term outcomes after focal cerebral ischemia in mice. CNS Neurosci. Ther. 2016, 22, 961–969. [Google Scholar] [CrossRef]
- Mutharasan, R.K.; Nagpal, V.; Ichikawa, Y.; Ardehali, H. microRNA-210 is upregulated in hypoxic cardiomyocytes through Akt- and p53-dependent pathways and exerts cytoprotective effects. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H1519–H1530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burstein, E.; Hoberg, J.E.; Wilkinson, A.S.; Rumble, J.M.; Csomos, R.A.; Komarck, C.M.; Maine, G.N.; Wilkinson, J.C.; Mayo, M.W.; Duckett, C.S. COMMD Proteins, a Novel Family of Structural and Functional Homologs of MURR1. J. Biol. Chem. 2005, 280, 22222–22232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, H.; Kang, Y.J. Role of copper in angiogenesis and its medicinal implications. Curr. Med. Chem. 2009, 16, 1304–1314. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Zhang, W.; Kang, Y.J. Copper affects the binding of HIF-1α to the critical motifs of its target genes. Metallomics 2019, 11, 429–438. [Google Scholar] [CrossRef] [PubMed]
- Martin, F.; Linden, T.; Katschinski, D.M.; Oehme, F.; Flamme, I.; Mukhopadhyay, C.K.; Eckhardt, K.; Tröger, J.; Barth, S.; Camenisch, G.; et al. Copper-dependent activation of hypoxia-inducible factor (HIF)-1: Implications for ceruloplasmin regulation. Blood 2005, 105, 4613–4619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujie, T.; Takahashi, A.; Takahashi, M.; Hara, T.; Soyama, A.; Makino, K.; Takahashi, H.; Yamamoto, C.; Kumagai, Y.; Naka, H.; et al. Transcriptional Induction of Cystathionine γ-Lyase, a Reactive Sulfur-Producing Enzyme, by Copper Diethyldithiocarbamate in Cultured Vascular Endothelial Cells. Int. J. Mol. Sci. 2020, 21, 6053. [Google Scholar] [CrossRef] [PubMed]
- Klevay, L.M. Copper and ischemic heart disease. Biol. Trace Element Res. 1983, 5, 245–255. [Google Scholar] [CrossRef]
- van de Sluis, B.; Muller, P.; Duran, K. Increased activity of hypoxia-inducible factor 1 is associated with early embryonic lethality in Commd1 null mice. Mol. Cell. Biol. 2007, 27, 4142–4156. [Google Scholar] [CrossRef] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sacco, A.; Martelli, F.; Pal, A.; Saraceno, C.; Benussi, L.; Ghidoni, R.; Rongioletti, M.; Squitti, R. Regulatory miRNAs in Cardiovascular and Alzheimer’s Disease: A Focus on Copper. Int. J. Mol. Sci. 2022, 23, 3327. https://doi.org/10.3390/ijms23063327
Sacco A, Martelli F, Pal A, Saraceno C, Benussi L, Ghidoni R, Rongioletti M, Squitti R. Regulatory miRNAs in Cardiovascular and Alzheimer’s Disease: A Focus on Copper. International Journal of Molecular Sciences. 2022; 23(6):3327. https://doi.org/10.3390/ijms23063327
Chicago/Turabian StyleSacco, Anna, Fabio Martelli, Amit Pal, Claudia Saraceno, Luisa Benussi, Roberta Ghidoni, Mauro Rongioletti, and Rosanna Squitti. 2022. "Regulatory miRNAs in Cardiovascular and Alzheimer’s Disease: A Focus on Copper" International Journal of Molecular Sciences 23, no. 6: 3327. https://doi.org/10.3390/ijms23063327
APA StyleSacco, A., Martelli, F., Pal, A., Saraceno, C., Benussi, L., Ghidoni, R., Rongioletti, M., & Squitti, R. (2022). Regulatory miRNAs in Cardiovascular and Alzheimer’s Disease: A Focus on Copper. International Journal of Molecular Sciences, 23(6), 3327. https://doi.org/10.3390/ijms23063327